
Abstract
Cyanobacterial blooms in drinking water supply affect its quality, which ultimately impacts ecosystem and public health. Thus, this cross-sectional study was conducted to perform a preliminary study on cyanotoxins via analysis of samples collected only once from two sites during the month of peak algal bloom and to subsequently prompt a comprehensive risk assessment in a major drinking water source, Legedadi Reservoir, of Addis Ababa, the capital city of Ethiopia. Samples were collected during peak algal bloom month (January 2018) from two sampling sites, near the dam (S1) and at the center of the reservoir (S2). Identification and enumeration of phytoplankton taxa were done and the measurement of common hepatotoxin (MCs and NOD) concentrations was conducted using liquid chromatography-tandem mass spectrometry. In the reservoir, cyanobacteria made up to 98% of total phytoplankton abundance, with Dolichospermum and Microcystis spp, dominating the phytoplankton community. In these first cyanotoxin analyses conducted for a drinking water supply source in Ethiopia, six major MC variants, namely MC-dmRR, MC-RR, MC-YR, MC-dmLR, MC-LR, and MC-LA, were detected in both algal seston and water samples. MC-LR was the most dominant MCs variant, while nodularin was not detected for both sampling sites. Extracellular total MC concentrations (μg L−1) of 453.89 and 61.63 and intracellular total MC concentrations (μg L−1) of 189.29 and 112.34 were recorded for samples from S1 and S2, respectively. The high concentrations of extracellular MCs, with MC-LR constituting the greatest proportion, indicate the extremely high potential public health risk for end-users.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In developing countries like Ethiopia, surface water bodies are under severe threat due to uncontrolled disposal of industrial waste and application of chemical fertilizers on agricultural lands located near water bodies [1]. Industrial wastewaters and agricultural runoff ultimately elevate nutrient levels and promote cultural eutrophication of lakes/reservoirs [2]. Eutrophication of drinking water source reservoir degrades water quality through the induction of hazardous cyanobacterial blooms [3]. Climate change-induced high temperature further enhances the dominance of cyanobacterial species in reservoirs/lakes [4].
Cyanobacterial blooms in a drinking water supply source reduce water quality by increasing turbidity, affecting taste and odor, and producing toxins, which eventually influence ecosystems and affect public health [5]. Different environmental factors affect cyanotoxin production. For instance, temperature, light intensity, pH, nutrients, salinity, ultraviolet radiation, wind, trace metals, and environmental pollutants influence cyanotoxin production. [6, 7] pointed out that nutrient limitation (specifically N-limitation) and light regulate mcy gene and hepatotoxin production. [8] reported that when the concentration of nitrate increases, MC production was increased in non-nitrogen-fixing species.
According to [9, 10], a shortage of trace metals (iron, zinc, copper, and magnesium) in aquatic systems promotes the production of intracellular cyanotoxins as a means of acquisition or storage. When there is a high level of trace metal that leads to toxification in an aquatic system, cyanobacteria produce extracellular toxins to form metal complexes and detoxify metals [11].
Hepatotoxins (MCs and NODs), which are the most common toxic substances associated with blooms, are similar in structure, mode of action and are produced non-ribosomally from mcy and nda gene clusters, respectively [12].
MCs are cyclic heptapeptides, while NODs are cyclic pentapeptides; however, they both have the common amino acid, ADDA (β-amino acid 3-amino-9-methoxy-2,6,8-trimethyl10-phenyldeca-4,6-dienoic acid), the most unusual substructure, which has not been reported except in MCs and NODs [13]. MC-LR and NOD-R are the most abundant congeners of MCs and NODs, respectively [13, 14]. MCs are stable and resistant to high temperature, extreme pH, sunlight and presence of common hydrolytic enzymes due to their cyclic structure [15]. These cyanotoxins are mostly associated with the acute intoxication of humans and livestock [3].
MCs are mainly liver toxicants causing inhibition of eukaryotic serine/threonine protein phosphatases 1 and 2A, which trigger extreme phosphorylation of cytoskeletal filaments leading to serious liver injury. Long-term exposure causes the promotion of liver tumors. Nodularins cause gastroenteritis, allergic irritation reactions, and liver diseases. Nodularin-R is the most notorious hepatotoxin that causes severe liver damage in humans and other animals [16, 17]. The WHO’s tolerable limits for Nodularins and Microcystins-LR concentrations (μg /L) in drinking water are 1.5 and 1.0, respectively [18]. The lethal dose 50 (LD50) of MC-LR is 25 to 150 μg/kg, while the LD50 for NOD-R is 50 to 150 μg/kg [19].
Under bloom condition, the problem becomes more pronounced in drinking water supply sources as conventional drinking water treatments are ineffective in removing these toxins [20].
Legedadi Reservoir, the subject of the study reported here, is the major drinking water supply source for Addis Ababa, the capital city of Ethiopia. Year-round occurrence and dominance of potentially toxic cyanobacterial species in this reservoir have been reported by [21]. However, efforts made to restore the reservoir using environmentally sound and safe restoration techniques are virtually absent, while the application of algaecides like copper sulfate has been a common practice in Legedadi Reservoir. The main drawback of using copper sulfate as an algaecide is its induction of cyanobacterial cell lysis with consequent release of intracellular toxins [22].
