
Abstract
[Al,F]-free M-BEA (M = Ti, Fe, Co, Ga) nanocrystals were synthesized via a green steam-assisted conversion route. Obtained samples had regular morphology, hierarchical structure and superior surface texture. A “seed-assisted micro-zone synchronous crystallization” model was firstly proposed to explain the crystallization process in a dense steam system.
Graphic abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
M-BEA (Beta zeolite with *BEA topology), a class of heteroatomic catalysts with directional catalytic function, can endow traditional Beta zeolite superior performance in various reactions, whether gas-phase reactions or liquid-phase reactions. Ti-BEA was the earliest M-BEA catalyst, and it broke through the limitation that titaniumsilicon zeolite was only used for gas-phase reactions. Currently, Ti-BEA zeolite has always performed well in green epoxidation reactions, such as aromatics hydroxylation [1], cyclohexanone ammoniation [2] and olefin epoxidation [3]. Differently, Fe-BEA zeolite was mainly used in gas-phase reactions, and was the most effective catalyst for NH3-SCR reaction (NOx selective catalytic reduction by NH3) [4, 5]. NH3-SCR reaction is the most efficient technology for the removal of NOx in diesel vehicle [6], therefore, designing efficient, stable and environmental friendly NH3-SCR catalysts is of great significance. As far as SCR reactions, if the NH3 is replaced by an alkane, NH3-SCR reaction becomes another reaction type, namely CH4, C3H8 or C4H10-SCR-NOx reactions [7]. In this case, Co-BEA becomes the effective catalyst, especially for propane-SCR reaction [8]. Ga-BEA was synthesized late, but it was greatly effective for alkane aromatization reaction, especially for propane aromatization [9, 10]. In addition, Sn-BEA [11], V-BEA [12] and other heteroatom M-BEA zeolites [13] also have their corresponding applications. In summary, M-BEA zeolites are indispensable for industrial application. But unfortunately, the synthesis of M-BEA catalysts have been under a difficult circumstance due to complicated synthetic procedures, high cost of materials and time, serious pollution and waste [3, 7, 14, 15]. Thus, it is meaningful to develop a green and effective method to accelerate its industrial process.
Nowadays, traditional hydrothermal synthesis is still the mainstream of M-BEA zeolite, which relies on mass transfer and thermal effects of much solvent to promote nucleation and growth process. But the low concentration of nutrients caused by much solvent are often more difficult for zeolitic nucleation and growth, so large amount of template agent (tetraethylammonium hydroxide, TEAOH) and long synthetic period (malar ratio TEAOH/SiO2 = 0.55, over 7 days [16]) are often the prerequisite for obtaining crystals with high crystallinity. Aluminum, as a nucleator, can facilitate the process of nucleation spontaneously [17, 18]. But the presence of aluminum usually leads to the formation of strong acid sites and decreases the selectivity of products [19,20,21], so that the advantages of real active metal sites are not fully developed. Afterwards, a post-modification strategy was developed to avoid the difficulty of nucleation, but obtaining Si-BEA precursor required highly concentrated acid to remove structural aluminum for constructing structural defects [22, 23], which provide a home for subsequent grafting of heteroatoms into silicon framework. But unfortunately, this post-modification treatment tends to deposit heteroatoms on the porous surface and cannot enter the framework, thus easily forming extra-framework species that are apt to lost during reactions. To fundamentally solve the aluminum deficiencies, hydrofluoric acid was applied as charge compensator to assist nucleation process [24, 25], but this introduction significantly decreases the alkalinity of and mobility of precursor, successively slowing down mass diffusion and nucleation rate, which leads to micro-level crystal and the inaccessibility of active sites. Therefore, M-BEA zeolites without aluminum and fluorine ([Al,F]-free), have barely been synthesized by one-step hydrothermal synthesis. In addition, low yield of products, low utilization of autoclaves, high reaction pressure and large surplus of wastewater are all the inherent disadvantages of hydrothermal synthesis. Considering all these unsatisfactory problems, it is strongly desirable to develop a more efficient and green route to synthesize M-BEA zeolites.
