
Abstract
The impact on the final morphology of ceria (CeO2) nanoparticles made from different precursors (commercial: cerium acetate/nitrate) and in house: cerium tri(methylsilyl)amide (Ce-TMSA)) via a microwave solid state reaction has been determined. In all instances, powder X-ray diffraction indicated that the cubic fluorite CeO2 phase (PDF# 04–004-9150, with the space group Fm-3 m) had formed. Scanning electron microscopy (SEM) images revealed spherical nanoparticles were produced from the Ce-TMSA precursor. The commercial acetate and nitrate precursors produced particles with irregular morphology. The roles of the precursor decomposition and binding energy in the synthesis of the nanocrystals with various morphologies, as well as a possible growth mechanism, were evaluated based on experimental and computational data. The formation of spherical shaped nanoparticles was determined to be due to the preferential single-step decomposition of the Ce-TMSA as well as the low activation energy to overcome decomposition. Due to the complicated decomposition of the commercial precursors and high activation energy the resulting particles adopted an irregular morphology. Highly uniform samarium doped ceria (SmxCe1-xO2-δ) nanospheres were also synthesized from Ce-TMSA and samarium tri(methylsilyl)amide (Sm-TMSA). The effects of reaction time and temperature, on the final morphology were observed through SEM. The rapid single-step decomposition of TMSA-based precursors as observed through thermogravimetric analysis (TGA) and confirmed through the calculation of potential energy surfaces and binding energies from density functional theory (DFT) calculations, indicated that nanoparticle formation follows LaMer’s classical nucleation theory.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Ceria (CeO2) nanomaterials have found widespread use in sensors [1, 2], absorbers, [3] and fuel cells as a solid electrolyte due to their inherent physical properties including, chemical inertness, ionic conductivity, high dielectric constant, and moderate band gap [4, 5]. The size of the Ce4+ cation (0.96 Å) and its inherent crystal structure allows for easy doping of CeO2 by trivalent lanthanide cations (Ln3+); the open structure of the cubic fluorite lattice can accommodate high levels of point defect disorder [6]. This property has led to the development of easily doped CeO2 materials like SmxCe1-xO2-δ and GdxCe1-xO2-δ. Doping CeO2 with Ln3+ cations introduces significant oxygen vacancies which can increase ionic conductivity over neat CeO2. There have been many reports on the synthesis of doped CeO2 materials [7,8,9,10,11], but among these new materials, samarium doped ceria (SmxCe1-xO2-δ) has emerged as one of the more promising doped CeO2 materials due to the similarities in ionic radii of Ce4+ (0.96 Å) and Sm3+ (1.09 Å); this allows for easy solid solution formation. Upon successful doping, SmxCe1-xO2-δ has reported one of the higher ionic conductivities and particularly low operating temperature requirements [12]. For example, Huang et al. showed a nearly two order of magnitude increase in ionic conductivity at 600 °C by doping CeO2 with 23% samarium [13].
It has been reported that the properties of CeO2-based ceramics are highly dependent on the size, shape, and crystalline phase of the particles [14,15,16]. For instance, Mai et al. observed an oxygen storage content of 554 μmol O g−1 for CeO2 nanorods compared to an oxygen storage content of 318 μmol O g−1 for CeO2 nanopolyhedra [16]. Ma et al. utilized a novel hydrothermal method to synthesize SmxCe1-xO2-δ nanorods, which due to their increased surface area, displayed a very high power density of 522 mW cm−2 [17]. Since the shape-phase-property relationship is important for various applications, it is critical to establish control over the morphology of CeO2 based materials (i.e., morphology, phase, shape, etc.) as a function of Ln3+ doping. Previous efforts to tailor the morphology of various CeO2-based nanoparticles have been investigated using commercially available precursors by altering specific synthesis variables, such as pH [18], precursors [19], and surfactants [20]. Recently, Wang et al. demonstrated the synthesis of CeO2 in the form of stacked nanoplatelets, nanorods, nanosquares, and round nanoplatelets using a solution-based synthesis in the presence of different mineralizers [21]. While morphological control of neat CeO2 and trivalent doped CeO2 has been realized by varying the pH, surfactant, and precursors in solution, to the best of our knowledge there has been no report on the systematic comparison of nonvolatile (commercial/hydrate/air-stable) vs volatile (in-house/non-hydrate/air-unstable) precursors for the production of neat and Ln3+-doped CeO2 nanoparticles.
