
Abstract
Malaria, a disease caused by one of the world’s fatal parasites Plasmodium falciparum, is responsible for over a million death annually. P. falciparum dihydroorotate dehydrogenase (PfDHODH) is a validated target of this deadly parasite. Quantitative structure–activity relationship and molecular docking in silico methods were employed in the discovery of unique PfDHODH inhibitors from the computational design derivatives of indolyl-3-ethanone-α-thioethers through models generation via a genetic function algorithm methods. The best model indicates good power of prediction with coefficient of determination, R2 = 0.9482, adjusted coefficient of determination (\({\text{R}}_{\text{adj}}^{2}\)) = 0.9288, Leave one out cross-validation coefficient (Q2) = 0.9201 and the external validation (\({\text{R}}_{\text{pred}}^{2}\)) = 0.6467. The contribution of every descriptor in the model was investigated through finding their mean effect to (pIC50) the activities of the compounds. With MATS5m (− 0.11725), RDF75m (− 0.12097), VE3_Dzp (0.14697), and MLFER_BH (1.08528) contributing more to the model, while AATSC8p (− 0.04833) and minHBa (0.05430) contributed the least to the model. Hence, the mean effect indicated MLFER_BH to be the most relevant descriptor, which aided the design of five derivatives of indolyl-3-ethanone-α-thioethers. All the designed antimalarial compounds were deeply docked within the binding region thereby forming several hydrogens and hydrophobic bonds leading to the generation of better binding affinity and high binding scores (− 156.181 kcal/mol) compared to the design template (− 138.201 kcal/mol) and the standard drug (− 128.467 kcal/mol). Furthermore, all the five designed antimalarial compounds were found to be better bonded to the binding pocket of PfDHODH than other compounds reported by other researchers.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Plasmodium is the causative organism of malaria, the most destructive disease worldwide [1], and is transferred between humans through infected Anopheles mosquitoes. The global cases of malarial were put at 228 million yearly with 405,000 deaths, with children under the ages of 5 are the most affected which account for 585,000 (67%) of all cases [2]. Murray and Perkins in 1996 [3], reported various species of Plasmodium, out of which the most savage of all the species of this genus is P. falciparum [4]. Malaria death rates may have decreased in recent years, the disease mortality figures are still on the high side even though the disease is preventable and treatable. This is largely due to loss of efficacy of antimalarial drugs in clinical use such as Chloroquine, Amodiaquine, Pamaquine, and Mefloquine, as a result of increasing drug resistance to the malarial problem [5].
Therefore, the development of new alternative agents that takes into account the problem associated with multiple drug resistance is highly necessary. Series of inquests on the inhibitory efficiencies of several derivatives against a target protein by testing different molecular structures. The inquest was centered on natural products as medicinal plants [6], marine organisms [7] or even bacteria [8] in addition to synthetic processes to prepare new heterocyclic [9] and organometallic [10] compounds. New indolyl-3-ethanone-α-thioethers derivatives were reported to have improved activity for inhibiting antimalarial action [11]. P. falciparum parasites rely mostly on nucleotide synthesis through the de novo pathway to provide the necessary precursor for DNA and RNA biosynthesis, unlike human cells that salvage preformed pyrimidine based as well as pyrimidine biosynthesis from the host cell through the de novo path. Plasmodium metabolic pathways are different from those of human hosts. Hence, aiming purine and pyrimidine metabolic pathways gives a promising route for unique drug development [12]. The oxidation of l-dihydroorotate (DHO) to produce orotate, is catalyzed by Dihydroorotate dehydrogenase enzyme as part of the fourth and rate-limiting step of the pyrimidine biosynthesis pathway [13].
Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) was vital for parasite growth and was proven to be an antimalarial drug target for the antimalarial agents [14]. Several agents of triazolopyrimidine, benzamide, naphthamide and urea were reported to inhibit PfDHODH [15]. The major challenges facing the use of these antimalarial drugs are lack of antimalarial vaccine and the resistance of P. falciparum to the available drugs. These challenges associated with antimalarial compounds lead to the development of a quantitative structure–activity relationship (QSAR) model. QSAR is the mathematical model relating the structure of compounds with their biological activities. This research was aimed at relating some structural features of indolyl-3-ethanone-α-thioethers derivatives with their biological activities through the use genetic function algorithm (GFA) calculations and hence, lead to the design of new antimalarial compounds. More also, the binding modes of the active sites of the hypothetical antimalarial compounds were investigated by performing the molecular docking of the designed compounds.