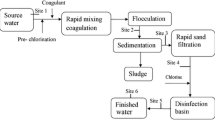
Thus, the application of copper sulfate in a reservoir supporting toxic cyanobacterial blooms can lead to enormous public health risk. Furthermore, the Legedadi Reservoir’s water supply system has conventional methods of water treatment including pre-chlorination (3.06 mg/l), coagulation (main coagulant polydimethyl-diallyl-ammonium-chloride), sedimentation, filtration, and post-chlorination (2.8 mg/l). This conventional treatment system is not effective in removing extracellular cyanotoxins [23], and the coagulants used in the treatment further induce cell lysis and consequent release of intracellular toxins [24]. All these factors incite high public health concern for using Legedadi Reservoir as drinking water supply source.
So far, few studies [25,26,27,28] have quantified MCs in a freshwater hydropower reservoir in Ethiopia. However, information on the concentrations of cyanobacterial toxins and associated public health risk in drinking water supply source reservoirs such as Legedadi Reservoir is nonexistent. Cyanobacterial blooms and cyanotoxins in drinking water sources are serious problems, particularly in countries like Ethiopia, where more than 62 million people lack access to safe water and alternative sources of drinking water supply are inadequate [29]. Thus, there was the need to undertake public health risk assessment of using water from Legedadi Reservoir. Therefore, this preliminary cross-sectional study on cyanotoxins was conducted via the analysis of samples collected once from two sites during the time of peak algal bloom, and to subsequently prompt a comprehensive risk assessment in a major drinking water source, Legedadi Reservoir.
2 Materials and methods
2.1 Study area
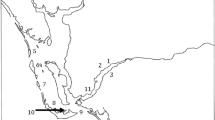
Legedadi Reservoir, one of the major drinking water supply sources for the capital city of Ethiopia, Addis Ababa, is located approximately 25 km east of Addis Ababa. It is found at an altitude of 2450 m, and geographical position of 9° 01′ N–9° 13′ N latitude and l38° 60′ E–39° 07′ E longitude (Fig. 1).

Location map of the study area with sampling sites
The reservoir was constructed in 1967 and is administered by Addis Ababa Water and Sewerage Authority (AAWSA). It had an initial storage capacity of 45.9 million cubic meters and surface area of 5.33 km2 [30]. The maximum and mean depths of the reservoir at 2466 meter above sea level are 30 and 4 m, respectively.
The reservoir has an outflow rate (to water treatment plant) of 126,666 (m3/day) and water retention time of 325 days [30]. The catchment of the reservoir has a total area of 205.7 km2. It is a sub-catchment of the Akaki River basin, which flows in a northeast–southwest direction and forms the northwest corner of the Awash River basin. The reservoir's catchment has an annual sediment yield of 845 t/km2/year. Expectedly, the huge sediment transport is accompanied by associated high nutrient load on the reservoir.
2.2 Sampling protocol
Two sampling sites were carefully selected considering the areas of frequent surface scum appearance and application of CuSO4 by Addis Ababa water and sanitation authority (AAWSA).
The first site (S1) was located near the dam (deepest part of the reservoir), while the second one (S2) was located at the center of the reservoir. Due to budget limitations, sample collection was conducted only once during the peak bloom month (January 2018) using a Van Dorn bottle sampler Horizontal Model (Eijkelkamp Soil & Water supplier) to document baseline information on the contamination of the reservoir water with cyanotoxins. Samples were taken from the surface and 0.25 m depth and mixed in equal proportions to produce a composite sample. For the identification of major cyanobacterial genera, the composite samples were properly mixed, and 1000 ml aliquots were dispensed into bottles and fixed with Lugol’s iodine (0.01% v/v).
2.3 In situ measurements
The transparency of the water (Secchi depth, ZSD) was estimated using a 20-cm-diameter black and white Secchi disk (United Scientific Supplies SCDSK1). Turbidity was measured using portable turbidity meter (model HI 93 703–11). Temperature and dissolved oxygen (DO) were measured using a portable digital oxygen meter (EUTECH instruments, model DO300), pH was measured using digital pH meter (HANNA instruments, model HI 9024), while conductivity was determined using multi-parameter meter (HQ40d). The specific conductivity (K25) was calculated according to [31].
2.4 Analysis of physicochemical parameters
Total suspended solids (TSS) were determined gravimetrically using a known volume of properly mixed sample, which was filtered on pre-weighed glass fiber filter paper (GF/F) and pre-dried at 105 °C to constant weight to measure total suspended solids [32]. The titration method was used to estimate total alkalinity (TA) and phenolphthalein alkalinity (PA). Water samples were titrated with 0.2 N H2SO4 using phenolphthalein and bromocresol green-methyl red indicators within a few hours after sample collection according to [33] and expressed in meq L−1. Except for total phosphorus (TP) and ammonia (NH3 + NH4+-N), water samples were filtered with GF/F and used for spectrophotometric determination of inorganic nutrients. The ascorbic acid method was used to determine the soluble reactive phosphate- phosphorus (SRP) and total phosphorus (TP) after persulfate digestion [32]. The sodium salicylate method was used to determine nitrate (NO3-N), while the molybdosilicate method was used to estimate silica (SiO2) [32]. Finally, the phenate method was used to estimate ammonia [33].
