
Abstract
5,8-Dibromo-13,13-bis(alkyl)dibenzofluorene derivatives functionalized with different alkyl molecules [M2a–M2f] and 2-tert butyl-9,10-di (p-hydroxyphenyl) anthracene [M1b], were designed to research the impact of molecular structure on the optical properties of polymer. Polymers were synthesized by aromatic nucleophilic substitution in moderate to strong yields (60–65%). After structural characterization through several techniques (e.g. FTIR, 1H, and 13C-NMR), special emphasis was put on the study of their optical properties through UV–Vis and photoluminescence spectroscopy. The polymers demonstrated strong thermal stability up to 350 °C and high glass transition temperature (108–133 °C). All of the polymers showed blue emission within a range of 416–433 nm and had a band gap of 2.87–2.99 eV. The electrochemical study reveals that, HOMO levels for polymers were estimated in the range of − 5.22 to − 5.26 eV and LUMO of − 2.23 to − 2.38 eV. The polymers demonstrated strong thermal stability and blue emission within a range of 416–433 nm. These polymeric materials have demonstrated high photoluminescence which may be useful for future display applications as a good source.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Blue-light-emitting materials are favoured among the groups of different optoelectronic materials due to their high thermal stability, high photoluminescence (PL) quantum yield and use as a host in white light emission [1,2,3,4]. Conjugated polymers were the basis for comprehensive research in strongly conjugated organic polymers. Many conjugated polymers with various optical, electrical, and magnetic properties have been synthesized for display applications [5,6,7]. Poly(phenylenevinylene) (PPV), poly(2,7-(9,9-dialkyl)fluorene)s (PFs) and their derivatives have gained considerable attention in recent decades as a promising substrate for full-color flat panel display [8]. Recent OLEDs (Organic Light Emitting Diodes) have made tremendous progress, allowing for the development of thin and light weight display at low applied voltage and high speed response with high brightness. However, under the present conditions the OLED needs to attain a light output with higher brightness or higher conversion efficiency. In fact, the OLED has a lot of reliability problems too. For example, the system undergoes changes associated with its long-term usage over time, or is degraded by oxygen-containing ambient gas, humidity and the like. Moreover, when the device is connected to a full color display, the OLED will emit blue, green, and red lights with high color purity.
Good applications of OLEDs can be found in displays and solid-state lighting [9,10,11,12] due to their dominance such as high power output, eye-friendly emission, light weight, mechanical efficiency, environmental reasons, etc. Polyfluorene derivatives have been of particular interest in recent years due to their excellent thermal stability, large band gap and high levels of solid-state quantum yields. But instability in blue emissions and high potential for ionization limited the use of fluorene derivatives as organoeletronics. During the manufacture and operation of the system, polyfluorene undergo aggregation and/or interchain excimers, resulting in red-shifted emissions and lower performance resulting in thermo- or electro-oxidative degradation of the polyfluorene backbone. The quest for new π-conjugated organic compounds with improved properties remains one of the promising directions in modern materials science chemistry for the production of high-efficiency OLEDs. In addition, a method is proposed for the use of dibenzofluorenes (DBFs) as display material to solve the above problem [13]. Harvey et al. first reported a synthesis of dibenzofluorene [14] with high melting points, glass transition temperatures relative to fluorene moiety. Organic materials based on dibenzofluorenes are demonstrated to have excellent properties for use in luminescent devices, including high stability and efficiency, and low excimer formation. In addition to emissive properties, the products may be advantageously designed to have the capability to transport hole or electron.
Even DBFs have a comparatively shallower HOMO stage. As a consequence, by using the DBFs as a host for the light-emitting layer, the driving voltage can be decreased by reducing the hole injection barrier from the injection/transport hole layer to the light-emitting layer. The carrier balance, which is one of the causes of OLED degradation, is improved by the use of DBFs to prolong the OLED’s life. The DBFs can shallow their HOMO level, while retaining the band gap dimension. Hence, the efficiency of moving energy from the host to the guest is also important. DBFs have a broad steric obstruction (with a non-planner structure) due to the distortion of the skeleton itself, thus inhibiting guest quenching concentration and may be doped at a high concentration which also inhibits excimer light emission. The DBFs also have large amorphism due to distortion of the skeleton itself and can thus form light emitting layers with adequate film characteristics [15,16,17,18,19,20,21,22,23].
Furthermore, due to its high quantum photoluminescent performance due to the orthogonal structure arrangement between phenyl substitutes and anthracene rings, diphenylanthracene is another commonly used moiety in LEPs [24]. Synthesis and sequence analysis of non-symmetric diphenylanthracenes were conducted [25]. Consequently, light-emitting diodes containing polyacene are required to have higher quantity of photoluminescence, blue emission and good system properties [26, 27].
In the present work, we report the synthesis of a series of polyDBF alkyl and allyl substitutes containing diphenylanthracene moiety. In addition, previous studies stated that the problem of low EL and color stability due to PF-associated aggregation was solved with the use of substituted dibenzofluorene monomer instead of fluorene. The crystallinity of synthesized polymers decreases due to the large steric group substitution which can make them ideal for display applications. This work explores the chemical structure, spectroscopic, thermal, photophysical, and electrochemical properties of these polymers.
2 Experimental section
2.1 Materials and methods
All chemicals and reagents were purchased from s. d. fine, Mumbai and Sigma Alderich and were used without any further purification. All of the solvents were used after purification according to conventional method. Moisture sensitive reactions were carried out in a dry reaction vessel under nitrogen atmosphere.