2 Materials and methods
2.1 Experimental dataset
Thirty-one indolyl-3-ethanone-α-thioethers derivatives were used as a data set. Their structural formula and antimalarial activity values against P. falciparum were obtained from the literature [11]. The activities of the compounds expressed as IC50 (μM) (50% inhibition growth concentration against the parasite P. falciparum) were converted to pIC50 (−Log10 IC50) as presented in Table 1.
2.2 Geometry optimization and calculation of descriptors
The 2D molecular structure of the data sets was produced from the molecular sketched in the Spartan14 [16]. These were subsequently converted to 3D by view module in the software. Conformational geometries were optimized using density functional theory (DFT) by invoking B3LYP [17] and a basis set of 6-311G* to provide a precise conformer relation throughout the compounds. The energy minimized structures were ported to PaDEL-Descriptor used to compute various descriptor classes ranging from 0D, 1D, 2D, and 3D-classes of Chemometric molecular descriptors [18].
2.3 Data pre-treatment
The pre-treatment of the molecular descriptors involves the removal of constant values descriptors together with those variables having high correlation coefficient values using “Data Pre-Treatment GUI 1.2” software that employs the V-WSP program [19, 20].
2.4 Model development and selection
The model was built using the Molegro Data Modeller software where the descriptors and activities of the compounds were imported into the Molegro worksheet. The software randomly separated the dataset into two sets by setting 74% of the data sets (23 compounds) as the training sets and 26% of the data set (08 compounds) as the test sets. After selecting the training sets, the modeling option of the toolbar was selected were where all the active descriptors were all selected while the invaluable descriptors were frozen, and the regression analysis was carried out to develop the model. The choice of models constructed was done based on the conditions of R2, Q2, and \(R_{\text{pred}}^{2}\) [21, 22].
2.5 QSAR model validation
The generated model was used on the test set to predict the activity of the set and the result obtained was analyzed for the existence of systematic error in the models [23]. In the absence of systematic error, the models was validated both internally and externally. Internal validation was done with the training set data only using the leave-one-out (LOO) cross-validation technique. In the LOO, the training set was altered by discarding a data set compound and using the remaining data to construct a model using the validating model descriptors. The new equation obtained was subjected to predicting the activity of the discarded compound. This cycle was redone down to when all the molecules of the data set had been removed a single time.
2.6 Descriptor relevance (mean effect)
The influence of the calculated descriptors towards activities of the generated model was measured in terms of the mean effect. The mean effect was obtained from Eq. 1.
where \(\beta_{j}\) conforms with the descriptor j’s coefficient, Dj conforms with each value of matrix descriptor in the training set and m conforms with the tally of model descriptors present and n stands for the tally of molecules used as training set [24].
2.7 Models applicability domain (AD)
The plot of standardized residuals against leverage values (William’s plot), was employed to interpret the relevant area of the model in terms of biological territory. The leverage strategies of applicability domain was utilized in this study [25] where compounds are assigned specific leverages based on their descriptor and is expressed as: \(h_{i } = x_{i}^{T} \left( {X^{T} X} \right)^{ - 1} x_{i}\), where \(x_{i}\), stand for the row-vector descriptor of the concern compound i, and the training set descriptor matrix was represented as n × k. The caution leverage (h*) represent the limit of typical values for anomalies of X and it’s expressed as: \(h^{*} = 3\left( {p + 1} \right)/n\) where n stands for the sum of training compounds, and p the sum of model descriptors present. The compounds that have their leverages \(h_{i}\) value greater than caution leverage (h*) and standardized residual values greater than within plus or minus three standard deviation units were seen as anomalies [24].
2.8 Molecular docking studies
The software, Molegro Virtual Docker (MVD) predicted protein–ligand interactions form on fresh exploration methods that blend differential transformation with a cavity prediction method [26]. High resolution 1.50 Å crystal structure of P. falciparum dihydroorotate dehydrogenase (PfDHODH) (PDB: 4ORI) was obtained from the protein data bank. It was first extracted and opened with Material Studio software where the protein was first treated by removing water molecules, ligand groups, ions and heteroatoms contained in the pdb files while hydrogen was added to the protein component and saved. The saved file was then imported into the Molegro Virtual Docker where the binding pocket was defined with the aid of Molegro Virtual Docker cavity detection algorithm and the docking was performed to predicting the binding mode of the ligand and the target protein in form of scoring function. The MolDock scoring function is originally Gehlhaar’s piecewise linear potential (PLP) that was expanded to including new hydrogen bonding and electrostatic terms [27,28,29].