Here we proposed a universal route for synthesizing heteroatomic M-BEA zeolites by introducing Ti, Fe, Co, Ga atoms into *BEA framework. This route was in a nearly dry steam environment without the presence of crystallization liquid, leading to a large increase in the concentration of nutrients, which greatly increased the nucleation rate and shortened the crystallization period. Thus the amount of template was reduced to TEAOH/SiO2 = 0.2 only after 2 days, far less than reported (TEAOH/SiO2 ≥ 0.55, 7–14 days). This method not only realized the synthesis of framework F-free and Al-free hierarchical M-BEA nanocrystals, but also greatly reduced production cost and environmental pollution. Very importantly, a model for the formation of M-BEA hierarchical structure was simultaneously proposed to explain the process of nucleation and growth in the dense-gel and steam system.
2 Experiment
2.1 Synthesis of M-BEA zeolites and pure Si-Beta seeds
Taking Co-BEA synthesis as an example, 0.3 g NaOH and 3 g TEAOH solution were dissolved with 20 g deionized water. Then, 0.17 g cobalt acetate solids were added into the clear solution and stirred for 20 min until the formation of milky white liquid. Then 0.3 g pure-Si Beta seeds were added and stirred uniformly, followed by the addition of 1.5 g fumed silica (A200). The resulting mixture was aged at room temperature under dynamic stirring conditions for 20 min. The final gel molar composition was SiO2:0.028C4H6O4Co:0.30NaOH:0.20TEAOH:44H2O. After aging, the gel was dried at 70 °C for 24 h. After dry and grind, measured 2.0 g dry gel was placed on a 10 mL raised Teflon holder and sealed in a 40 mL Teflon-lined stainless steel autoclave involving 0.5 g of deionized water. The autoclave was sealed and heated at 418 K for 48 h, then the as-prepared zeolite Co-BEA was obtained after washing and drying. To achieve the Co-BEA products, the as-prepared samples were ion exchanged in 1.0 mol/L NH4NO3 solution three times at 90 °C for 2 h, followed by calcining at 550 °C for 6 h. Schematic diagram was shown in Scheme S1.
The synthetic procedure of Ti-BEA, Fe-BEA and Ga-BEA samples was same as above except the difference of heteroatom sources. Detailed synthesis and physico-chemical property of pure Si-BEA seeds were all shown in our previous study [26].
3 Results and discussion
3.1 Roles of pure Si-Beta seeds
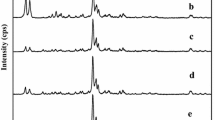
During the steam-assisted crystallization of M-BEA (M = Ti, Fe, Co, Ga) crystals, due to the absence of nucleation-promoting agents (Al3+ or F−), the presence of seed crystals is particularly pivotal for the isomorphous substitution of Ti, Fe, Co, Ga into *BEA framework. XRD patterns in Fig. 1 exhibit perfect crystallization of M-BEA samples with 10 wt% seed crystals, giving a series of peaks at 7.7°, 16.5°, 21.4°, 22.4°, 25.3°, 26.8°, 29.5°, 30.5° associated with integral *BEA structure [27]. In contrast, the no-seeded sample displays no diffraction peaks, except a weak peak at 2θ = 22.4° degree (Fig. S1a, ESI†). It is more likely to be the result of spontaneous nucleation of templated precursor after crystallization process, because the calcined no-seeded sample shows typical isotherm for Langmuir adsorption, giving HK pore size distribution at 0.6–0.7 nm (Fig. S1b). These results suggest that the no-seeded sample already exists some microporosity from *BEA nucleation before the formation of crystalline channels, indicating that tetraethylammonium cations (TEA+) have been inserted into the amorphous dry precursor by the spontaneous dispersion during crystallization [28, 29]. However, just with so little template (TEAOH/SiO2 = 0.2), the number of crystal nucleus needed for perfect crystallization is far insufficient, so it is necessary to introduce sufficient seeds to construct sufficient nucleus sites depending on non-spontaneous nucleation process. When seeds addition is increased to 10%, the crystalline channels can completely constructed, which can be proved by the large specific surface area and pore volume of the four M-BEA zeolites (Table 1). Anyway, for the SAC synthesis of M-BEA zeolites, the spontaneous nucleation process induced by template agent and the non-spontaneous nucleation process complementally promote the isomorphous substitution of Ti, Fe, Co, Ga into *BEA framework.