2.2 Synthesis of monomers
2.2.1 Synthesis of 2-t-butyl-9,10-di (p-hydroxyphenyl) anthracene monomer
2.2.1.1 Preparation of 2-tert butyl-9,10-di (p-methoxyphenyl) anthracene [M1a]
To synthesize the Grignard reagent, the solution of p-bromo anisole (32 mmol) in THF has been applied to nitrogen purged magnesium solution (32 mmol) in THF at room temperature. Suspension of 2-tert butyl anthraquinone (3.2 mmol) in THF was added drop wise after stirring the reaction mixture for half an hour, holding temperature between 0 and 5 °C. When addition had been completed, the mixture temperature was raised to 60 °C and kept stirring for 2 h. This was followed by cooling the mixture in ice, to which 30 mL of 20% HCl and 3 g of SnCl2·2H2O were added. The mixture was heated to 60 °C for additional one hour. The collected substance has been refined, washed with water and diethyl ether respectively.
The synthesized compound was well characterized by various techniques (Data included in supporting information).
2.2.1.2 Preparation of 2-tert butyl-9,10-di (p-hydroxyphenyl) anthracene [M1b]
A 2-tert butyl-9,10-di (p-methoxyphenyl) anthracene was de-methylated by refluxing it with HBr (48%) in glacial acetic acid for 24 h at 120 °C in an inert nitrogen atmosphere. The crude product was filtered, washed with water to remove acetic acid and recrystallization carried out in ethanol to get crystalline light yellow solid [8, 22,23,24,25, 28,29,30,31,32,33]. The yield of the product was 86%.
NMR Signals and FTIR signals of 2-tert butyl-9,10-di (p-hydroxyphenyl) anthracene: Solid, 1H NMR (300 MHz, CDCl3, δ ppm): 6.94–7.7 (m, 13H, Ar–H); 3.98 (s, 2H, –OH); 1.3 (s, 9H, 3-CH3); 13C NMR: (75 MHz, CDCl3, δ in ppm) 116.8–157.2 (aromatic carbon); 41.4 (tert carbon); 31.7 (–CH3); IR (cm−1): 3422 (–OH stretch); 3057 (C–H stretch); 1608–1401 (C=C stretch); 811–895 (phenyl group); Mass spectrum: m/z 418 (M+); Anal.Calcd for C30H26O2: C 86.16%; H 6.18%; O 7.66%; Found: C 86.29%; H 6.12%; O 7.59%; M.P-197 °C.
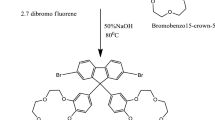
2.2.2 Synthesis of 5,8-dibromo-13,13-bis(alkyl)dibenzofluorene monomer [2]
2.2.2.1 Preparation of 13,13-dialkyl-dibenzofluorene
Under nitrogen atmosphere, dibenzofluorene (1 g, 0.004 mmol), 50% aq. NaOH and a catalytic amount of tertiary butyl ammonium iodide (TBAI) (0.14 g, 0.004 mmol) were added to a flask. Alkyl bromide (3.5 ml, 0.02 mmol) was added slowly to the flask and then heated for 5 h at 100–110 °C. The reaction mixture was cooled and extracted chloroform then organic layer was washed twice with water and dried over anhydrous Na2SO4. Purification of product was done by silica gel column chromatography using PET ether as a solvent [27, 34].
2.2.2.2 Preparation of 5,8-dibromo-13,13-dialkyl-dibenzofluorene
A mixture of 13,13-dialkyl-dibenzofluorene (1 g, 0.003 mmol) and NBS (1.3 g, 0.007 mmol) in DMF was stirred at room temperature for 12 h and then extracted with chloroform. The separated organic layer was washed with brine and dried over anhydrous Na2SO4. The crude product obtained was purified using column chromatography (eluent: petroleum ether: chloroform 9:1) [18].
The similar reactions were carried out with different alkyl bromide derivatives like ethyl bromide [M2a], propyl bromide [M2b], butyl bromide [M2c], pentyl bromide [M2d], hexyl bromide [M2e], allyl bromide [M2f].
2.2.2.3 NMR signals, FTIR signals and elemental analysis of monomers
5,8-Dibromo-13,13-Bis(ethyl)-dibenzofluorene[M2a]: Solid, 1H NMR (300 MHz, δ ppm, CDCl3) 1.09 (t, 6H, –CH3); 2.867 (q, 4H, –CH2); 7.42–8.8 (m, 10H, Ar-ring); 13C NMR (75 MHz, δ ppm, CDCl3)-8.47; 32.67; 61.49; 119.28; 124.75; 127.01; 127.85; 128.48; 129.49; 130.44; 134.95; 139.80; 144.82; IR (KBr cm−1): 3048 (S); 2963 (S); 2928 (W); 1584 (S); 1453 (m); 816 (m); 754 (S); MS (m/z) 480; Anal. Calcd for C25H20Br2: C, 62.5%; H, 4.167% Found: C, 62.34%; H, 4.05%; M.P-210–212 °C.
5,8-Dibromo-13,13-Bis(propyl)-dibenzofluorene [M2b]: Solid, 1HNMR (300 MHz, δ ppm, CDCl3) 0.24 (t, 6H, –CH3); 0.46 (m, 2H, –CH2); 2.67 (m, 2H, –CH2); 7.2–8.5 (10H, Ar-ring); 13C NMR (75 MHz, δ ppm, CDCl3) 14.19; 17.08; 42.43; 60.40; 119.29; 127.74; 124.78; 128.083; 129.183; 129.520; 129.942; 134.921; 139.235; 145.78; IR (KBr cm−1): 3047 (S); 2952 (S); 2926 (W); 1583 (S); 1459 (m); 908 (S); 816 (m); 754 (S); MS (m/z) 508; Anal. Calcd for C27H24Br2: C, 63.77%; H, 4.72% Found: C, 63.64%; H, 4.55%; M.P-215–218 °C.