A broad variety of synthesis processes has been utilized to make CeO2-based nanomaterials including: hydrothermal methods [22], solvothermal methods [23], co-precipitation [24], sol–gel [23], and microemulsion [25]. While many of these methods have produced high quality materials with good shape and size control, they are reliant on secondary mechanisms/catalysts such as surfactants and pH. Furthermore, they can involve aging steps or complex surfactant washing procedures that make them unattractive for large scale synthesis. As an alternative to solution-based techniques, solid-state reactions have the potential for direct preparation of economically viable, high-purity, stoichiometric metal oxide nanoparticles while avoiding many of the aforementioned complexities [26]. Solid-state reactions can also be improved upon by utilizing microwave heating. Compared to conventional heating methods, microwave heating is more efficient, has a more homogenous heating profile, and can lead to higher yields [27].
Given the limited investigations of traditional vs non-traditional precursors to produce faceted nanoparticles, a systematic approach was taken to produce CeO2 and SmxCe1-xO2-δ nanoparticles by decomposing traditional cerium/samarium acetate hydrate (Ln-acetate) and cerium/samarium nitrate hexahydrate (Ln-nitrate) precursors, as well as nontraditional samarium/cerium tri(methylsilyl)amide (Ln-TMSA) precursors via microwave-assisted solid state in ambient conditions. This report details the synthesis, characterization, and growth mechanism of CeO2 and SmxCe1-xO2-δ nanoparticles. The effect of different precursors to produce SmxCe1-xO2-δ nanoparticles as well as the effects of reaction temperature, time, and initial molar ratio were investigated. Details of the possible growth mechanism of spherical nanoparticle of CeO2 and SmxCe1-xO2-δ nanoparticles are explained through thermogravimetric analysis (TGA) and density functional theory (DFT) calculations that reinforce the classic LaMer growth mechanism.
2 Experimental
2.1 Materials and methods
All syntheses were performed under-atmospheric conditions, unless otherwise stated. Cerium(III) nitrate hexahydrate, samarium(III) nitrate hexahydrate, cerium(III) acetate hydrate and samarium(III) acetate hydrate, cerium(III) chloride, samarium(III) chloride, and potassium bis(trimethylsilyl)amide (KNR2), toluene, tetrahydrofuran (THF) and hexanes, were purchased from Sigma-Aldrich, USA. Cerium(III) TMS and samarium(III) TMS were prepared in-house using established literature routes [28]. All reagents were of analytical grade and used without further purification.
2.2 Characterization methods
Analytical data were collected on dry, free-flowing powders. Powder X-ray diffraction (PXRD) of the products were conducted on a zero-background holder by a Siemens D500 Diffractometer equipped with a graphite monochromator with Cu Kα radiation operating at 45 kV and 40 mA. The samples were scanned at a rate of 4°/s in the 2θ range of 20°–100°. The patterns were analyzed with the JADE 9.6.0 software with ICDD PDF-4 database. Raman spectra were obtained using a Witec Raman Confocal Microscope with an excitation laser operating at 532 nm. X-ray fluorescence (XRF) spectra were obtained using a Thermo Scientific ARL Quant’X EDXRF Analyzer. For all scanning electron microscopy (SEM) analyses, the samples were dispersed on carbon tape and examined with a Hitachi S-5200 Scanning Electron Microscope under an accelerating voltage of 5 kV and a current of 7A. Differential scanning calorimetry (DSC) and TGA were measured using a Mettler Toledo TGA/DSC 3 + Thermogravimetric Analyzer equipped with a high temperature (HT) furnace. The samples were heated a rate of 5 °C/min in the temperature range of 25–500 °C.
2.3 Computational Details
To complement experimental TGA analysis, gas-phase electronic structure calculations were performed for Ln-TMSA and Ln-acetate (representative of the commercial precursors) using Gaussian 09 [29] at the DFT level with the hybrid B3LYP functional (Becke's three parameter hybrid exchange functional with Lee–Yang–Parr correlation function) [30,31,32]. The small-core effective core potential (ECP) MWB28 was used for the Ce and Sm atoms [33]. All other atoms were described using the 6-31G basis set [32, 34, 35] as part of the complete basis set model described by Petersson et al. [36]. The ECP MWB28 core potentials include a scalar relativistic correction and has been previously applied to Ce- and Sm-based molecules resulting in agreement with experimental structures [37, 38]. Molecular structures were selected from solved crystal structures and optimized in unrestricted calculations. All atoms were allowed to relax during optimization. Agreement in the metrical data between the crystal structures and DFT optimized molecules (Table S1) validates the quality of the DFT models and is discussed in the results. Potential energy surface (PES) scans were performed to identify the strength of the Ln-TMSA interaction and the presence of intermediate states during disassociation. The Ln-N distance was stretched by 0.05 Å, and then the structure was relaxed. No other constraints were placed on the system during relaxation. This process was repeated iteratively until the Ln-N distance reached 5.0 Å. In addition to the PES scans, dissociation energy (ED) data was collected for complete removal of the trimethylsilyl amide (TMSA) ligand. ED data was calculated by removing a TMSA ligand from the Ln-TMSA molecule and relaxing the resulting structure as well as the lone TMSA ligand. ED was calculated as the difference between the molecule (ELn-TMSA), the energy of a lone TMSA ligand (ETMSA), and the energy of the precursor without a third TMSA ligand (ELn-TMSA-TMSA):
2.4 Preparation of Ln-TMSA precursors
Separate solutions of LnCl3 in THF and KNR2 in THF were prepared in inert atmosphere. The LnCl3 solution was cooled and progressively treated with the KNR2 solution. The final mixture was stirred overnight, filtered with hexanes, extracted with toluene, and then stripped again to yield Ln-TMSA powder.