2.5 Identification and enumeration of phytoplankton taxa
Samples were well mixed and 100 ml aliquots were transferred to 1000 ml measuring cylinders and kept in the dark for 24 h. Then, 90 ml of the supernatant was carefully drawn off. The remaining 10 ml was mixed properly and 1 ml was placed in a Sedgewick-Rafter cell and allowed to settle. Identification to genus/species level was done using various identification guides, e.g., [34, 35]. According to [36], cell abundance was estimated by counting cells of algal units encountered in 40–50 grids.
where N is the number of cells or units counted, A is area of field (area of each grid, 1mm2), D is depth of Sedgwick-Rafter chamber (1 mm), and F is number of fields/grids counted.
2.6 Estimation of phytoplankton biomass
Phytoplankton biomass was estimated as chlorophyll-a (Chl-a) concentration (μg L−1) determined using a phytoplankton analyzer (PHYTO-PAM, Heinz Walz GmbH, Effeltrich, Germany).
2.7 Identification and quantification of cyanotoxins
Water samples were filtered using 47 mm GF/F filters. The seston retained by the filters was covered with aluminum foil and placed in Petri dishes and then kept in a freezer until analysis was conducted. The filtered water samples were kept in a freezer over night before analyses were conducted.
2.7.1 Extraction of MCs and NOD from algal seston
Extraction of MCs and NOD was done according to [37]. First, known volume of sample was filtered through a glass microfiber filter papers (47 mm GF/F filters). Then, the filter papers that retained algal seston were folded in half and put in a 10-ml glass tube and freeze-dried on a freeze-drier (Alpha 1–2 LD, Martin Christ Gefriertrocknung sanlagen GmbH, Osterode am Harz, Germany) for two hrs. After that, 2.5 ml of 75% methanol-25% Millipore water (v/v) was used to extract sample three times in a water bath at 60 °C using the glass pipette squeeze method. Then, 7.5 ml samples on the speedvac tube were dried up using a Speedvac (Savant SPD121P, Thermo Scientific, Waltham, MA, USA) centrifuge pre-heat at 50 °C. Then, 300 μl of 100% MeOH was used to reconstitute the sample three times after well vortexed and mixed. Afterward, the reconstituted 900μL samples were transferred to 2 ml Eppendorf vials with a cellulose-acetate filter (0.2 μm, Grace Davison Discovery Sciences, Deerfield, IL, USA) and centrifuged for 5 min at 16,000 × g (Galaxy 16DH, VWR, International). Finally, filtrates were transferred to amber glass vials and caps were tightly closed and stored in a freezer till LC–MS/MS analysis. Prior to LC–MS/MS analysis, calibration standards were prepared [38].
2.7.2 Extraction of extracellular microcystins and nodularin
Filtrates of water samples filtered using 47 mm GF/F filters and stored in a freezer were dried in a Speedvac (Savant SPD121P, pre-heat centrifuge at 50 °C). Then, 300 μl of 100% MeOH was used to reconstitute the sample three times after well vortexed and mixed. Afterward, the reconstituted 900μL samples were transferred to 2-ml Eppendorf vials with a cellulose-acetate filter (0.2 μm, Grace Davison Discovery Sciences, Deerfield, IL, USA) and centrifuged for 5 min at 16,000 × g (Galaxy 16DH, VWR, International). Finally, filtrates were transferred to amber glass vials and caps were tightly closed and stored in a freezer till LC–MS/MS analysis. Prior to LC–MS/MS analysis, calibration standards were prepared [38].
2.7.3 LC–MS/MS analyses
The concentrations of nine MC variants (dm-7-MC-RR, MC-RR, MC-YR, dm-7-MC-LR, MC-LR, MC-LA, MC-LY, MC-LW, and MC-LF) and nodularin (NOD) were examined by LC–MS/MS according to the procedure of [39]. LC–MS/MS analyses were done using an Agilent 1200 LC and an Agilent 6410A QQQ (Agilent Technologies, Santa Clara, CA, USA). MCs variants were separated on an Agilent Eclipse XDB-C18 4.6_150 mm, 5 m columns. Calibration standards were obtained from DHI LAB Products (Hørsholm, Denmark) [37] (supplementary material). The sample was injected with a flow rate of 0.5 ml/min; the column temperature was 40 °C. Eluents were Millipore water with 0.1% formic acid (v/v, Eluent A) and acetonitrile with 0.1% formic acid (v/v, Eluent B) that were run using an elution program of 0–2 min 30% B, 6–12 min 90% B, with a linear increase of B between 2 and 6 min and a 5-min post run at 30% B. Detailed information on MS/MS settings for each MC variant and NOD are shown in (supplementary material). Recovery of sample and analysis was estimated by spiking a cyanobacterial matrix in triplicates: The recovery rate was 100% for MC-LA, 99.5% for DM-7-MC-RR, 96.1% for MC-RR, 75% for MC-YR, 77.8% for DM-7-MC-RR, 79.1 for MC-LR, 72.8% for MC-LY, 53.5% for MC-LW, and 63.9% for MC-LF. Furthermore, repeatability, the limit of detection and limit of quantification of LC–MS/MS analyses are described in [38].