5,8-Dibromo-13,13-Bis(butyl)-dibenzofluorene[M2c]: Solid, 1HNMR (300 MHz, δ ppm, CDCl3) 0.41 (t, 6H, –CH3); 0.2 (m, 4H, –CH2); 0.89 (m, 4H, –CH2); 2.76 (m, 4H, –CH2); 7.08–8.40 (m, 10H, Ar–H); 13C NMR (75 MHz, δ ppm, CDCl3) 13.52; 22.69; 5.75; 39.90; 60.17; 119.35; 124.38; 127.72; 128.06; 129.10; 129.46; 129.72; 134.92; 139.68; 145.68; IR (KBr cm−1): 3050 (W); 2950 (S); 2927 (W); 1583 (S); 1457 (m); 807 (m); 744 (S); MS (m/z) 536; Anal. Calcd for C29H28Br2: C, 64.92%; H, 5.22% Found: C, 64.78%; H, 5.05%; M.P-255–257 °C.
5,8-Dibromo-13,13-Bis(pentyl)-dibenzofluorene[M2d]: Solid, 1H NMR (300 MHz, δ ppm, CDCl3)-0.5 (t, 6H, –CH3); 0.2 (m, 4H, –CH2); 0.83 (m, 8H, –CH2); 2.74 (m, 4H, –CH2); 7.2–8.2 (m, 10H, Ar–H); 13C NMR (75 MHz, δ ppm, CDCl3)-13.88; 22.39; 23.26; 31.85; 40.07; 60.44; 119.317; 124.745; 127.73; 128.09; 129.16; 129.45; 129.70; 134.93; 139.30; 145.71; IR (KBr cm−1): 3052 (W); 2947 (S); 2925 (W); 1582 (S); 1465 (m); 1072 (W); 817 (m); 755 (S); MS (m/z) 564; Anal. Calcd for C31H32Br2: C, 65.95%; H, 5.67% Found: C, 65.34%; H, 5.35%; M.P-231–234 °C,
5,8-Dibromo-13,13-Bis(hexyl)-dibenzofluorene[M2e]: Solid, 1H NMR (300 MHz, δ ppm, CDCl3) 0.58 (t, 6H, –CH3); 0.25 (m, 4H, –CH2); 0.85 (m, 12H, –CH2); 2.69 (m, 4H, –CH2); 7.2–8.35 (m, 10H, Ar–H); 13C NMR (75 MHz, δ ppm, CDCl3)-14.10; 22.34; 23.7; 29.7; 31.5; 40.3; 60.25; 124.53; 125.25; 125.65; 125.97; 126.51; 128.40; 129.55; 129.88; 133.62; 138.32; IR (KBr cm−1): 3052 (W); 2951 (S); 2923 (W); 1588 (S); 1466 (m); 1086 (W); 805 (m); 736 (S); MS (m/z) 592; Anal. Calcd for C33H36Br2: C, 66.89%; H, 6.08% Found: C, 66.69%; H, 6.05%; M.P-225–226 °C.
5,8-Dibromo-13,13-Bis(allyl)-dibenzofluorene[M2f]: Solid, 1H NMR (300 MHz, δ ppm, CDCl3) 3.6 (d, 4H, –CH2); 4.9 (m, 2H, trans 2 proton); 4.2 (d, 2H, cis 2 proton); 4.5 (d, 2H, trans 2 proton); 7.1–8.4 (10H, Ar–H); 13C NMR (75 MHz, δ ppm, CDCl3)-39.32; 43.50; 116.14; 118.18; 123.19; 124.69; 127.17; 128.46; 129.28; 130.41; 133.10; 133.33; 138.86; 144.05; IR (KBr cm−1): 3051 (W); 2925 (W); 1591 (S); 1459 (m); 1057 (W); 812 (m); 742 (S); MS (m/z) 502; Anal. Calcd for C27H18Br2: C, 64.54%; H, 3.58% Found: C, 64.43%; H, 3.60%; M.P-205–206 °C.
2.3 General polymerization
A mixture of 5,8-dibromo-13,13-dialkyldibenzofluorene monomer (0.231 g, 0.001 mol) and 2-tert butyl-9,10-di(p-hydroxyphenyl)anthracene monomer (0.362 g, 0.001 mol) was taken in a three-necked, round bottom flask. Co-solvent of toluene and dioxane (1:2) and potassium carbonate was used as a base in this mixture and 12 h of reaction mixture was refluxed. Upon completion of polymerization, excess 5,8-dibromo-13,13-dialkyldibenzofluorene was added for end capping of the hydroxyl group. The temperature of the reaction was then lowered to room temperature and applied to water. The polymer was extracted using dichloromethane and precipitated in methanol. The obtained polymer was dried in an oven [27, 28]. The brown colored polymers were obtained with a yield of 60–65%.
DBF-diphenylacene polymer series have been synthesized using the same reaction scheme;
2.3.1 NMR signals, FTIR signals, elemental analysis, molecular weight of polymers
Poly[2-t-butyl-9,10-diphenylanthracene-co-13,13-bis(ethyl)dibenzofluorene] Poly[TBDPA-EDBF] [P1]: Solid, 1H NMR (300 MHz, δ ppm, CDCl3): 1.192 (s, 9H, –C(CH3)3); 0.9–2.77 (m, alkyl proton); 7.45–8.8 (m, ArH); 13C NMR (75 MHz, δ ppm, CDCl3): 8.77; 31.2; 32.57; 42.92; 61.79; 120.28; 125.75; 126.01; 126.85; 127.48; 129.19; 131.44; 135.05; 139.99; 145.82; IR (KBr cm−1): 3055 (s) [C–H, aromatic]; 1606.70 (s) [C=C, aromatic]; 2929.87 (s) [aliphatic C–H]; 1504.48 (b) [C–C, aromatic]; 1041.56 (s) [C–O–C, ether linkage]; Mw/Mn: (15,220/10,080) = 1.51, Anal calcd for: C770H616O14: C, 91.67; H 6.11; O 2.22%. Found: C 91.57, H 6.17; O 2.26%.