XRD patterns of Ti-BEA, Fe-BEA, Co-BEA and Ga-BEA samples with 10% seeds/SiO2 mass ratio at 145 °C for 2 days
3.2 Formation of square morphology
Notably, all the M-BEA samples achieved by SAC method show a nanosized regular square morphology, although there exists some differences in crystal size and thickness (Fig. 2). The acquisition of the square morphology is closely related to the crystallization kinetics and growth environment of zeolites. Under a more alkaline environment, zeolite prefers to grow along (110) plane in comparison with (001) plane, as a result, the M-BEA samples finally exhibit square shape [26, 30]. And for different M-BEA samples, since different heteroatom sources introduced into the growth systems, naturally, the crystallization environment are different, resulting in the difference of crystal size along [110] and [001] dimensions. In addition, as quite different from hydrothermal or solvent-free synthesis [31, 32], SAC method has unique advantage in zeolitic nucleation, growth and assembly process [33], so obtained crystals are nanocrystal with nanometer scale. During steam-assisted crystallization, the rate and number of nuclei increased significantly as the dry gel was tightly packed by the hot steam. Importantly, the local limiting effect caused by the dense gel system significantly limits the excessive growth of M-BEA nanocrystals, and finally guarantees the samples present at the nanometer level [34]. Accordingly, the size of Ti, Fe, Co, Ga-BEA nanocrystals are about 450 × 450 × 100 nm, 450 × 450 × 100 nm, 600 × 600 × 100 nm, 250 × 250 × 200 nm, respectively.

SEM images of Ti-BEA, Fe-BEA, Co-BEA and Ga-BEA samples with 10% seeds/SiO2 mass ratio at 145 °C for 2 days
3.3 Construction of hierarchical structure
Interestingly, hierarchical structure can be directly constructed without secondary treating and pore-enlarging additives, just by this simple steam-assisted conversion method. The rough surface of the obtained particles (Fig. 2) indicate that they are the aggregates composed of small highly intergrown nanocrystals with abundant mesopores [35, 36], furthermore, the TEM (Fig. 3) images and nitrogen sorption test (Fig. 4) provide strong evidences for the existence of this hierarchical structure. The nitrogen isotherms (Fig. 4) show that all the M-BEA samples exhibit typical type IV isotherms [26, 37], firstly display a steep increase at a very low relative pressure (P/P0 < 0.1), indicating the intrinsic well-constructed microspores stemming from the *BEA structure. And distinct type H4 hysteresis loops originated from the capillary condensation appear at 0.4 < P/P0 < 1.0 [33], which usually occur in some adsorbents with both micropores and mesopores or solids containing slit fissures, evidencing the hierarchical structure containing both of microspores and mesopores. Moreover, the observation of dense porous channels in TEM images (Fig. 3) is another powerful example of hierarchical structure, which echo the superior textural properties (Table 1), especially, large external surface area (163.53, 128.68, 173.97, 109.11 m2 g−1), mesoporous pore volume (0.21, 0.20, 0.14, 0.08 cm3 g−1) and average pore size (about 4–10 nm).