2.5 Microwave-assisted solid-state synthesis of SmxCe1-xO2-δ powders
CeO2 and SmxCe1-xO2-δ nanoparticles were prepared via solid state microwave radiation of the respective metal precursors (Ln-nitrate, Ln-acetate, Ln-TMSA) in the presence of atmospheric air. All reactions involving acetate and nitrate precursors were prepared and performed in ambient conditions, while the Ln-TMSA precursors were handled in a glove box prior to heating in ambient conditions. In a standard synthesis method, the precursor was ground in a mortar to insure a homogenous mixture. After mixing the necessary precursors, the reaction was heated in a Microwave Research and Application BP-210 microwave oven under atmospheric conditions at 750 °C, using air as the reaction media. For the formation of SmxCe1-xO2-δ various ratios and reaction conditions of Ce-TMSA and Sm-TMSA, shown in Table 1, were attempted to increase the Sm3+ dopant concentration and control the physical nature of the material (size, morphology, and crystallinity). After the reactions were conducted as described the isolated materials were collected and analyzed without further modifications.
3 Results and discussion
The robust and diverse physical properties of CeO2 and SmxCe1-xO2-δ have led to numerous investigations detailing the morphological impact on their optical and electronic properties [39, 40]; however, there are no reports on understanding the generation of the aforementioned materials using traditional vs non-traditional precursors in a microwave without the presence of a liquid solution or surfactant. Due to the shape-dependent properties of nanomaterials and the void in literature on microwave-assisted solid-state synthesis of various precursors, it was of interest to investigate the effect of traditional and non-traditional precursors, on the properties of the resultant CeO2 and SmxCe1-xO2-δ nanoparticles. Therefore, a systematic study was undertaken, and the results discussed below focus on the crystal structure and particle morphology of resulting nanoparticles. A growth mechanism is also described based on the (i) decomposition profiles, (ii) calculated dissociation energies, and (iii) calculated potential surfaces of precursor materials.
3.1 Formation of spherical cerium oxide nanoparticles
PXRD data, shown in Fig. 1a, was collected on the powders isolated from reacting the Ce-precursors (acetate, nitrate, and TMSA) at 750 °C for 10 min. For each sample the major peaks of the PXRD patterns were indexed to the cubic fluorite CeO2 phase (PDF# 04–004-9150) with the space group Fm-3 m and no other impurities can be observed in the patterns. SEM images of products generated from the microwave reaction of the individual Ce-precursors are shown in Fig. 1b-d. Figure 1b, c show SEM images of the product formed from Ce-acetate and Ce-nitrate, respectively. For these systems, highly irregular CeO2 nanoparticles are present as well as agglomerated particles. The irregular morphology of the acetate and nitrate-based CeO2 indicates that there are adsorbed surface species during the conversion reaction, and that transport of ions on the surfaces is not rapid enough to form regular facets based on crystal symmetry. However, Fig. 1d shows that decomposition of the Ce-TMSA precursor yields CeO2 nanoparticles with a highly defined spherical morphology. In contrast, Ce-TMSA processed in a conventional furnace with identical conditions yielded a minute amount of spherical nanoparticles with the majority exhibiting irregular morphology, as shown in Figure S1. The round spherical morphology agrees with a rapid decomposition process achieved through rapid microwave heating, where cation transport to the nuclei is diffusion limited. These variations in growth kinetics and nucleation will be discussed more in depth in the growth mechanism section.