Poly[2-t-butyl-9,10-diphenylanthracene-co-13,13-bis(propyl)dibenzofluorene] Poly[TBDPA-PrDBF] [P2]: Solid, 1H NMR (300 MHz, δ ppm, CDCl3): 1.86 (s, 9H, –C(CH3)3); 0.24–2.9 (m, alkyl proton); 6.5–8.12 (m, ArH);13C NMR (75 MHz, δ ppm, CDCl3):12.19; 15.08; 31.33; 40.43; 41.11; 61.40; 118.29; 127.74; 124.78; 128.18; 129.98; 130.52; 132.94; 133.92; 141.23; 147.78; IR (KBr cm−1): 3058.94 (s) [C–H, aromatic]; 1602.39; (s) [C=C, aromatic]; 2931.80 (s) [aliphatic C–H]; 1507.66 (b) [C–C, aromatic]; 1033.85 (s) [C–O–C, ether linkage]; Mw/Mn: (14,473/11,220) = 1.29, Anal calcd for: C855H720O15: C, 91.44; H 6.42; O 2.14%. Found: C 91.50, H 6.39; O 2.11%.
Poly[2-t-butyl-9,10-diphenylanthracene-co-13,13-bis(butyl)dibenzofluorene]Poly[TBDPA-BuDBF] [P3]: Solid, 1H NMR (300 MHz, δ ppm, CDCl3): 1.45 (s, 9H, –C(CH3)3); 0.2–2.47 (m, alkyl proton); 7.08–8.40 (m, ArH); 13C NMR (75 MHz, δ ppm, CDCl3):12.52; 21.69; 24.75; 31.29; 39.99; 40.18; 61.27; 118.35; 123.88; 128.02; 129.16; 130.10; 130.46; 131.72; 135.32; 139.88; 146.68; IR (KBr cm−1): 3049 (s) [C–H, aromatic]; 1589.34 (s) [C=C, aromatic]; 2956.23 (s) [aliphatic C–H]; 1492.90 (b) [C–C, aromatic]; 1034.71 (s) [C–O–C, ether linkage]; Mw/Mn: (14,425/10,088) = 1.43, Anal calcd for: C767H676O13: C, 91.24; H 6.70; O 2.06%. Found: C 91.12, H 6.79; O 2.09%.
Poly[2-t-butyl-9,10-diphenylanthracene-co-13,13-bis(pentyl)dibenzofluorene] Poly[TBDPA-PeDBF] [P4]: Solid, 1H NMR (300 MHz, δ ppm, CDCl3): 1.286 (s, 9H, –C(CH3)3); 0.96–3.1 (m, alkyl proton); 7.2–8.9 (m, ArH);13C NMR (75 MHz, δ ppm, CDCl3):13.98; 23.49; 25.26; 32.01; 33.95; 41.01; 42.07; 61.14; 118.817; 125.845; 128.13; 129.21; 128.96; 130.45; 130.17; 136.03; 141.2; 144.91; IR (KBr cm−1): 3038.82 (s) [C–H, aromatic]; 1604.26 (s) [C=C, aromatic]; 2933.73 (s) [aliphatic C–H]; 1510.70 (b) [C–C, aromatic]; 1041.56 (s) [C–O–C, ether linkage]; Mw/Mn: (20,019/12,060) = 1.66, Anal calcd for: C915H840O15: C, 91.04; H 6.97; O 1.99%. Found: C 91.20, H 6.87; O 1.93%.
Poly[2-t-butyl-9,10-diphenylanthracene-co-13,13-bis(hexyl)dibenzofluorene] Poly[TBDPA-HxDBF] [P5]: Solid, 1H NMR (300 MHz, δ ppm, CDCl3): 1.321 (s, 9H, –C(CH3)3); 0.25–2.69 (m, alkyl proton); 7.2–8.35 (m, ArH); 13C NMR (75 MHz, δ ppm, CDCl3):12.10; 21.34; 25.17; 28.97; 30.98; 32.05; 40.88; 41.03; 60.25; 124.53; 126.15; 127.15; 128.07; 126.51; 128.40; 128.85; 129.88; 132.92; 140.22;IR (KBr cm−1): 3049.22 (s) [C–H, aromatic]; 1614.66 (s) [C=C, aromatic]; 2914.08 (s) [aliphatic C–H]; 1510.70 (b) [C–C, aromatic]; 1042.91 (s) [C–O–C, ether linkage]; Mw/Mn: (14,296/10,816) = 1.38, Anal calcd for: C819H780O13: C, 90.87; H 7.21; O 1.92%. Found: C 90.75, H 7.23; O 2.02%.
Poly[2-t-butyl-9,10-diphenylanthracene-co-13,13-bis(allyl)dibenzofluorene] Poly[TBDPA-ADBF] [P6]: Solid, 1H NMR (300 MHz, δ ppm, CDCl3): 1.386 (s, 9H, –C(CH3)3); 3.732(d, 4H, –CH2); 5.323(m, 2H, trans 2 proton); 3.994(d, 2H, cis 2 proton); 4.921(d, 2H, trans 2 proton); 6.974–8.368 (m, 25H, ArH); 13C NMR (75 MHz, δ ppm, CDCl3):32.07; 39.32; 39.99; 44.80; 117.64; 116.88; 122.10; 124.69; 126.67; 128.46; 129.98; 129.91; 134.20; 134.33; 138.89; 147.01;IR (KBr cm−1): 3051.41 (s) [C–H, aromatic]; 1606.13 (s) [C=C, aromatic]; 2957.83 (s) [aliphatic C–H]; 1502.15 (b) [C–C, aromatic]; 1038.25 (s) [C–O–C, ether linkage]; Mw/Mn: (17,298/11,160) = 1.55, Anal calcd for: C855H660O15: C, 91.94; H 5.91; O 2.15%. Found: C 91.78, H 6.05; O 2.17%.