TEM images (left) and high resolution images of a Ti-BEA, b Fe-BEA, c Co-BEA, d Ga-BEA samples

Nitrogen isotherms with pore size distribution of a Ti-BEA, b Fe-BEA, c Co-BEA, d Ga-BEA samples
3.4 Evidences of heteroatoms entering framework
To confirm that different heterometallic atoms have indeed grown in zeolitic silicon matrix, UV–Vis, FT-IR and XPS tests jointly provide strong evidences for different M-BEA samples. For Ti-BEA samples, the absorbance at near 230 nm (Fig. 5a) corresponds to tetrahedrally coordinated Ti species inserted into framework, and the lower binding energy values of ~ 458 and ~ 464 eV in XPS spectra (Fig. 6a) are assigned to 2p3/2 and 2p1/2 photoelectrons of tetrahedrally coordinated framework Ti(IV) species [38, 39]. UV–Vis spectra (Fig. 5b) of Fe-BEA exhibit a strong band at near 242 nm, which is assigned to Fe3+ at isolated tetrahedral framework sites (charge transfer band), and the weak band at 304 nm is attributed to d–d transitions of the Fe3+ ion in tetrahedral symmetry [40, 41]. The XPS photoelectrons also (Fig. 6b) show a main peak at 711.2 eV and satellite peak at the high binding energy side (724.9 eV), which are all considered to be the characteristic for Fe3+ species. Beyond that, the only peak at 532.5 eV (Fig. S2 left, ESI†) corresponds to the oxygen species involved in the formation of either Si–O–Fe or Si–O–Si linkages of the zeolitic framework [42]. Figure 5c shows the UV–Vis spectra of Co-BEA samples, and all visible peaks associated with Co2+ species can be matched with the literature [43]. The peaks at 539, 591, 640 nm are evidence that Co has been successfully inserted into matrix, and the triplet is representative of Co2+ ions present in the vicinity of a six-member ring at the intersection of two straight channels of *BEA structure. The Co 2p2/3 XPS photoelectrons in Fig. 6c exhibit a main peak at 782.6 eV and a satellite peak, which are also characteristic peaks of Co2+ species in the matrix [43, 44]. Figure 5d shows the FT-IR spectra in the structural region of *BEA type zeolites, where the Ga-BEA sample exhibits the asymmetric T–O stretch band at 1172 cm−1, while the pure Si-BEA sample does not. This finding can be rationalized by considering that the isomorphous substitution of Si by Ga in framework [45]. Except for this, the two feature bands representing the *BEA structure appear the redshift to the lower wavenumber region due to the longer bond length of Ga–O than that of Si–O, from 533, 575 to 513, 571 cm−1, respectively. The main absorption band at 238 nm in UV–Vis spectra (Fig. S2 right, ESI†) further indicate that the Ga element is in the presence of the 4-coordinated Ga species in the framework, which is correspond with the peak at about ~ 20.5 eV attributed to the 4-coordinated framework of Ga species [45, 46].

UV–Vis spectra for a Ti-BEA, b Fe-BEA, c Co-BEA and FT-IR spectra for d Ga-BEA samples (left)

XPS of a Ti-BEA, b Fe-BEA, c Co-BEA, d Ga-BEA samples
In summary, the isomorphous substitution of Si by M (Ti, Fe, Co, Ga) in the *BEA framework can be fully implemented, and the corresponding M species also exist in a good coordination environmental, instead of kinds of metal oxides in extra-framework. These results suggest that the steam-assisted conversion route is an effective and universal method for the incorporation of heteroatom into zeolitic framework.
3.5 “Seed-assisted micro-zone synchronous crystallization” model
As an effective and green method of synthesizing zeolites, nowadays, no clear model can be proposed to explain why the hierarchical structure can be directly constructed by the steam-assisted conversion method. Here, we proposed a “seed-assisted micro-zone synchronous crystallization” model to explain the formation of hierarchical heteroatomic M-BEA zeolites in a dense steam environment (Scheme 1). From a microscopic molecular perspective, this model gives a detailed explanation of the formation of mesoporous and microporous structure from a dense precursor gel, so as to provide the guidance for one-step synthesizing heteroatomic hierarchical zeolites, in the case where neither crystallization aids such as aluminum or fluorine nor pore-enlarging agents required.