PXRD spectra a confirms that all precursors lead to highly crystalline cerium oxide. SEM images of nanoparticles synthesized from b Ce-acetate c Ce-nitrate and d Ce-TMSA precursors synthesized at a reaction temp of 750 °C and a reaction time of 10 min
3.2 Formation of Samarium-Doped Ceria Nanospheres
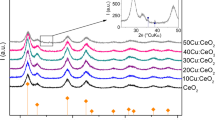
Due to the serendipitous generation of spherical CeO2 nanoparticle from microwave irradiation of the Ce-TMSA precursor, SmxCe1-xO2-δ was synthesized by combining Ce-TMSA and Sm-TMSA precursors. SmxCe1-xO2-δ was generated with an initial Ce:Sm molar ratio of 4:1,3:1, 2:1 and a reaction time of 10 min at 1000 °C. PXRD data, shown in Fig. 2a, were collected on the powders isolated from the different reaction conditions. The crystalline product was indexed to the cubic fluorite phase (PDF# 04–004-9150), with no secondary phase present (Sm2O3; cubic la-3 phase). Upon addition of Sm3+, the PXRD patterns exhibit a Sm3+ concentration-dependent downfield shift relative to CeO2 for all the major reflections, which can be seen clearly in Fig. 2b. The shift to lower angles is due to lattice strain from size mismatch cause by the uptake of differently sized Sm3+ ions, suggesting the successful incorporation of Sm3+ in the cubie lattice of CeO2 [41]. As shown in Fig. 2c, production of SmxCe1-xO2-δ yielded spherical nanoparticles, indicating that uptake of Sm3+ ions does not affect the final morphology.

a PXRD spectra of SmxCe1-xO2-δ prepared with variable starting Ce:Sm ratios. The PXRD inset in b shows the downfield shift in the spectra of CeO2 upon the addition of samarium ions. c SEM images of SmxCe1-xO2-δ nanoparticles shows spherical morphology
In order to confirm the uptake of Sm3+ ions in the cubic fluorite CeO2 lattice as well as to ascertain any structural changes, SmxCe1-xO2-δ powders were analyzed via Raman spectroscopy and XRF (Figure S2). Representative Raman spectra for as synthesized CeO2 and SmxCe1-xO2-δ are shown in Fig. 3. The major peak for both powders is the triply degenerate F2g band at ~ 460 cm−1 which is assigned to the CeO2 cubic fluorite structure [42]. This Raman active mode is attributed to the symmetric breathing of oxygen around the central cerium ions [43]. The symmetry of this breathing mode means that they are sensitive to disorder in the oxygen lattice that can occur through doping. Upon Sm3+ incorporation in CeO2, a broad absorption band in the region of 520–630 cm−1 emerges as oxygen vacancies are introduced and disorder is introduced into the oxygen sublattice. Additionally, the F2g peak becomes asymmetric, broadens, and shifts to lower frequencies due to increased oxygen vacancy and changes in the lattice structure [44]. This result is consistent with other reports of SmxCe1-xO2-δ [45, 46].

Raman spectra of CeO2 and SmxCe1-xO2-δ
3.3 Effect of synthesis parameters on nanoparticles size
To understand the effect of temperature on the formation of SmxCe1-xO2-δ, Ln-TMSA based nanoparticles with an initial Ce:Sm ratio of 4:1 of were investigated at 500 °C, 750 °C, and 1000 °C for 10 min, respectively. SEM images of the product from this study are shown in Fig. 4a-c. At all temperatures spherical nanoparticles were obtained, however, there is variation in final nanoparticle size. To quantify the effect of temperature on the size dispersion of the product, the diameter of > 200 particles were analyzed and plotted in histograms for each reaction, shown in Fig. 4d-f. At a final processing temperature of 500 °C the majority of the nanoparticles have diameters between 10–50 nm, at 750 °C the average diameter increases to 75–125 nm, and at 1000 °C most particles have diameters between 175–250 nm. It is worth noting that the particle distributions follow a log-normal distribution as illustrated by the overlays on the histograms.

SEM images of SmxCe1-xO2-δ nanoparticles synthesized with a starting Ce:Sm ratio of 4:1, a reaction time of 10 min, at a temperature of a 500 °C b 750 °C and c 1000 °C. The corresponding particle size distributions are shown in d–f
Additionally, the effect of reaction time on the formation of SmxCe1-xO2-δ nanoparticles generated with an initial Ce:Sm ratio of 2:1 at 750 °C was investigated for 5, 10, and 20 min, respectively. SEM images of the final products are shown in Fig. 5a-c. After analyzing > 200 nanoparticles, it is evident that longer reaction times increases the average final nanoparticle size, shown visually in Fig. 5d-f. With all other conditions kept constant, there should be identical nucleation events and the particle size distribution should be identical upon reaching the final dwell temperature. However, if the longer the reaction proceeds at the final reaction temperature, more growth will occur, particularly in the form of Ostwald ripening [47]. According to the Gibbs-Thompson description of nucleation, dissolution can occur as the reaction proceeds due to the curvature radius of formed particles. These smaller particles can then be re-dissolved into solution and incorporated into larger particles [48].