3 Results and discussion
3.1 Characterization
FTIR spectra of all Poly[DBF]s were recorded in KBr discs on a spectrophotometer of the Perkin-Elemer 1600 series. 1H and 13C NMR spectra were recorded on a Bruker AMX-300 spectrometer in CDCl3 and chemical shift reported in ppm values with TMS (tetramethylsilane) as an internal standard. The elemental analysis of C, H and N was performed by a microanalytical technique on the Euro Vector EA 3000 instrument. Thin Layer Chromatography analysis was conducted to confirm the purity of the compound on silica gel G (merck) coated plates. Melting points were determined by model Thermo cal, Analab scientific instrument Pvt Ltd. Molecular weights of the synthesized monomers were determined by GC–MS shimadzu GCMS/QP 2010 and for polymers by Perkin Elmer series 200 GPC equipped with an isocratic pump, a solvent degasser, a column oven, a refractive index (RI) detector, and Chromatographic Column PLgel 10 m Mixed-B, 300μ 7.5 mm [tetrahydrofuran (THF) 40 °C, flow rate of 1 mL min−1 and polystyrene was used for calibration. X-ray diffraction technique has studied the compound’s crystalline behaviours. The experiments were carried out using shimadzu XRD-7000 X-ray diffractrometer. The XRD pattern was obtained using Cu (Kα) (λ = 1.542 Ǻ) radiation that was monochromatised with a curved Graphite crystal monochromator and nickel filter. Shimadzu UV-2100 spectrophotometer and Perkin-Elmer Instruments LS55 Luminescence Spectrometer used to study absorption and emission spectra of synthesized compound with wavelength range 200–800 nm. Using Hitachi S-4800 field emission scanning electron microscope (Japan), SEM was investigating the surface morphological properties of polymers (Figs. 1, 2).

Synthesis of 2-t-butyl-9,10-di (p-hydroxyphenyl) anthracene monomer (1)

Synthesis of 5,8-dibromo-13,13-bis(alkyl)dibenzofluorene monomer (2)
3.2 Structural characterization
The structural characterization of the monomers and the polymers was done by 1H, 13C NMR, and FTIR spectroscopy. The first important feature of the polymer infrared spectra was the absence of absorption peaks at 3200–3500 cm−1 suggesting completion of polymerization and termination of the TBDPA (2-tert butyl-9,10-di (p-hydroxyphenyl) anthracene) terminal hydroxy group monomers. Another important feature of the spectra was the absence of a band at 696.96 cm−1, indicating the removal during polymerization of Br atoms of alkyl substituted dibenzofluorene monomer suggesting the removal of HBr molecule during polymerization. Due to C–O–C stretching vibrations of ether binding on the main polymer backbone, the high absorption peak in the range 1033.85–1042.81 cm−1 was observed in the spectra. Formation of polymers via removal of HBr molecule was confirmed by this observation. Due to the frequency of C=C vibration of conjugated aromatic rings the broad band in the range 1589.34–1614.66 cm−1 was observed in the infrared spectra (Fig. 3).

Synthesis of polymers (P1–P6)
3.3 Optical properties
In dilute THF solution the solution-phase UV–Vis absorption and emission spectra were performed. Until calculation, the solvent THF was purged with nitrogen to prevent the risk of concentration quenching, and the concentration was around 10 ppm. Optical analysis on the basis of their absorption and emission spectra was carried out for the polymers. Figure 4a, b reflect normalized UV–Visible and PL spectra of synthesized polymers and summarize their data in Table 1. The peaks in the region of 264–270 nm were observed in the UV absorption range, which were characteristic peaks for diphenylanthracene [35] and another peak in the region of 370–396 nm due to the transition from dibenzofluorene in the backbone of the main polymer [36].

a Normalised overlay absorption spectra of polymers in THF. b Normalised overlay emission spectra of polymers in THF
Analysis of photoluminescence showed that high intensity peaks were obtained for synthesized polymers in the wavelength range of 416–433 nm that is characteristic of blue emission [17]. The polymers’ emission strength indicated the polymers would be extremely luminescent. P1, P2, P3 and P4, P5 emissions were found to be 432 nm, 431 nm, 433 nm, 429 nm, 433 nm respectively, while P6 emissions were found to be 416 nm (Fig. 4b). The solution of polymers showed a strong luminescence on UV excitation as shown in (Table 1).
It was also noted that wide emission spectrum was exhibited for the group of synthesized polymers dibenzofluorene which enhances the displacement of the geometry of equilibrium from its ground state at the excited states [37, 38].
Thin films for all polymers were prepared at room temperature by spin-casting (1500 rpm) on quartz plates in THF solutions (10 ppm). In solid states, both polymers displayed blue light under UV excitation. Table 1 listed representative data for the UV–visible and PL polymer spectra (P1–P6) as films. Such polymers’ absorption spectra and emission spectra (thin film) were slightly red-shifted in solution with respect to their respective spectra, which was an indication of no apparent molecular conformational transition from solution to solid state.
The PL quantum yield of the synthesized polymers was determined by using 9,10-diphenylanthracene in cyclohexane as reference standard. The data regarding quantum yield is listed in Table 1. The polymer solutions display PL quantum yield within 0.29–0.67 range. The optical band gap was in the 2.94–2.97 eV range.
Alkyl substituted dibenzofluorene monomer emission was observed at approximately 385–397 nm, while di(p-hydroxyphenyl)acene monomers at a range of 441–450 nm were obtained. The λmax emission for polymers was comparable to that for monomers that showed more resemblance to the λmax emission of di (p-hydroxyphenyl)acene monomers.