Model of “seed-assisted micro-zone synchronous crystallization” related to the formation of hierarchical structures
The formation of hierarchical M-BEA material is depicted in Scheme 1. Here, the hierarchical M-BEA zeolites are generated in a multistep process: (1) homogenizing of raw materials (silicon, heteroatomic sources and template agent) in alkaline medium and dry them. (2) Transferring dry nutrients into autoclave and beginning crystallization. During crystallization, every dry gel particles will be surrounded by steam under the high temperature and pressure, further forming numerous small microcrystalline units covered with thin liquid film [50] as well as carrying out micro-zone synchronous reaction. Specifically, the steam-assisted micro-zone synchronous reaction can be divided into three processes, namely, steam works in three main processes (3): Step 1, formation of nucleation centres with the help of both local concentration fluctuations and seed-assisted nucleation. Specifically, once the crystallization process begins, steam will invade the dense dry particles and wrap every dry particles in a tiny water capsule to form numerous micro-zone reaction units. Under the action of water initiation in this capsules (local concentration fluctuations), the seeded and templated dry gel begin to dissolve and active, subsequently the titanium-silicon nutrients in dry gel begin synchronously migrate and further deposit to the pre-existing seed surface, forming numerous nucleation centres under the guidance of seeds (seed-assisted nucleation). So far, the nucleation stage has been completed by the steam assistance. Step 2, successive transformation of these nucleus centres into nanocrystals with pseudo-*BEA structure [47,48,49]. Specifically, the titanium-silicon nutrients continue synchronously migrate to the nucleus centres, further polymerize and grow to form isolated nanocrystals with pseudo-*BEA structure. In every microcrystalline units, both the number and size of nanocrystals increase significantly due to continued synchronous migration of nutrients. When the titanium-silicon nutrients is used up, the areas originally located titanium-silicon nutrients form into empty areas, which are responsible for the eventual construction of mesoporous structures. So far, the nanocrystals with pseudo-*BEA structure are formed. Step 3, formation of aggregated networks by condensation at contact points of numerous nanocrystals surfaces. In this step, large number of nanocrystals (from different microcrystalline units) start to aggregate into a network by condensation at contact points of the particle surfaces. Due to the low mobility of nanocrystals in the nearly dry environment, the aggregation phenomenon actually carries out along the edge of the empty areas instead of inside the empty areas. Therefore, the original empty areas finally are connected irregular intermediate voids, eventually establishing the mesoporous structure. The aggregated network gradually expand more broaden until the steam completely disappears, thus, the hierarchical zeolites have a certain size. Finally, as the remove of OSDAs by calcination, intrinsic micropores are not only exposed but the hierarchical structure is also stronger, and the hierarchical structure completely is constructed.
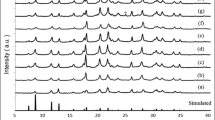
To provide experimental data for above model, the crystallization and morphology of intermediate products were tested and shown in Figs. 7 and 8, respectively (taking the crystallization process of Ti-BEA nanocrystals as an example). In general, zeolite crystallization process was divided into two stages, including the rapid nucleation stage and the slow growth stage. After an overall analysis for crystallization and morphology, we suggested that the first 12 h were the rapid nucleation stage in the entire crystallization period (48 h), which mainly underwent nucleation reactions. The remaining 36 h was the stage where nutrients attach to the crystal nucleus and further grew slowly, including the migration, attachment and polymerization of nutrients. The specific process was as follows:

XRD patterns of intermediate product during the crystallization process of titaniumsilicon dry gel

SEM images of intermediate product during the crystallization process of titaniumsilicon dry gel
When the dry gel [corresponding to (A) in Scheme 1] had not been crystallized, it exhibited an amorphous state because no characteristic peak appeared in the XRD pattern (Fig. 7a). This result was consistent with the SEM image in Fig. 8a, where these dry gels were in an cohesive amorphous state, and a small amount of square seeds were mixed in it. These amorphous substances were the “nutrient library” for the subsequent nucleation process and growth process, and continuously supply nutrients for crystallization reaction. The existence of seeds provides ready-made nucleation sites for subsequent nucleation reactions.
After 6 h of crystallization, the most important characteristic peak (22.4°) of *BEA structure began to appear. Correspondingly, some crystal nuclei began to appear in the amorphous substances in the SEM image (Fig. 8b). The above facts indicated that the nucleation reaction was in progress under the induction of seeds. The state of the substance at this time corresponded to (B) in Scheme 1. After 12 h of crystallization, the basic diffraction peaks began to appear at 7.7°, 21.4°, 22.4°, 26.8°, 30.5° (Fig. 7c), indicating that the crystal nuclei at this time had basic *BEA structure, but it is only the “prototype” of the perfect *BEA structure. This “prototype” corresponded to the pseudo-*BEA structure in Scheme 1. (C). Figure 8c exhibited the morphology of intermediate products at this time: as nucleation reaction continued, the crystal nucleus grew further from the amorphous gel. As the size increased, its basic square morphology began to appear. This structural and morphological feature suggested that nucleation state had been ended, and then entered the stage of crystal growth.