3 Results
3.1 Physicochemical parameters
The clarity of the reservoir, expressed in terms of mean Secchi depth (cm), was 11 and 10 at S1 and S2, respectively. The mean levels of turbidity of the reservoir (NTU) were 311 and 315 at S1 and S2, respectively, while those of TSS (mg L–1) were 0.24 at both S1 and S2. The mean water temperature (°C) of S1 and S2 were 17.86 and 17.5 respectively. The reservoir was well oxygenated, with mean DO concentrations (mg L−1) of 7.22 and 6.85 at S1 and S2, respectively.
The mean concentrations (µg L−1) of SRP at S1 and S2 were 166.6 and 130, respectively (Table 1) while the mean total phosphorous (TP) concentrations (µg L−1) were 597 and 605 at S1 and S2, respectively (Table 1). The mean concentrations (µg L−1) of nitrate at S1 and S2 were 276 and 314, respectively, while mean concentration (µg L−1) of ammonia was 3.06 at S1 and 3.09 at S2.
3.2 Major phytoplankton taxa
Cyanobacteria constituted 98% and 93.5% of total phytoplankton species at S1 and S2, respectively. Cryptophyta and Chlorophyta were the second and third most abundant groups at both sampling sites, with percentage contributions of 0.73% and 0.64% at S1 and 3.9% and 1.66% at S2, respectively (Fig. 2).

Relative abundance of major algal groups
Eleven species of potentially toxic cyanobacteria were identified in samples at the two sampling sites of the reservoir (Table 2). The most dominant cyanobacterial genera in this study were Dolichospermum and Microcystis (Fig. 3), with cell densities of 6,951,000 cells L−1 and 1,918,000 cells L−1, respectively. The percentage contributions of the dominant species, Dolichospermum flos-aquae, Dolichospermum spiroides and Microcystis aeruginosa, to the total cyanobacterial abundance were 48.3%, 14.3%, and 12.5%, respectively (Fig. 4a).

Relative abundance of the major cyanobacterial species expressed as percentage of the total cyanobacterial abundance

Total microcystins concentrations in relation to the total abundance of microcystin-producing species
3.3 Cyanotoxins
LC–MS/MS analyses of cyanobacterial extracts revealed the occurrence of substantial levels of extracellular MCs (μg L−1) of 453.89 and 61.63 and of intracellular MCs (μg L−1) of 189.29 and 112.34 at S1 and S2, respectively (Table 3). The observed results indicate that total extracellular MCs concentration at S1 was higher than the total intracellular MC concentration at S2 (Fig. 4b).
Six variants of MC were identified in Legedadi Reservoir, the main MC variants being MC-dmRR, MC-RR, MC-YR, MC-dmLR, MC-LR, and MC-LA. The concentrations of all MC's variants except MC-LA were higher at S1 than at S2.
MC-LR was the most dominant MC variant, with percentage contributions to the total MC concentrations (μg L−1) of 72.6% and 74.9% in water and 75.2% and 59.4% in algal seston at S1 and S2, respectively. Among the different MC variants, MC-YR was the second most dominant MC's congener in water and algal seston samples at both sampling sites. The percentage contributions of MC-YR to total MCs were 12.7% and 15.3% in water samples and 9.3% and 13.2% in algal seston samples, at S1 and S2, respectively. Nodularin was not detected in any of the samples analyzed.
4 Discussion
The present results show that the reservoir had unusually high concentration of nutrients during the sampling time. The levels of nutrients recorded during the present study were high compared to those commonly recorded for tropical lakes including those reported previously for this study reservoir [21] and Koka Reservoir in Ethiopia [26, 28] during the sampling time. The present high levels of nutrients are associated with the high anthropogenic disturbance occurring in the catchment areas of the reservoir and the consequent nutrient-rich runoff to the reservoir. Moreover, the abundance and dominance of cyanobacterial species observed in this study have resulted primarily from the availability of high levels of nutrients. Total phosphorus and low water clarity were identified as the most important environmental factors associated with phytoplankton community structure [40].
In the case of high reservoir turbidity, the conflicting results were reported. Some studies have suggested that cyanobacteria can out-compete other phytoplankton species in turbid systems [41, 42], while others claim that the high level of non-algal turbidity reduces cyanobacterial growth [43, 44]. Yet, the gas vacuoles in cyanobacteria give them a competitive advantage to rendering them buoyant thereby enabling them to regain their vertical position where efficient photosynthesis is possible. Therefore, turbidity in water bodies cannot adversely affect cyanobacteria.