SEM images of SmxCe1-xO2-δ δ nanoparticles synthesized with a starting Ce:Sm ratio of 2:1, a reaction temperature of 750 °C, and a reaction time of a 5 min b 10 min and c 15 min. The corresponding particle size distributions are shown in d–f
3.4 Decomposition mechanism of SmxCe1-xO2-δ precursors
In solid-state reactions, the mechanism by which precursors breakdown plays a major role in the final morphology and properties of the resulting product. To help understand the effect of different precursors on final nanoparticle morphology, the thermolysis properties of Ln-precursors were studied using TGA, shown in Fig. 6. The commercial Ln-acetate/nitrate precursors display multi-step decomposition steps as reported in literature and displayed in Table S2 [49,50,51,52]; Ln-TMSA precursors display a single step decomposition. Additionally, Ln-TMSA exhibits a much lower decomposition temperature; Sm-TMSA decomposes at ~ 200 °C, but Sm-acetate and Sm-nitrate do not decompose fully until > 400 °C. The simpler decomposition mechanism and lower decomposition temperature of Ln-TMSA are indicators for classical nucleation behavior that can ultimately lead to well defined spherical nanoparticles.

TGA analysis of a cerium-based precursors and b samarium-based precursors
To help identify particle nucleation mechanisms, DFT calculations can provide insight into the breakdown mechanism of Ln-precursors. The presence of a single step decomposition in Ln-TMSA precursors suggests that TMSA ligands are removed from the Ce/Sm center without the formation of intermediate states. To evaluate the effect of TMSA ligand removal, a potential energy scan (PES) scan was performed on Ce-TMSA by iteratively stretching the Ce-N bond in 0.05 Å steps and allowing the structure to completely relax. The PES shown in Fig. 7, does not indicate the formation of significant intermediate states as indicated by the Lorentz potential like behavior. The small change in slope at ~ 3.6 Å is a result of the methyl groups loosely interacting with the Ce center but is not a strong enough interaction to suggest the existence of an intermediate structure.

PES scan of Ce-TMSA molecule. Insets are snapshots of Ce-TMSA molecule with Ce-N distances of 2.30 Å and 4.95 Å. Atom colors: Ce (pink), Si (yellow), C (grey), N (blue), and H (white)
The effect of intermediate structures can be observed quantitatively by calculating the ED of the first decomposition step for Ln precursors. Shown in Table 2 are the pathways and ED for the first decomposition step of Ln-TMSA and Ln-acetate precursors calculated as the difference between the relaxed energies of the products and reactants shown schematically in Fig. 8. For the cerium precursors, Ce-TMSA displays a lower binding energy of -4.348 eV as compared to -1.574 for Ce-acetate. Similarly, Sm-TMSA displays a lower binding energy of -3.334 as compared to -1.598 for Sm-acetate. The intermediate structures formed during acetate decomposition result in higher binding energies that make it difficult for molecular species to become available for particle nucleation. Nitrate decomposition also displays a multi-step profile; nitrates are known in molten salt chemistry to break down into nitrite ions before complete decomposition. Due to this behavior, it is expected that the binding energies would be in a similar range as that of the Ln-acetate precursors.

Schematic of the first step of decomposition for Ln-acetate and Ln-TMSA precursors. Atom colors: Ln (pink), C (grey), H (white), Si (yellow), N (blue)
According to the TGA data and relevant calculations, it is apparent that the single step decomposition of Ln-TMSA and subsequent formation of spherical nanoparticles falls in line with LaMer’s classical nucleation theory, shown schematically in Fig. 9. In step 1, the concentration of Ln monomers increases at a steady rate as TMSA ligands begin to separate from the metal center. In step 2, the saturation of monomers overcomes the energy barrier at some critical solute concentration (\({\mathrm{C}}_{\mathrm{min}}^{\mathrm{nu}})\) to achieve a burst nucleation event. In step 3, crystallization occurs as the concentration of monomers decreases due to continued growth of stable nuclei. In step 4, nuclei continue to grow due to continued monomer diffusion and Ostwald ripening. Spherical nanoparticles are formed due to rapid nucleation followed by the diffusion of readily available monomer species leaving no time for faceted surfaces to form around nuclei. In the case of nitrate and acetate-based precursors, the multi-step decomposition profiles are indicative of a sol–gel polymerization nucleation mechanism that leads to irregular morphologies as facets form semi-randomly on nuclei surfaces [48].