The last 36 h was for the crystal growth, this state was relatively slower than nucleation state, due to including subsequent migration, attachment and polymerization processes. During the 36 h, the crystallinity of Ti-BEA nanocrystals gradually increased, and perfect Ti-BEA nanocrystals would be formed when the crystallization ended (Figs. 7d–f, 8d–f). When the crystallization reaction progressed to 18 h (Fig. 8d), a large number of square nanocrystals began to grow, with the nutrients synchronously migrated, attached, and polymerized to the surface of the crystal nucleus. In this process, the number of independent square crystals increased with the number of amorphous substances decreasing [corresponding to (D) in Scheme 1]. When the time increased to 20 h, the nanocrystals grew further, and the square shape became more regular and the crystal size also became more uniform (Fig. 8e). After 48 h, the dry nutrients were completely consumed, and the perfect square Ti-BEA nanocrystals were synthesized. After calcination, its hierarchical structure came out (TEM images in Fig. 8f). The specific microscopic formation of hierarchical structure has been shown in Scheme 1.
3.6 Greenness of the SAC method
SAC method is a green synthesis method, and the greenness is reflected on entire synthesis process: (1) the green of raw materials. SAC method replaces poisonous and expensive organic silicon/heteroatomic sources with innocuous and cheap inorganic silicon/heteroatomic sources; (2) the green of synthesis steps. SAC method makes full use of steam penetration, greatly increases the concentration of “nutrients” in crystallization system, and significantly accelerates the nucleation and crystallization efficiency, so the crystallization time only takes 2 days; (3) in terms of raw material utilization, the amount of organic template is much lower than the current reported, and entire synthesis steps does not need corrosive hydrofluoric acid and other poisonous substances. Thus the raw material utilization is greener; (4) SAC method is greener in environmental protection. SAC method relies on steam-assisted crystallization instead of a large amount of solvent, so there is almost no wastewater discharge after the crystallization; (5) the product yield of SAC method is close to 100%, and the product has the advantages of high crystallinity, regular morphology, excellent texture properties, and hierarchical pore structure. In summary, the SAC method is greener than traditional hydrothermal synthesis.
In addition, SAC method is less poisonous in raw materials, synthetic processes and waste discharge than traditional hydrothermal method: Firstly, SAC method replaces the highly poisonous organosilicon and heteroatomic sources with the non-poisonous inorganic silicon and low-poisonous inorganic heteroatomic sources. Moreover, the whole synthesis process does not require highly poisonous hydrofluoric acid; Secondly, in terms of the synthesis steps, SAC method innovatively mixes the poisonous organic template agent into the dry gel in advance, thus avoiding the decomposition of organic template agent and the evaporation of poisonous gas; Finally, in terms of wastewater discharge, SAC method is basically no poisonous wastewater after crystallization, so it does not involve the discharge of poisonous wastewater.
4 Conclusions
Here this work proposed a steam-assisted conversion route for synthesizing M-BEA zeolites. Compared traditional hydrothermal process, this method is more greener, efficient and lower-cost for synthesizing M-BEA zeolites, and can greatly meet the characteristics of M-BEA zeolites on a large-scale industrialization. Moreover, this study proposed a “seed-assisted micro-zone synchronous crystallization” model, in order to explain the crystallization process in steam system. Promisingly, the strategy provided here may be not only suitable for single metal atom into silicon matrix, but also may be applicable to composite heteroatomic *BEA zeolites, or even ternary heteroatomic zeolites, such as [Ti,Ga]-BEA, [Fe,Co]-BEA, [Ti,Fe,Ga]-BEA et al., to enrich the kinds of active sites inside the crystals.