3 Results and discussion
3.1 QSAR results
After meticulous authentication and inspection, the selected model alongside its validation parameter is presented below;
The selection was by virtue of the significance of the parameters as it has the largest value of \({\text{R}}^{ 2} = 0. 9 4 8 2,\;{\text{R}}_{\text{Adj}}^{2} = 0. 9 2 8 8,\;{\text{Q}}_{\text{cv}}^{2} = {\text{of }}0. 9 20 1\;{\text{and}}\;{\text{R}}_{\text{ext}}^{2} = 0. 6 4 6 7\). The internal, as well as the external validation parameters of the model, were in agreement with the minimum standard for a dependable and powerful model. An increase in physicochemical parameters of descriptors MATS5m, VE3_Dzp, minHBa, and RDF75m will increase inhibitory activities of indolyl-3-ethanone-α-thioethers derivatives against P. falciparum dihydroorotate dehydrogenase since their coefficients are positive. Likewise, descriptors with negative coefficients such as AATSC8p and MLFER_BH implies that inhibitory activities of indolyl-3-ethanone-α-thioethers derivatives will increase against PfDHODH enzyme with decreasing values of the descriptors.
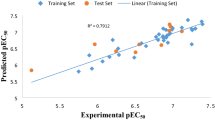
The plot of experimental activities versus the predicted activities of the data sets is shown in Fig. 1 and the accuracy of the best model was confirmed as the predicted R2 value concords with R2 = 0.8494 reported graphically. The predictive strength of the model is in the high linearity of the plot. Table 2 compares the predictive pIC50 and that of the experimental pIC50 with the residual values very low confirming the predictability of the mode. External model validation shows an accurate relation among the experimental and predicted pIC50 of the test set.

Experimental activity plotted against predicated activity for training and tests sets of the model
The relevance of the descriptors as well as the correlation between them was reflected in Table 3. Out of the six descriptors in the model, the mean effect statistical analysis (Table 3) revealed that the descriptors such as AATSC8p (Average centered Broto-Moreau autocorrelation—lag 8/weighted by polarizabilities), MATS5m (Moran autocorrelation—lag 5/weighted by mass), RDF75m (Radial distribution function—075/weighted by relative mass), minHBa (Minimum E-States for (strong) Hydrogen Bond acceptors), VE3_Dzp (Logarithmic coefficient sum of the last eigenvector from Barysz matrix/weighted by polarizabilities), and MLFER_BH (overall or summation solute hydrogen bond basicity) were reported in order of increasing contribution strength. From Table 3, MLFER_BH (overall or summation solute hydrogen bond basicity) [30] descriptor with mean effect value of 1.08528 was revealed to have contributed most toward the QSAR model development.
3.2 Applicability domain of the model
A close observation of the applicability domain for the training as well as the test set objects (Fig. 2) shows that no compound appeared beyond the warning value (h* = 0.913). Indicating lack of outliers (h > h* = 0.913), i.e. no compound (either a training or test sets) is a typical anomaly within the cut off value of ± 3.0σ. Hence, the model was considered to have as good predictions.

Williams plot for an external validation of activities of indolyl-3-ethanone-α-thioethers derivatives. Cut-off value h* = 0.913
3.3 In-silico design of antimalarial compound
The molecule with serial number 22 in the dataset (Table 1) was used as a template (Fig. 3) to design its several hypothetic novel derivatives. The template was chosen because of it relatively high activity i.e. pIC50 = 7.0458, very good AD’s leverage value and excellent standardized residual. The design of the derivatives was guided by the information obtained from the descriptors contained in the model. For example, MLFER_BH descriptor as earlier explained was found to be the most influential descriptor given the magnitude of its mean effect (Table 3). Therefore, the addition of electrophiles (electron-withdrawing groups) to the template will increase the antimalarial activity of the novel compounds. The template was modified through the addition and removal of a variety of substituents such as –Br, –Cl, and –NO2 groups. The five compounds designed (Table 4) were found to have better activities than all the experimental compounds (Table 1). Also, three of the designed compounds; 1-(5-bromo-1H-indol-3-yl)-2-((4-nitrophenyl)thio)ethanone (pIC50 = 7.8893), 2-((4-chlorophenyl)thio)-1-(5-nitro-1H-indol-3-yl)ethanone (pIC50 = 7.9520) and 1-(5-nitro-1H-indol-3-yl)-2-((4-nitrophenyl)thio)ethanone (pIC50 = 8.2129) were found to have better activities than the standard drug (chloroquine) (pIC50 = 7.5528), with compound 1-(5-nitro-1H-indol-3-yl)-2-((4-nitrophenyl)thio)ethanone (pIC50 = 8.2129) having the overall better activity.