Schematic for SmxCe1-xO2-δ spherical nanoparticle formation based on LaMer nucleation theory
4 Conclusions
In conclusion SmxCe1-xO2-δ nanoparticles were produced using a microwave-assisted solid-state synthesis. By varying reaction conditions like the molar ratio of reactants, the reaction temperature, and reaction time, spherical nanoparticles with sizes ranging from 20 nm up to 600 nm were produced. Based on TGA analysis and DFT calculations, SmxCe1-xO2-δ nanoparticles produced with TMSA based precursors grow according to classical LaMer nucleation theory leading to the formation of spherical nanoparticles. The results of this research indicate that microwave-assisted solid-state synthesis can be used to make doped Ceria nanoparticles, but more research is needed in order to control the homogeneity of these particles. Solid state synthesis can be readily scaled up, easily controlled, and does not need solvent or washing, making it an attractive option for producing rare earth oxide-based materials. Future studies will focus on the optimization of SmxCe1-xO2-δ nanoparticles as well as the introduction of additional rare earths.
References
Stefanik TS, Tuller HL (2001) Ceria-based gas sensors. J Eur Ceram Soc 21:1967–1970. https://doi.org/10.1016/S0955-2219(01)00152-2
Subbiah DK, Kulandaisamy AJ, George RB et al (2018) Nano ceria as xylene sensor – Role of cerium precursor. J Alloys Compd 753:771–780. https://doi.org/10.1016/j.jallcom.2018.04.248
Álvarez-Asencio R, Corkery RW, Ahniyaz A (2020) Solventless synthesis of cerium oxide nanoparticles and their application in UV protective clear coatings. RSC Adv 10:14818–14825. https://doi.org/10.1039/d0ra01710h
Vangelista S, Piagge R, Ek S et al (2017) Atomic Layer Deposition of Cerium Dioxide Film on TiN and Si Substrates: Structural and Chemical Properties. MRS Adv 2:3005–3010. https://doi.org/10.1557/adv.2017.404
Chiu F-C, Lai C-M (2010) Optical and electrical characterizations of cerium oxide thin films. J Phys D Appl Phys 43:075104. https://doi.org/10.1088/0022-3727/43/7/075104
Omar S, Nino JC (2013) Consistency in the chemical expansion of fluorites: A thermal revision of the doped ceria. Acta Mater 61:5406–5413. https://doi.org/10.1016/j.actamat.2013.05.029
Bondioli F, Ferrari AM, Lusvarghi L et al (2005) Synthesis and characterization of praseodymium-doped ceria powders by a microwave-assisted hydrothermal (MH) route. J Mater Chem 15:1061–1066. https://doi.org/10.1039/b415628e
Kumar A, Babu S, Karakoti AS et al (2009) Luminescence properties of europium-doped cerium oxide nanoparticles: Role of vacancy and oxidation states. Langmuir 25:10998–11007. https://doi.org/10.1021/la901298q
Kumar SA, Kuppusami P, Yen-Pei F (2020) Structural, morphological and electrical properties of Sm-Gd Co-doped ceria thin films for micro-solid oxide fuel cells. Mater Lett 275:128110. https://doi.org/10.1016/j.matlet.2020.128110
Esther Jeyanthi C, Siddheswaran R, Kumar P et al (2014) Structural and spectroscopic studies of rare earths doped ceria (RE La, Sc, Yb:CeO2) nanopowders. Ceram Int 40:8599–8605. https://doi.org/10.1016/j.ceramint.2014.01.076
Esther Jeyanthi C, Siddheswaran R, Medlín R et al (2014) Electrochemical and structural analysis of the RE3+:CeO2 nanopowders from combustion synthesis. J Alloys Compd 614:118–125. https://doi.org/10.1016/j.jallcom.2014.05.208
Kosinski MR, Baker RT (2011) Preparation and property-performance relationships in samarium-doped ceria nanopowders for solid oxide fuel cell electrolytes. J Power Sources 196:2498–2512. https://doi.org/10.1016/j.jpowsour.2010.11.041
Huang W, Shuk P, Greenblatt M (1997) Hydrothermal Synthesis and Properties of Ce1-xSmxO2-x/2 and Ce1-xCaxO2-x Solid Solutions. Chem Mater 9:2240–2245. https://doi.org/10.1021/cm970425t
Trovarelli A, Llorca J (2017) Ceria Catalysts at Nanoscale: How Do Crystal Shapes Shape Catalysis? ACS Catal 7:4716–4735. https://doi.org/10.1021/acscatal.7b01246
Zhou F, Zhao X, Xu H, Yuan C (2007) CeO2 spherical crystallites: Synthesis, formation mechanism, size control, and electrochemical property study. J Phys Chem C 111:1651–1657. https://doi.org/10.1021/jp0660435