In the reservoir, there was a higher level of total extracellular MCs than total intracellular MCs. This extracellular MCs concentration could even be much higher when considering the argument by [28] that the procedure currently in use does not enable us to accurately and exclusively determine the two fractions, as MCs get bound to surfaces of suspended particulate matter and are subsequently retained by the filter paper. These particle-bound MCs, which are part of the extracellular fraction, are not available for inclusion in the measurement of MCs in the filtrate. This eventually results in an underestimation of the extracellular MCs concentration. The total concentration of extracellular MCs at S1 was more than twice that of the intracellular MCs. The higher concentration of MCs at S1 may have resulted from the application of CuSO4 by AAWSA in the reservoir at S1. CuSO4 induces cell lysis and the consequent release of MCs. In a healthy cyanobacterial bloom, the highest proportion (98%) of microcystins is retained within the cells [45], while cell damage and death will lead to release of intracellular toxins to the environment [13].
The presence of such high concentrations of extracellular toxins in the reservoir and the ineffectiveness of the conventional water treatment process in removing the cyanotoxins from treated water causes a huge public health risk to the end-user. Furthermore, the primary oxidants such as potassium permanganate and chlorine could oxidize cyanotoxins in water bodies to form harmful by-products of trihalomethanes and mutagenic furanones [46].
The concentrations of MCs in this study are noticeably greater than those reported for most African lakes and reservoirs, including Lake Chivero, Zimbabwe [0.1–1.6 μg L−1; [47], Ugandan freshwaters [0.02–10 μg L−1; [48] and Koka Reservoir in Ethiopia [1.71 to 33 μg L−1, [28]. Freshwater cyanobacterial species including Dolichospermum, Fischerella, Gloeotrichia, Microcystis, Nodularia, Nostoc, Oscillatoria, and Planktothrix produce MCs, which are common and tremendously toxic secondary metabolites in water bodies [49]. Therefore, any of cyanobacterial species identified in this study could be responsible for the observed MC concentration in the reservoir. To specifically identify which cyanobacterial genus was responsible for the toxin production, further molecular analysis of mcy encoding gene is obligatory. However, the few studies conducted on cyanotoxins in Koka Reservoir in Ethiopia have associated cyanotoxins production mainly with Microcystis and Dolichospermum spp. [25,26,27,28].
The largest portion of total MCs was constituted by MC-LR, which fundamentally causes a great concern for the end-user as MC-LR has been identified as the most potent toxin and chemically stable in water [50]. Acute exposure to MC-LR leads to massive intra-hepatic hemorrhage, liver swelling, and death, whereas chronic exposure causes genotoxicity and carcinogenicity [51]. World health organization has also categorized MC-LR as “probably carcinogenic for humans" [52].
Conventional treatment systems cannot effectively remove the stable cyclic structure of MC-LR [53]. Therefore, the occurrence of high levels of extracellular MC-LR in the reservoir and the ineffectiveness of the treatment system in removing the toxin from the water indicate the high potential public health risk for the end-users.
The concentration of extracellular MC-LR was about 330 times higher than the permissible level established for drinking water [1 μg L−1, [18]. MC-LR is relatively persistent in the aquatic environment, after algaecide treatment [54]. Jones and Falconer [54] observed the presence of MC-LR up to 21 days after algaecide treatment of Microcystis aeruginosa bloom. This fact reflects the serious health risk associated with the use of Legedadi Reservoir as a source of drinking water supply as year-round occurrence and dominance of cyanobacteria has been observed in the reservoir. The use of CuSO4 by water quality managers to control the year-round occurrence of cyanobacterial bloom has exacerbated the problem.
Arginine-containing microcystins congeners (MC-dmRR and MC-RR) were detected at lower concentrations in algal seston and water samples from S1. Furthermore, they were not detected in samples from S2. The use of GF/C filter paper during sample extraction could be responsible for the observed low concentration of MC-dmRR and MC- RR. According to [55], compared to those samples whose filtration involved use of filter papers of other grades, in GF/C filtered samples, all microcystins that contained arginine were present at significantly lower concentrations because arginine-containing microcystins adhere to GF/C filters.
Nodularin was not detected in samples from both sites. This could be due to the fact that the genus Nodularia, the most common NOD producer, was not encountered in samples examined. Moreover, nodularin is also more commonly detected in brackish water than freshwater water [56]. Yet, [57] detected trace amounts of NOD in the absence of Nodularia sp. and nodularin was also detected in freshwater bodies dominated by microcystis spp. [58, 59].
Public health risk assessment of Legedadi Reservoir indicated the occurrence of extracellular MC-LR concentrations (μg L−1) of 329.07 and 46.2 at S1 and S2, respectively, and intracellular MC-LR concentrations (μg L−1) of 142.37 and 67.12 at S1 and S2, respectively. These concentrations are much higher than the WHO guideline value for drinking water. The occurrence of high concentration of MC-LR near the raw water intake site (S1) and the application of chlorine in the water treatment process twice, at pre-chlorination (3.06 mg/L) and post-chlorination (2.8 mg/L), induces cyanobacterial cell lysis and the consequent release of MCs to the treated water could immensely augment the public health risk. This reservoir provides 52% of the domestic water supply for 2.75 million people. Therefore, the public health risk of using Legedadi Reservoir as a source of drinking water supply is great. Furthermore, the local communities in the catchment area of the reservoir directly use the reservoir water without further treatment for domestic purposes and this causes great concern for the health of local inhabitants.