Designed template, compound 22, 2-((4-bromophenyl)thio-1-(5-chloro-1H-indol-3-yl)ethanone, with pIC50 = 7.0458, a leverage value of 0.240182, and standardized residual value of − 0.12544
3.4 Molecular docking studies results of indolyl-3-ethanone-α-thioethers derivatives
PfDHODH is an enzyme in the mitochondrial that catalyzes it reactions in the presents of both Flavin mononucleotide (FMN) and coenzyme Q (CoQ). Two half-reactions are required for the catalysis to result: catalytic oxidation of dihydroorotate by FMN, followed by catalytic reoxidation of FMN by CoQ. In PfDHODH, the catalytic domain (β/α-barrel fold in the inner membrane space) is formed by amino-acid residues 162–565. And the residues to the N-terminus of this domain is saddled with anchoring the protein to the inner mitochondrial membrane [15]. Majority of DHODH inhibitors are attached to the assumed CoQ binding site, which is located adjacent to FMN between the β/α-barrel domain and the N-terminal α-helical membrane domain. The differences in amino acid sequence between the Plasmodium and human enzyme, in the inhibitor-binding site was identified to build the species-selectivity of this inhibitors including triazolopyrimidine-based PfDHODH inhibitors [31, 32]. The docking analysis showcased the preferred binding-conformation of designed derivatives (ligands) to the CoQ binding site of the target protein. These confirmation aid in predicting the nature and strength of interaction between the ligands and the target molecule. The structure of P. falciparum dihydroorotate dehydrogenase (PfDHODH) with the target site as indicated in Fig. 4. The docking result of the designed derivatives, template and standard drug were displayed in Table 5. The MolDock Score of design derivatives are 22A (− 136.818 kcal/mol), 22B (− 133.376 kcal/mol), 22C (− 141.336 kcal/mol), 22D (− 124.645 kcal/mol), and 22E (− 134.756 kcal/mol) as revealed in Table 5. The designed compounds all have a higher binding affinity that design template with the exception of compound 22D that has its binding energy lower than that of the standard drug. Compound 22C (− 141.336 kcal/mol) was found to have the highest binding affinity as such is more compatible with the receptor than its co-designed compounds as well as even the standard drug. Compounds 22A and 22C are the most active ligands as reflected by their docking affinities in Table 5. Various interactions between these compounds and the target protein as shown in Fig. 5 revealed that for compound 22A, which include but not limited to two hydrogen bonding interactions, one each for conventional hydrogen bonding and carbon-hydrogen bonding both between amino acid residue, HIS56 and carbonyl oxygen of the inhibitor, distance of 2.80 Å, and 2.26 Å respectively in addition of several other hydrophobic interactions. Compound 22C has two conventional hydrogen bonds, the first between amino residue TYR356 and carbonyl oxygen atom of the inhibitor, distance 2.33 Å and the second between hydrogen atom of indole ring with SER305, distance 2.30Å in addition to four hydrophobic interactions (an amide-Pi stacked between ALA55 and benzene ring, distance 4.31 Å; and three Alkyl, between indole ring and VAL143, distance 4.28 Å and between benzene ring and ALA143 and ILE360, distance 4.37 Å and 3.74 Å respectively). These interactions show the binding role of oxygen, hydrogen and carbon atoms as well as their inhibitory capacities.