Mai HX, Sun LD, Zhang YW et al (2005) Shape-selective synthesis and oxygen storage behavior of ceria nanopolyhedra, nanorods, and nanocubes. J Phys Chem B 109:24380–24385. https://doi.org/10.1021/jp055584b
Ma Y, Wang X, Li S et al (2010) Samarium-doped ceria nanowires: Novel synthesis and application in low-temperature solid oxide fuel cells. Adv Mater 22:1640–1644. https://doi.org/10.1002/adma.200903402
Hajizadeh-Oghaz M, Razavi RS, Ghasemi A (2016) The Effect of Solution pH Value on the Morphology of Ceria-Yttria Co Stabilized Zirconia Particles Prepared Using the Polymerizable Complex Method. J Clust Sci 27:469–483. https://doi.org/10.1007/s10876-015-0942-2
Barkam S, Ortiz J, Saraf S et al (2017) Modulating the Catalytic Activity of Cerium Oxide Nanoparticles with the Anion of the Precursor Salt. J Phys Chem C 121:20039–20050. https://doi.org/10.1021/acs.jpcc.7b05725
Chowdhury S, Lin KS (2011) Synthesis and characterization of 1D ceria nanomaterials for CO oxidation and steam reforming of methanol. J Nanomater. https://doi.org/10.1155/2011/157690
Wang D, Kang Y, Doan-Nguyen V et al (2011) Synthesis and oxygen storage capacity of two-dimensional ceria nanocrystals. Angew Chemie Int Ed 50:4378–4381. https://doi.org/10.1002/anie.201101043
Muñoz FF, Acuña LM, Albornoz CA et al (2015) Redox properties of nanostructured lanthanide-doped ceria spheres prepared by microwave assisted hydrothermal homogeneous co-precipitation. Nanoscale 7:271–281. https://doi.org/10.1039/c4nr05630b
Amarsingh Bhabu K, Theerthagiri J, Madhavan J et al (2016) Cubic fluorite phase of samarium doped cerium oxide (CeO2)0.96Sm0.04 for solid oxide fuel cell electrolyte. J Mater Sci Mater Electron 27:1566–1573. https://doi.org/10.1007/s10854-015-3925-z
Shehata N, Samir E, Gaballah S (2016) New optical sensor for peroxides using neodymium-doped-ceria nanoparticles via fluorescence-quenching technique. Sens Actuators B Chem 231:341–348. https://doi.org/10.1016/j.snb.2016.03.036
Liu J, Ye J, Liu P et al (2011) Synthesis of monodisperse samarium-doped ceria nanocrystals via a microemulsion-mediated hydrothermal method with secondary light irradiation treatment. Mater Lett 65:143–145. https://doi.org/10.1016/j.matlet.2010.10.013
Ealias AM, Saravanakumar MP (2017) A review on the classification, characterisation, synthesis of nanoparticles and their application. IOP Conf Ser Mater Sci Eng. https://doi.org/10.1088/1757-899X/263/3/032019
Gawande MB, Shelke SN, Zboril R, Varma RS (2014) Microwave-assisted chemistry: synthetic applications for rapid assembly of nanomaterials and organics. Acc Chem Res 47:1338–1348. https://doi.org/10.1021/ar400309b
Donald B, Ghotra JS, Road ME, et al (1973) Low co-ordination numbers in lanthanide and actinide compounds. Part I. The preparation and characterization of tris{bis(trimethylsilyl)-amido}lanthanides. J Chem Soc Dalt Trans pp 1021–1023
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ort and DJF (2016) Gaussian 09, Revision A. 02
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789. https://doi.org/10.1103/PhysRevB.37.785
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652. https://doi.org/10.1063/1.464913
Francl MM, Pietro WJ, Hehre WJ et al (1982) Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J Chem Phys 77:3654–3665. https://doi.org/10.1063/1.444267
Dolg M, Stoll H, Preuss H (1989) Energy-adjusted ab initio pseudopotentials for the rare earth elements. J Chem Phys 90:1730–1734. https://doi.org/10.1063/1.456066
Hariharan PC, Pople JA (1973) The influence of polarization functions on molecular orbital hydrogenation energies. Theor Chim Acta 28:213–222. https://doi.org/10.1007/BF00533485
Hehre WJ, Ditchfield K, Pople JA (1972) Self-consistent molecular orbital methods. XII. Further extensions of gaussian-type basis sets for use in molecular orbital studies of organic molecules. J Chem Phys 56:2257–2261. https://doi.org/10.1063/1.1677527