5 Conclusion
This preliminary study revealed the occurrence of high concentrations of intracellular and extracellular MCs in the reservoir. The high concentrations of total extracellular MCs, especially at S1, could be associated with the application of CuSO4 and the associated cell lysis. Additionally, the high levels of intracellular and extracellular MC in the reservoir indicate the high potential public health risk associated with the use of the Legedadi Reservoir as a source of drinking water supply. The concentrations of intracellular and extracellular microcystins (total MCs and MC-LR) are much higher than the permissible level set by WHO for drinking water (1 μg L−1). Moreover, the occurrence of high concentrations of extracellular MCs constituted largely by MC-LR at both sites and the inefficiency of the conventional treatment system in removing microcystins from treated water are suggestive of the high potential public health risk.
Despite the limited data, our findings provide information on the possible public health risk of MC in the drinking water source. The results are expected to serve as an alarm for the water quality manager of the reservoir signaling the need to undertake year-round water quality monitoring of cyanotoxins and to look for effective and environmentally safe algal bloom control methods and water treatment system that can effectively remove cyanotoxins from the treated water.
The study is a cross-sectional survey and could not designate the observed toxin levels as maximum or minimum concentrations. Therefore, systematic monitoring and analysis of MC throughout the year for at least a couple of years along with quantitative assessment of dominant species connected to toxin production are required.
Code availability
Not applicable.
References
GebreMariam Z, Desta Z (2002) The chemical composition of the effluent from Awassa Textile Factory and its effects on aquatic biota. SINET 25(2):263–274
He X, Liu YL, Conklin A, Westrick J, Weavers LK, Dionysiou DD, Lenhary JL, Mouser PJ, Szlag D, Walker HW (2016) Toxic cyanobacteria and drinking water: impacts, detection and treatment. Harmful Algae 54:174–193
Codd GA, Morrison LF, Metcalf JS (2005) Cyanobacterial toxins: risk management for health protection. Toxicol Appl Pharmacol 203:264–272
Paerl HW, Hall NS, Peierls BL, Rossignol KL (2014) Evolving paradigms and challenges in estuarine and coastal eutrophication dynamics in a culturally and climatically stressed world. Estuaries Coasts 37(2):243–258
Shen XQ, Zhu J, Cheng L, Zhang J, Zhang Z, Xu X (2011) Enhanced algae removal by drinking water treatment of chlorination coupled with coagulation. Desalination 271:236–240
Neilan BA, Pearson LA, Muenchhoff J, Moffitt MC, Dittmann E (2013) Environmental conditions that influence toxin biosynthesis in cyanobacteria. Environ Microbiol 15:1239–1253
Kaplan A, Harel M, Kaplan-Levy RN, Hadas O, Sukenik A, Dittmann E (2012) The languages spoken in the water body (or the biological role of cyanobacterial toxins). Front Microbiol 3:138:1–138:11
Downing TG, Sember CS, Gehringer MM, Leukes W (2005) Medium N: P ratios and specific growth rate comodulate microcystin and protein content in Microcystis aeruginosa PCC7806 and M. aeruginosa UV027. Microb Ecol 49:468–473
Polyak Y, Zaytseva T, Medvedeva N (2013) Response of toxic cyanobacterium Microcystis aeruginosa to environmental pollution. Water Air Soil Pollut 224:1–14
Yeung ACY, D’Agostino PM, Poljak A, McDonald J, Bligh MW, Waite TD, Neilan BA (2016) Physiological and proteomic responses of continuous cultures of Microcystis aeruginosa PCC 7806 to changes in iron bioavailability and growth rate. Appl Environ Microbiol 82:5918–5929
Martínez-Ruiz EB, Martínez-Jerónimo F (2016) How do toxic metals affect harmful cyanobacteria? An integrative study with a toxigenic strain of Microcystis aeruginosa exposed to nickel stress. Ecotoxicol Environ Saf 133:36–46
Thangavelu B, Jang-Seu K (2014) Impact of environmental factors on the regulation of cyanotoxin production. Toxins 6:1951–1978
Carmichael WW, Boyer GL (2016) Health impacts fromcyanobacteria harmful algae blooms: implications for the North American Great Lakes. Harmful Algae 54:194–212
Chen Y, Shen D, Fang D (2013) Nodularins in poisoning. Clin Chim Acta 425:18–29
Chorus I, Bartram J (1999) Toxic cyanobacteria in water. E & FN Spon, London
Dai R, Liu H, Qu J, Ru J, Hou Y (2005) Cyanobacteria and their toxins in Guanting Reservoir of Beijing, China. Hazard Mater 153:470–477. https://doi.org/10.1016/j.jhazmat.2007.08.078