Ribbon diagram showing the indolyl-3-ethanone-α-thioethers binding site on PfDHODH. Indolyl-3-ethanone-α-thioethers is displayed as IEαT, FMN, and L-orotate

2D and 3D docking poses showing interactions of compounds 22A and 22C in the binding sites of PfDHODH
3.5 Conclusion
QSAR techniques applied to an antimalarial derivative, indolyl-3-ethanone-α-thioethers relate the molecular structures of the compounds and their antimalarial activities. Genetic Function Algorithm (GFA) was used to produce a predictive, reliable and robust model. The internal and external validation, R2 values for the model were found to be 0.9482 and 0.6467 respectively. The descriptors responsible for the antimalarial activities revealed by the model are AATSC8p, MATS5m, VE3_Dzp, minHBa, MLFER_BH, and RDF75m. With MLFER_BH having the greatest influence on the activity as revealed by the mean effect. These descriptor was decisive in the design five hypothetical derivatives of indolyl-3-ethanone-α-thioethers with better activity against PfDHODH. The analysis of the docking studies carried out between these potential inhibitors and their target protein (PfDHODH) shows how design compounds inhibit PfDHODH by acting on the binding site. The most active hypothetical inhibitor of P. falciparum dihydroorotate dehydrogenase (PfDHODH) with docking score of − 141.336 kcal/mol interact with active site TYR356 and SER305 which play a decisive role in inhibiting the target protein. The findings of this study could represent good drug candidates for the treatment of malaria.
References
Mistry SN, Drinkwater N, Ruggeri C, Kannan Sivaraman K, Loganathan S, Fletcher S, Drag M, Paiardini A, Avery VM, Scammells PJ, McGowan S (2014) Two-pronged attack: Dual inhibition of Plasmodium falciparum M1 and M17 metalloaminopeptidases by a novel series of hydroxamic acid-based inhibitors. J Med Chem 57:9168–9183. https://doi.org/10.1021/jm501323a
World Health Organization (2019) World malaria report. http://apps.who.int/malaria/media/world-malaria-report-2019. Accessed 4 May 2020
Murray MC, Perkins ME (1996) Chapter 15. Chemotherapy of malaria. In: Annual reports in medicinal chemistry, vol 31, pp 141–150
Newton P, DPhil BM, White N (1999) MALARIA: new developments in treatment and prevention. Ann Rev Med 50(1):179–192
Guantai EM, Ncokazi K, Egan TJ, Gut J, Rosenthal PJ, Smith PJ, Chibale K (2010) Design, synthesis and in vitro antimalarial evaluation of triazole-linked chalcone and dienone hybrid compounds. Bioorg Med Chem 18(23):8243–8256. https://doi.org/10.1016/j.bmc.2010.10.009
Muthaura CN, Keriko JM, Derese S, Yenesew A, Rukunga GM (2011) Investigation of some medicinal plants traditionally used for treatment of malaria in Kenya as potential sources of antimalarial drugs. Exp Parasitol 127(3):609–626. https://doi.org/10.1016/j.exppara.2010.11.004
Wright AD, König GM, Angerhofer CK, Greenidge P, Linden R (1996) Desqueyroux-Faúndez. J Nat Prod 59:710e716
Papireddy K, Smilkstein M, Kelly JX, Shweta T, Salem SM, Alhamadsheh M, Haynes SW, Challis GL, Reynolds KA (2011) Antimalarial activity of natural and synthetic prodiginines. J Med 54:5296–5306
Calas M, Ouattara M, Piquet G, Ziora Z, Bordat Y, Ancelin ML, Escale R, Vial H (2007) Potent antimalarial activity of 2-aminopyridinium salts, amidines, and guanidines. J Med Chem 50(25):6307–6315. https://doi.org/10.1021/jm0704752
Bellot F, Coslédan F, Vendier L, Brocard J, Meunier B, Robert A (2010) Trioxaferroquines as new hybrid antimalarial drugs. J Med Chem 53(10):4103–4109. https://doi.org/10.1021/jm100117e
Svogie AL, Isaacs M, Hoppe HC, Khanye SD, Veale CGL (2016) Indolyl-3-ethanone-α-thioethers: a promising new class of non-toxic antimalarial agents. Eur J Med Chem 114:79–88. https://doi.org/10.1016/j.ejmech.2016.02.056
Cassera MB, Zhang Y, Hazleton KZ, Schramm VL (2011) Purine and pyrimidine pathways as a target in Plasmodium falciparum. Curr Top Med Chem 11:2103–2115