3.4 Gel permeation chromatography (GPC)
GPC technique was used to calculate average molar mass (Mn) of the copolymers. Table 2 shows the molecular weight data for the synthesized polymers Mn and Mw. The number-average molecular weights (Mn) determined by the use of THF as eluent is around 10,080 with a polymer dispersion (PD) of 1.51 for P1; 11,220 with a PD of 1.29 for P2; 10,088 with a PD of 1.43 for P3; 12,060 with a PD of 1.66 for P4; 10,816 with a PD of 1.38 for P5 and 11,160 with a PD of 1.55 for P6 as against polystyrene as a standard. Number average molecular weight (Mn) was used for the further analyses of certain parameters. Molar mass was used to measure the degree of polymerization (DP). The average molecular weight (Mn) and weight average molecular weight (Mw) were used to calculate the polydispersity index (PDI) which was about one indicating uniform and one polymer length [39]. This observation suggested that the PolyDBFs synthesized were non-branching form.
3.5 Thermal studies
Thermogravimetric analysis in N2 atmosphere has studied the thermal properties of polymers (P1–P6), and the results are tabulated in Table 2. The thermal analysis showed that polymers were up to a varying degree relatively stable. The thermograms showed the incremental weight loss due to degradation by fragmentation, with rising temperatures. Below 100 °C there was no substantial weight loss for the polymers suggesting lack of moisture (free moisture or bond moisture) in the polymer chain. Polymers P1–P5 undergoes a single-step degradation process observed in the 30–1000 °C range and followed by a weight loss of 94–99%. The two-step process of degradation was observed for P6, the first step was observed in the 30–320 °C range due to a weight loss of 44.60%.The second step was observed in the range of 320–1000 °C owing to the weight loss of 54%. During repeated heating/cooling DSC cycles for P1–P6, which presumably resulted from the rigidity of the polymer chains, there was no detection of any melting point signals. In the DSC curve, distinct glass transition temperatures (Tg) were observed in the 108–133 °C range, which were higher than those recorded in the literature for polyfluorenes (Tg is 80 °C) and PPV (Tg is 100 °C) [40, 41]. The existence of the rigid dibenzofluorene unit can be derived from the value of Tg, which improves the polymer’s molecular rigidity and restricts its segmental mobility. It is necessary to establish polymer light emitting diodes PLEDs from materials that have a relatively high Tg value to avoid the problems associated with the formation of aggregates and excimers upon heat exposure [30]. Hence the introduction of alkyl dibenzofluorene unit into the polymeric backbone results in improved thermal stability and contributes to higher Tg which was ideal for polymer acting as an active layer in display applications.
3.6 X-ray diffraction studies
XRD can be used to determine strains, desired orientations, crystallographic structures and crystalline material grain size. Figure S1 (supporting information) shows the XRD patterns of the polymers. One of the polymer’s important properties was the crystallinity of polymer. Polymer occurs in both amorphous and crystalline form. The polymer’s crystallite size is used to define the polymer character and degree of crystallinity present in the polymer sample [31,32,33, 42]. The polymers’ crystallite sizes were determined using the Deby-Scherrer relationship and found within 0.2251–3.6155 range as shown in Table S1 (supporting information). The polymer X-ray diffractogram exhibited well-resolved peaks of varying intensity. Peaks were slightly higher in intensity in some of the diffractograms but the measured crystallite size was less than 4 Ǻ. Also, some expanding region was found under the peaks and this indicated that in nature the polymers were semi-crystalline (Fig. 5).

Overlay thermograms of polymers
3.7 Electrochemical studies
Cyclic voltammetry (CV) was used to analyze the electrochemical behaviour of the resulting polymers. The polymer CV curve (P1–P6) shown in Fig. 6 and the corresponding CV data are listed in Table 3. The oxidation potential (Eox) of the polymers was calibrated to the ferrocene/ferrocenium redox couple (Fc/Fc+), which was determined to be 0.38 V under the same experimental conditions. The redox potential of Fc/Fc+ was supposed to have an absolute energy level of 4.8 eV relative to a vacuum. Based on the onset potential of oxidation and reductions, the energy levels of HOMO and LUMO were calculated in relation to ferrocene energy level. The EHOMO values were calculated from oxidation potential peak and were in the range of 5.22–5.26 eV and LUMO energy level were estimated on the basis of the relationship ELUMO = EHOMO − Egopt. The LUMO values were in the range of 2.23–2.38 eV.

Cyclic voltammograms of polymers (P1–P6) in DCM using Ag/Ag+ electrode calibrated with fc/fc+ and TBAPF6 [tertiary butyl ammonium hexafluorophospate (0.1 m)] as an electrolyte. The scan rate used was 50 mv S−1
Unlike other conjugated polymers, polymers displayed reversible partial oxidation (p-doping) and partial reduction (n-doping) processes [43,44,45]. The high EHOMO values indicated that, in future high voltage display applications, the synthesized polymers used to be a suitable material [43].
3.8 Surface morphology
For SEM analysis, samples were dispersed in a suitable solvent using bath sonication and drop cat on Si wafers to view under the microscope. The polymers SEM microphotographs are shown in Figure S3 (Supporting Information). The images consisted of a large, irregular crystal with trace particles. Furthermore, the microcrystal SEM photomicrographs showed platy rough structure, amorphous globular structure and rock shaped structure. In SEM micrographs the microscopic and macroscopic separation of the phase was not observed. SEM analysis found that in nature polymer P1 was more crystalline while in nature remaining polymers P2–P6 were partially crystalline.