Petersson GA, Bennett A, Tensfeldt TG et al (1988) A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J Chem Phys 89:2193–2218. https://doi.org/10.1063/1.455064
Ramezanzadeh B, Bahlakeh G, Ramezanzadeh M (2018) Polyaniline-cerium oxide (PAni-CeO2) coated graphene oxide for enhancement of epoxy coating corrosion protection performance on mild steel. Corros Sci 137:111–126. https://doi.org/10.1016/j.corsci.2018.03.038
Xie H, Wang J, Qin Z et al (2014) Octacoordinate metal carbonyls of lanthanum and cerium: experimental observation and theoretical calculation. J Phys Chem A 118:9380–9385. https://doi.org/10.1021/jp504079k
Gnanam S, Rajendran V (2013) Influence of various surfactants on size, morphology, and optical properties of CeO2 nanostructures via facile hydrothermal route. J Nanoparticles 2013:1–6. https://doi.org/10.1155/2013/839391
Suresh R, Ponnuswamy V, Mariappan R (2013) Effect of annealing temperature on the microstructural, optical and electrical properties of CeO 2 nanoparticles by chemical precipitation method. Appl Surf Sci 273:457–464. https://doi.org/10.1016/j.apsusc.2013.02.062
Hos JP, McCormick PG (2003) Mechanochemical synthesis and characterisation of nanoparticulate samarium-doped cerium oxide. Scr Mater 48:85–90. https://doi.org/10.1016/S1359-6462(02)00351-2
Weber WH, Hass KC, McBride JR (1993) Raman study of CeO2: second-order scattering, lattice dynamics, and particle-size effects. Phys Rev B 48:178–185. https://doi.org/10.1103/PhysRevB.48.178
Fernández-García M, Martínez-Arias A, Iglesias-Juez A et al (2000) Structural characteristics and redox behavior of CeO2-ZrO2/Al2O3 supports. J Catal 194:385–392. https://doi.org/10.1006/jcat.2000.2931
McBride JR, Hass KC, Poindexter BD, Weber WH (1994) Raman and x-ray studies of Ce1-xRExO2-y, where RE=La, Pr, Nd, Eu, Gd, and Tb. J Appl Phys 76:2435–2441. https://doi.org/10.1063/1.357593
Deng T, Zhang C, Xiao Y et al (2015) One-step synthesis of samarium-doped ceria and its CO catalysis. Bull Mater Sci 38:1149–1154. https://doi.org/10.1007/s12034-015-0994-9
Wang SF, Yeh CT, Wang YR, Wu YC (2013) Characterization of samarium-doped ceria powders prepared by hydrothermal synthesis for use in solid state oxide fuel cells. J Mater Res Technol 2:141–148. https://doi.org/10.1016/j.jmrt.2013.01.004
Baronov A, Bufkin K, Shaw DW et al (2015) A simple model of burst nucleation. Phys Chem Chem Phys 17:20846–20852. https://doi.org/10.1039/c5cp01745a
Varanda LC, Souza CGS, Moraes DA et al (2019) Size and shape-controlled nanomaterials based on modified polyol and thermal decomposition approaches. A brief review. Acad Bras Cienc. https://doi.org/10.1590/0001-3765201920181180
Chicago W OF THE RARE OTHER In a recent study involving the dete ~ ination of the solubilitics of some rare earth and other metal nitrates in organic soIvents1e2 , it was found that little was All of the rare earth nitrate hexahydrates decomposed in the same genera. p 15
Melnikov P, Arkhangelsky IV, Nascimento VA et al (2014) Thermolysis mechanism of samarium nitrate hexahydrate. J Therm Anal Calorim 118:1537–1541. https://doi.org/10.1007/s10973-014-4067-x
Arii T, Kishi A, Ogawa M, Sawada Y (2001) Thermal decomposition of cerium(III) acetate hydrate by a three-dimensional thermal analysis. Anal Sci 17:875–880. https://doi.org/10.2116/analsci.17.875
Patil KC, Chandrashekhar GV, George MV, Rao CNR (1968) Infrared spectra and thermal decompositions of metal acetates and dicarboxylates. Can J Chem 46:257–265. https://doi.org/10.1139/v68-040
Funding
Sandia National Laboratories is a multimission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525. SAND2020-7474 C.
Author information
Authors and Affiliations
Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the Supplementary Information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bregman, A., Rimsza, J., Ringgold, M. et al. The role of precursor decomposition in the formation of samarium doped ceria nanoparticles via solid-state microwave synthesis. SN Appl. Sci. 3, 341 (2021). https://doi.org/10.1007/s42452-021-04288-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-021-04288-y