Melaram R (2019) Microcystins and daily sunlight: predictors of chronic liver disease and cirrhosis mortality. J Environ Sci Public Health 3(3):379–388. https://doi.org/10.26502/jesph.96120070
WHO (World Health Organization) (2017) Guidelines for drinking-water quality, 4th edn. World Health Organization, Geneva
Kuiper-Goodman T, Falconer I, Fitzgerald J (1999) Human health aspects. In: Chorus I, Bartram J (eds) Toxic cyanobacteria in water: a guide to their public health consequences, monitoring and management. E & FN Spon Publishers, London, p 36
Pestana CJ, Reeve PJ, Sawade E, Voldoire CF, Newton K, Praptiwi R, Newcombe G (2016) Fate of cyanobacteria in drinking water treatment plant lagoon supernatant and sludge. Sci Total Environ 5:173. https://doi.org/10.1016/j.scitotenv.2016.05.173
Sirage A (2006) Water quality and phytoplankton dynamics in Legedadi Reservoir, Ethiopia. M.Sc. Thesis, Addis Ababa University
Merel S, Walker D, Chicana R, Snyder S, Baurès E, Thomas O (2013) State of knowledge and concerns on cyanobacterial blooms and cyanotoxins. Environ Int 59:303–327. https://doi.org/10.1016/j.envint.2013.06.013
Hoeger SJ, Hitzfeld BC, Dietrich DR (2005) Occurrence and elimination of cyanobacterial toxins in drinking water treatment plants. Toxicol Appl Pharm 203:231–242
Lam AKY, Prepas EE, Spink D, Hrudey SE (1995) Chemical control of hepatotoxic phytoplankton blooms: implications for human health. Water Res 29:1845–1854
Willén E, Ahlgren G, Tilahun G, Spoof L, Neffling M, Meriluoto J (2011) Cyanotoxin production in seven Ethiopian Rift Valley lakes. Inland Waters 1(2):81–91
Major Y, Kifle D, Spoof L, Meriluoto J (2018) Cyanobacteria and microcystins in Koka reservoir (Ethiopia). Environ Sci Pollut Res 25:26861–26873
Zewde TW, Johansen JA, Kefile DK, Demissie BT, Hansen JH (2018) Toxicon concentrations of microcystins in the muscle and liver tissues of fi sh species from Koka reservoir Ethiopia: a potential threat to public health. Toxicon 153:85–95
Tilahun S, Kifle D, Zewde TW, Johansen JA, Demissie TB, Hansen JH (2019) Toxicon temporal dynamics of intra-and extra-cellular microcystins concentrations in Koka reservoir (Ethiopia): implications for public health risk. Toxicon 168:83–92. https://doi.org/10.1016/j.toxicon.2019.06.217
https://doi.org/10.1007/s11160-014-9366-6. Accessed 5 Oct 2020
DARAL-OMRAN (2011) Urban water supply and sanitation project: consultancy service for master plan review, catchment rehabilitation and awareness creation for Geffersa. Federal Democratic Republic of Ethiopia, Legedadi
Talling J (1965) The chemical composition of African lake waters. Int Rev Ges Hydrobiol 50:421–463
APHA (1992) Method 4500-CL: standard methods for the examination of water and wastewater 552(4)
Wetze RG, Likens GE (2000) Limnological analyses. (C. E. Services, Ed.) (3rd edn). Springer, New York
Cronberg K (2004) Some nostocalean cyanoprokaryotes from lentic habitats of eastern and southern Africa. Nova Hedwigia 78:71–106
Komárek J, Anagnostidis K (2005) Cyanoprokaryota 2: Oscillatoriales. In: Büdel B, Gärtner G, Krienitz L, Schagerl M (eds) SüsswasserfloravonMitteleuropa 19.2. Elsevier, Heidelberg
Hotzel CR (1999) A phytoplankton methods manual for Australian freshwaters
Lürling M, Faassen EJ (2013) Dog poisonings associated with a Microcystis aeruginosa Bloom in the Netherlands. Toxin 5:556–567
Elisabeth JF, Lürling M (2013) Occurrence of the microcystins MC-LW and MC-LF in Dutch surface waters and their contribution to total microcystin toxicity. Mar Drugs 11:2643–2654
Kondo F, Ikai Y, Oka H, Okumura M, Ishikawa N, Harada KI, Matsuura K, Murata H, Suzuki M (1992) Formation, characterization, and toxicity of the glutathione and cystein conjugates of toxic hepatopeptide microcystins. Chem Res Toxicol 5:91–596
Lu H, Campbell E, Campbell DE, Wang C, Ren H (2017) EPA public access. Atmos Environ 23:248–258. https://doi.org/10.1016/j.hal.2017.06.001
Scheffer M, Rinaldi S, Gragnani A, Mur LR, van Nes E (1997) On the dominance of filamentous cyanobacteria in shallow turbid lakes. Ecology 78:272–282
Burkholder JM, Larsen LM, Glasgow HBJ, Mason KM, Gama PPJ (1998) Influence of sediment and phosphorus loading on phytoplankton communities in an urban piedmont reservoirs. Lake Reserv Manag 14:110–121
Cuker BE, Gama PT, Burkholder J (1990) Type of suspended clay influences lake productivity and phytoplankton community response to phosphorus loading. Limnol Oceanogr 35:822–830
Andrew RMC, DeNoyelles MF, Dzialowski J, Smith VH, Wang SH (2011) Effects of non-algal turbidity on cyanobacterial biomass in seven turbid Kansas reservoirs, lake and reservoir management. Lake Reserv Manag 27(1):6–14