4 Conclusion
In summary, by aromatic nucleophilic substitution reaction we have synthesized and characterized blue light emitting alkyl and allyl substituted polydibenzofluorenes containing moiety of diphenylanthracene. All of these polymers are in fact non-hygroscopic and solids of stable brown color are soluble in common organic solvents such as CH2Cl2, CHCl3, THF and tolueneFor synthesized polymers, UV–Visible intense absorption peaks were observed between 264 and 396 nm and PL maximum at 416–433 nm with shoulder peak at 360–380 nm (P1–P5). Due to existence of H-bonding in allyl unit even the expansion of П-conjugation in polymer P6 exist single band in emission spectrum. The polymer solutions display a PL quantum yield within 0.29–0.67 range. The optical band gap was in the 2.94–2.97 eV range. Due to the dibenzofluorene unit’s large steric hindrance, aggregation of polymers was reduced, resulting in nearly equal PL emissions in both the solution and solid states. The polymers displayed excellent color stability and showed very high quantum yield in blue emission. Because of its combinations of strong quantum yield, thermal stability and excellent color stability, the resulting polymers display strong PL properties with blue emission. From the electrochemical study of polymers P1–P6, The HOMO levels were estimated in the range of − 5.22 to − 5.26 eV and LUMO of − 2.23 to − 2.38 eV. Additionally, for synthesized polymers, the gel permeation chromatography analysis showed reasonable poly-dispersity index in (PDI) values ranging from 1.29 to 1.66. This polymer displayed good temperature stability up to 350 °C and high glass transition temperature (108–133 °C) compared to existing polyfluorene. We suggest these polymers have the potential to achieve the target of sustainable light emitting device technology for solid state lightening and displays.
References
Berggren M, Inganäs O, Gustafsson G, Rasmusson J, Andersson MR, Hjertberg T, Wennerström O (1994) Light-emitting diodes with variable colours from polymer blends. Nature 372:444
Ye J, Chen Z, Fung M-K, Zheng C, Ou X, Zhang X, Yuan Y, Lee C-S (2013) Carbazole/sulfone hybrid d-π-A-structured bipolar fluorophores for high-efficiency blue-violet electroluminescence. Chem Mater 25:2630
Rothberg LJ, Lovinger AJ (1996) Status of and prospects for organic electroluminescence. J Mater Res 11:3174
Virgili T, Lidzey DG, Bradley DD (2000) Efficient energy transfer from blue to red in tetraphenylporphyrin-doped poly(9,9-dioctylfluorene) light-emitting diodes. Adv Mater 12:58–62
Wang W-Z, Zhu R, Fan Q-L, Xu H, Hou L-T, Cao Y, Huang W (2006) Synthesis, photophysics, and electroluminescence of poly(dibenzofluorene) s. Macromol Rapid Commun 27:1142–1148
Ego C, Grimsdale AC, Uckert F, Yu G, Srdanov G, Müllen K (2002) Triphenylamine-substituted polyfluorene—a stable blue-emitter with improved charge injection for light-emitting diodes. Adv Mater 14:809–811
Schilling B, Kaufmann DE, (1998) Suzuki coupling of chiral 1,1′-binaphthyl systems − new synthetic routes to functionalize the 2- and 2,2′-positions. Eur J Org Chem 4:701–709
Burroughes JH, Bradley DDC, Brown AR, Marks RN, Mackay K, Friend RH, Burn PL, Holmes AB (1990) Light-emitting diodes based on conjugated polymers. Nature 347:539–541
Reineke S, Lindner F, Schwartz G, Seidler N, Walzer K, Lüssem B, Leo K (2009) White organic light-emitting diodes with fluorescent tube efficiency. Nature 459(7244):234–238
Chapran M, Angioni E, Findlay NJ, Breig B, Cherpak V, Stakhira P, Tuttle T, Volyniuk D, Grazulevicius JV, Nastishin YA et al (2017) An ambipolar BODIPY derivative for a white exciplex OLED and cholesteric liquid crystal laser toward multifunctional devices. ACS Appl Mater Interfaces 9(5):4750–4757
Leo K (2011) Organic light-emitting diodes: efficient and flexible solution. Nat Photonics 5(12):716–718
Cherpak V, Stakhira P, Minaev B, Baryshnikov G, Stromylo E, Helzhynskyy I, Chapran M, Volyniuk D, Tomkuté-Lukšiené D, Malinauskas T et al (2014) Efficient “Warm-White” OLEDs based on the phosphorescent bis-cyclometalated iridium(III) complex. J Phys Chem C 118(21):11271–11278
Xu B, Holdcroft S (1993) Molecular control of luminescence from poly(3-hexylthiophenes). Macromolecules 26:4457–4460
Harvey RG (1991) A new general synthesis of polycyclic aromatic compounds based on enamine chemistry. J Org Chem 56:1210–1217
Ritchie J, Joe A, Crayston Jonathan PJ, Markhamb I, Samuel DW (2006) Effect of meta-linkages on the photoluminescence and electroluminescence properties of light-emitting polyfluorene alternating copolymers. J Mater Chem 16:1651–1656
Tartaglia S, Pace F, Scafato P, Rosini C (2008) A new case of induced helical chirality in a bichromophoric system: absolute configuration of transparent and flexible diols from the analysis of the electronic circular dichroism spectra of the corresponding di(l-naphthyl) ketals. Org Lett 10:3421–3424
Tian Y, Uchida K, Kurata H, Hirao Y (2014) Design and synthesis of new stable fluorenyl-based radicals. J Am Chem Soc 136:12784–12793
Kitamura C, Tanigawa Y, Kobayashi T (2012) 17,17-Dialkyltetrabenzo [a,c,g,i] fluorenes with extremely high solid-state fluorescent quantum yields: relationship between crystal structure and fluorescent properties. Tetrahedron 68:1688–1694
Schroeder K, Haase D, Saak W, Luetzen A, Schellenberg J (2006) Tetrabenzo[a,c,g,i]fluorenyllithium and η5-Tetrabenzo[a,c,g,i]fluorenyltitanium complexes. Organometallics 25:3824–3836