Pavadai E, El Mazouni F, Wittlin S, de Kock C, Phillips MA, Chibale K (2016) Identification of new human malaria parasite Plasmodium falciparum dihydroorotate dehydrogenase inhibitors by pharmacophore and structure-based virtual screening. J Chem Inf Model 56(3):548–562. https://doi.org/10.1021/acs.jcim.5b00680
Nigussie D, Beyene T, Shah NA, Belew S (2015) New targets in malaria parasite chemotherapy: a review. Malaria Contr Elimin S1:S1–007
Phillips AM, Rathod KP (2010) Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect Disorders Drug Targets 10(3):226–239
Shao Y, Molnar LF, Jung Y, Kussmann J, Ochsenfeld C, Brown ST (2006) Advances in methods and algorithms in the modern quantum chemistry program package. Phys Chem Chem Phys 8:3172
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation energy formula into a functional of the electron density. Phys Rev 37:785–789
Yap CW (2011) PaDEL-Descriptor: open-source software to calculate molecular descriptors and fingerprints. J Comput Chem 32(7):1466–1474
Panagos P, Meusburger K, Ballabio C, Borrelli P, Alewell C (2014) Soil erodibility in Europe: a high-resolution dataset based on LUCAS. Sci Total Environ 479(480):189–200. https://doi.org/10.1016/j.scitotenv.2014.02.010
Roy K, Kar S, Ambure P (2015) On a simple approach for determining the applicability domain of QSAR models. Chemometr Intell Lab Syst 145:22–29. https://doi.org/10.1016/j.chemolab.2015.04.013
Golbraikh A, Tropsha A (2002) Beware of q2! J Mol Graph Model 20:269–276
Afantitis A, Melagraki G, Sarimveis H, Igglessi-Makorpoulon O, Kollias G (2009) A novel QSAR model for predicting the inhibition of CXCR3 receptor by 4-N-aryl-[1,4] diazepane ureas. Eur J Med Chem 44:877–884
Roy K, Das RN, Ambure P, Aher RB (2016) Beware of error measures. Further studies on the validation of predictive QSAR models. Chemometr Intell Lab Syst 152:18–33
Minovski N, Župerl S, Drgan V, Novič M (2013) Assessment of applicability domain for multivariate counter-propagation artificial neural network predictive models by minimum Euclidean distance space analysis: a case study. Anal Chim Acta 759:28–42
Gramatica P, Giani E, Papa E (2007) Statistical external validation and consensus modeling: a QSPR case study for K oc prediction. J Mol Graph Model 25(6):755–766
Babu PA, Colluru VTSS, Anaparthy N (2012) In-silico characterization of ECE-1 inhibitors. Comput Biol Med 42(4):446–457. https://doi.org/10.1016/j.compbiomed.2011.12.013
Gehlhaar DK, Verkhivker G, Rejto PA, Fogel DB, Fogel LJ, Freer ST (1995) Proceedings of the fourth international conference on evolutionary programming, 1995, pp 615–627
Gehlhaar DK, Bouzida D, Rejto PA (1998) Proceedings of the seventh international conference on evolutionary programming, 1998, pp 449–461
Yang JM, Chen CC (2004) GEMDOCK: a generic evolutionary method for molecular docking. Proteins Struct Funct Bioinf 55:288–304
Platts JA, Butina D, Abraham MH, Hersey A (1999) Estimation of molecular linear free energy relation descriptors using a group contribution approach. J Chem Inf Comput Sci 39(5):835–845. https://doi.org/10.1021/ci980339t
Deng X, Gujjar R, ElMazouni F, Kaminsky W, Malmquist NA, Goldsmith EJ, Rathod PK, Phillips MA (2009) J Biol Chem 284:26999–27009
Deng X, Kokkonda S, El Mazouni F, White J, Burrows JN, Kaminsky W, Charman SA, Matthews D, Rathod PK, Phillips MA (2014) J Med Chem 57:5381–5394
Funding
The authors for this research did not receive any funding concerning this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical standard
No human or animal subjects were involved in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ibrahim, Z.Y., Uzairu, A., Shallangwa, G. et al. QSAR and molecular docking based design of some indolyl-3-ethanone-α-thioethers derivatives as Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) inhibitors. SN Appl. Sci. 2, 1170 (2020). https://doi.org/10.1007/s42452-020-2955-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-2955-1