Chorus JI, Falconer IR, Salas HJ, Bartram J (2000) Health risks caused by freshwater cyanobacteria in recreational waters. Toxicol Environ 3:323–347
De la Cruz AA, Antoniou MG, Pelaez M, Hiskia A, Song W, O’Shea KE, He X, Dionysiou DD (2011) Can we effectively degrade microcystins? Implications for impact on human health status. Anti-Cancer Agents Med Chem 11:19–37
Mhlanga AHL, Day J, Cronberg G, Chimbari M, Siziba N (2006) Cyanobacteria and cyanotoxins in the source water from Lake Chivero, Harare, Zimbabwe, and the presence of cyanotoxins in drinking water. Afr J Aquat Sci 31:165–173
Okello W, Kurmayer R (2011) Seasonal development of cyanobacteria and microcystin production in Ugandan freshwater lakes. Lake Reserv Manag 16:123–135
Ibelings BW, Chorus I (2007) Accumulation of cyanobacterial toxins in freshwater ‘seafood’ and its consequences for public health: a review. Environ Pollut 150:177–192
Tsuji K, Naito S, Kondo F, Ishikawa N, Watanabe MF, Suzuki M, Harada KI (1994) Stability of microcystins from cyanobacteria: effect of light on decomposition and isomerization. Environ Sci Technol 28:173–177
Dittmann E, Wiegand C (2006) Cyanobacterial toxins—occurrence, biosynthesis and impact on human affairs. Mol Nutr Food Res 50:7–17
Rosse Y, Baan R, Straif K, Secretan B, El Ghissassi F, Cogliano V (2006) Carcinogenicity of nitrate, nitrite, and cyanobacterial peptide toxins. Lancet Oncol 7:628–629
Lahti K, Hiisvirta L (1989) Removal of cyanobacterial toxins in water treatment processes: review of studies conducted in Finland. Water Supply 7:149–154
Jones GJ, Falconer IR (1994) Factors affecting the production of toxins by cyanobacteria. Final report to the land and water resources. Research and Development Corporation, Canberra
Shelley R, Jonathan P, Susanna A, Wood DR, Dietrich DPH, Prinsep MR (2015) The effect of cyanobacterial biomass enrichment by centrifugation and GF/C filtration on subsequent microcystin measurement. Toxins 7(3):821–834. https://doi.org/10.3390/toxins7030821
Domaizon I, Anneville O (2014) Nodularin and cylindrospermopsin: a review of their effects on fish Nodularin and cylindrospermopsin: a review of their effects on fish. Rev Fish Biol Fisher 25(1):1–19
Bui T, Dao T, Vo T, Lürling M (2018) Warming affects growth rates and microcystin production in tropical bloom-forming microcystis strains. Toxins 10:123. https://doi.org/10.3390/toxins10030123
Lürling M, Faassen EJ (2012) Controlling toxic cyanobacteria: effects of dredging and phosphorus-binding clay on cyanobacteria and microcystins. Water Res 46:1447–1459. https://doi.org/10.1016/j.watres.2011.11.008
Graham JL, Loftin KA, Meyer MT, Ziegler AC (2010) Cyanotoxin mixtures and taste-and-odor compounds in cyanobacterial blooms from the Midwestern United States. Environ Sci Technol 44:7361–7368
Acknowledgements
We thank Addis Ababa water and sanitation authority for assistance during the sampling. We would also like to thank the Aquatic Ecology and Water Quality Management Group at Wageningen University for providing all the laboratory facilities for toxin analysis, SIDA Project, for covering travel and accommodation costs to Wageningen University and Addis Ababa University thematic research Projects (Project No. TR/012/2016 and RD/PY-609/2016) for covering the cost of sampling in Legedadi Reservoir.
Author information
Authors and Affiliations
Contributions
HH, ML, conception and design, HH, WB, investigation, ML data curation, HH, DK, SL, ML data analysis, HH, draft the manuscript, DK, SL edit the manuscript, HH revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethics approval
Not applicable.
Availability of data and material
All data and material used in this manuscript are available.
Consent to participate
All authors have made substantial contributions to conception, design, investigation, data curation, data analysis, drafting, editing, and revising it critically, give final approval of the version to be published.
Consent for publication
All authors have contributed from conception to approval of the final version and agreed to be accountable for all aspects of the work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Habtemariam, H., Kifle, D., Leta, S. et al. Cyanotoxins in drinking water supply reservoir (Legedadi, Central Ethiopia): implications for public health safety. SN Appl. Sci. 3, 328 (2021). https://doi.org/10.1007/s42452-021-04313-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-021-04313-0