Kim YH, Kwon SK, Yoo DS, Rubner MF, Wrighton MS (1997) A novel, bright blue electroluminescent polymer: a diphenylanthracene derivative. Chem Matter 9:2699–2701
Kim Y-H, Ahn J-H, Shin D-C, Kim J-H, Park Y-W, Choi D-S, Kim Y-K, Kwon S-K (2001) Synthesis and characterization of new poly(terphenylene vinylene) derivative as blue emitting material. Bull Korean Chem Soc 22:1181–1182
Barve KA, Raut SS, Mishra AV, Patil VR (2011) Synthesis and studies of blue light emitting polymers containing triphenylamine-substituted fluorene and diphenylanthracene moiety. J Appl Poly Sci 122:3483–3492
Raut SS, Patil VR (2013) Synthesis and Spectral Studies of 6,13-di (p-hydroxyphenyl) pentacene and 6,13-di (p-hydroxynapthyl) pentacene. Polycl Aroma Comp 33:127–137
Raut SS, Barve KA, Bayes GS, Patil VR (2010) Synthesis of water-soluble polyesters containing carboxy-functional groups in the polymers chain and study of their metal complexes. J Inorg Organomet Polym 20:343–355
Hasija DC, Gopalakrishnan J, Mishra AV, Ghase VD, Patil VR (2020) Exploring copper as a catalyst for cost effective synthesis of polyfluorenes: an alternative to platinum and palladium. SN Appl Sci 2:569. https://doi.org/10.1007/s42452-020-2344-9
Yang J, Cheng H, Frost RL (2011) Synthesis and characterisation of cobalt hydroxy carbonate Co2CO3 (OH)2 nanomaterials. Spectrochim Acta Part A Mol Biomol Spectrosc 78:420–428
Ioannis K, Vladimir YR, Jan CG, Ronald F, Alexander EL, Elisabeth H (2011) Arylamino-functionalized fluorene-and carbazole-based copolymers: color-tuning their CdTe nanocrystal composites from red to white. J Poly Sci Part A Poly Chem 49:392–402
Chia-Hung C, So-Lin H, Dinakaran K, Mao-Yuan C, Kung-Hwa W (2005) Synthesis and characterization of luminescent polyfluorenes incorporating side-chain-tethered polyhedral oligomeric silsesquioxane units. Macromolecule 38:745–751
Do-Hoon H, Moo-Jin P, Ji-Hoon L, Nam-Sung C, Hong-Ku S, Changhee L (2004) Synthesis and light-emitting properties of polyfluorene copolymers containing a hydrazone derivative as a comonomer. Synth Met 146:145–150
Chou CH, Hsu SL, Dinakaran K, Chiu MY, Wei KH (2005) Synthesis and characterization of luminescent polyfluorenes incorporating side-chain-tethered polyhedral oligomeric silsesquioxane units. Macromolecules 38:745–751
Lee YZ, Chen X, Chen SA, Wei PK, Fann WS (2001) Soluble electroluminescent poly(phenylene vinylene) s with balanced electron-and hole injections. J Am Chem Soc 123:2296–2307
Peng Z, Zhang J (1999) New oxadiazole-containing conjugated polymer for single-layer light-emitting diodes. Chem Mater 11:1138–1143
Miteva T, Meisel A, Knoll W, Nothofer HG, Scherf U, Muller DC (2001) Improving the performance of polyfluorene-based organic light-emitting diodes via end-capping. Adv Mater 13:565–570
Redecker M, Bradley DDC, Inbasekaran M, Wu WW, Woo EP (1999) High mobility hole transport fluorene-triarylamine copolymers. Adv Mater 11:241–246
Tokito S, Tanaka H, Noda K, Okada A, Taga Y (1997) Thermal stability in oligomeric triphenylamine/tris(8-quinolinolato) aluminum electroluminescent devices. Appl Phys Lett 70:1929–1931
Sharma BK, Shaikh AM, Agarwal N, Kamble RM (2016) Synthesis, photophysical and electrochemical studies of acridone-amine based donor–acceptors for hole transport materials. RSC Adv 6:17129–17137
Roiter Y, Minko S (2005) AFM single molecule experiments at the solid–liquid interface: in situ conformation of adsorbed flexible polyelectrolyte chains. J Am Chem Soc 127:15688–15689
Rananware M, Ghase V, Patil VR (2018) Recent development of crown-substituted polyfluorenes for blue light-emitting devices in organic electronics. Poly Bull 76(3):1277–1294
Kadu RK, Patil VR (2017) New strategy for synthesis of polyphenylene substituted dendronized monomers containing fluorene unit and the study of their properties. Poly Arom Compd 37:1–12
Chalke RM, Patil VR (2017) New approaches towards the synthesis and characterization of alkoxy substituted spirobifluorenes and spirosilabifluorenes for organic optoelectronics. J Macromol Sci Part A 54:556–564
Chalke RM, Patil VR (2017) Novel methoxy spirobifluorene and alkyl substituted diphenylacene based organic blue light emitting polymers for application in organic electronics. Polymer 123:355–365
Gopalakrishnan J, Hasija DC, Patil VR (2020) Synthesis, photophysical studies of blue-light-emitting alternating polymers from substituted 9-sila fluorenes and substituted p-phenylacene. Chem Sel 5(8):2577–2580. https://doi.org/10.1002/slct.201904422
Grice AW, Bradley DDC, Bernius MT, Inbasekaran M, Wu WW, Woo EP (1998) High brightness and efficiency blue light-emitting polymer diodes. Appl Phys Lett 73:629–631
MacDiarmid AG, Mammone RJ, Kaner RB, Porter SJ, Pethig R, Heeger AJ, Rosseinsky DR (1528) Charge storage and charge transport in conducting polymers: solitons, polarons and bipolarons. Phil Trans R Soc Lond A 314(1528):3–15
Zhao L, Zhao L, Xu Y, Qiu T, Zhi L, Shi G (2009) Polyaniline electrochromic devices with transparent graphene electrodes. Electrochimica Acta 55(2):491–497
Acknowledgements
The author (VDG) thank to Department of Chemistry, University of Mumbai for providing Departmental Research Fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ghase, V.D., Hasija, D.C., Rananaware, M.M. et al. Alkyl and allyl substituted polydibenzofluorene: blue emitters for future display applications. SN Appl. Sci. 2, 1186 (2020). https://doi.org/10.1007/s42452-020-2940-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-2940-8