References
Callanan LH, Burton RM, Mullineux J (2012) Effect of semi-batch reactor configuration on aromatic hydroxylation reactions. Chem Eng J 180:255. https://doi.org/10.1016/j.cej.2011.11.033
Xu L, Li N, Wu P (2013) Clean synthesis of amides over bifunctional catalysts of rhodium-loaded titanosilicates. ChemCatChem 5:2462. https://doi.org/10.1002/cctc.201300126
Jappar N, Xia QH, Tatsumi T (1998) Oxidation activity of Ti-Beta synthesized by a dry-gel conversion method. J Catal 180:132. https://doi.org/10.1006/jcat.1998.2266
Dmitrii E, Doronkin Yu, Aleksandr V Alexei (2007) 309 nature of active sites in a Fe-Beta catalyst for NOx selective catalytic reduction by NH3. Mendeleev Commun 6:309. https://doi.org/10.1016/j.mencom.2007.11.001
Kogel M, Monnig R, Turek T (1999) Simultaneous catalytic removal of NO and N2O using Fe-MFI. J Catal 182:470. https://doi.org/10.1006/jcat.1998.2371
Giles R, Cant NW, Kogel M (2000) The effect of SO2 on the oxidation of NO over Fe-MFI and Fe-ferrierite catalysts made by solid-state ion exchange. Appl Catal B 2:75. https://doi.org/10.1016/S0926-3373(99)00136-8
Čapek L, Dědeček J, Wichterlová B (2004) Co-beta zeolite highly active in propane–SCR-NOx in the presence of water vapor: effect of zeolite preparation and Al distribution in the framework. J Catal 2:352. https://doi.org/10.1016/j.jcat.2004.08.001
Ohtsuka H, Tabata T, Okada O (1998) A study on the roles of cobalt species in NOx reduction by propane on Co-Beta. Catal Today 42:45. https://doi.org/10.1016/S0920-5861(98)00075-3
Schueth F, Spichtinger R (1991) Zeolite beta and isomorphously substituted Ga-beta as a catalyst for propane aromatization-infrared spectroscopic investigations. Am Chem Soc Div Pet Chem Prepr 4:677
Arnold A, Hunger M, Weitkamp J (2001) Dry gel synthesis of [Ga]beta-type zeolites and their quantitative characterisation by NMR spectroscopy. Chem Ing Tech 12:1588. https://doi.org/10.1002/1522-2640(200112)73:123.3.CO;2-8
Chang C, Cho H, Wang Z (2015) Fluoride-free synthesis of a Sn-BEA catalyst by dry gel conversion. Green Chem 17:2943. https://doi.org/10.1039/c4gc02457e
Zhang W, Hou W, Meng T (2017) Direct synthesis of V-containing all-silica beta-zeolite for efficient one-pot, one-step conversion of carbohydrates into 2,5-diformylfuran. Catal Sci Technol 7:6050. https://doi.org/10.1039/c7cy01834g
Winoto HP, Fikria ZA, Ha JM (2019) Heteropolyacid supported on Zr–Beta zeolite as an active catalyst for one-pot transformation of furfural to γ-valerolactone. Appl Catal B 24:1588. https://doi.org/10.1016/j.apcatb.2018.09.031
Kameoka S, Suzuki T, Kunimori K (2000) Selective catalytic reduction of N2O with methane in the presence of excess oxygen over Fe-BEA zeolite. Chem Commun 9:745. https://doi.org/10.1039/b001321h
Phadke Neelay M, Van der Mynsbrugge Jeroen (2018) Characterization of Isolated Ga3+ Cations in Ga/H-MFI Prepared by Vapor-Phase Exchange of H-MFI Zeolite with GaCl3. ACS Catal 8:6106. https://doi.org/10.1021/acscatal.8b01254
Wang BR, Peng XX, Yang JM (2019) Nano-crystalline, hierarchical zeolite Ti-Beta: hydrothermal synthesis and catalytic performance in alkenes epoxidation reactions. Microporous Mesoporous Mater 278:30. https://doi.org/10.1016/j.micromeso.2018.11.019
Serrano DP, van Grieken R (2001) Crystallization mechanism of all-silica zeolite beta in fluoride medium. Microporous Mesoporous Mater 46:35. https://doi.org/10.1016/S1387-1811(01)00272-4
Zhang JL, Wu ZJ, Dou T (2016) Synthesis of hierarchical zeolite Beta with low organic template content via the steam-assisted conversion method. Chem Eng J 291:82. https://doi.org/10.1016/j.cej.2016.01.088
Goa Y, Wu P, Tatsumi T (2004) Catalytic performance of [Ti, Al]-beta in the alkene epoxidation controlled by the postsynthetic ion exchange. J Phys Chem B 108:8401. https://doi.org/10.1021/jp048917y
Antunes MM, Lima S, Neves P (2015) One-pot conversion of furfural to useful bio-products in the presence of a Sn, Al-containing zeolite beta catalyst prepared via post-synthesis routes. J Catal 329:522. https://doi.org/10.1016/j.jcat.2015.05.022
Sazama P, Pilar R, Mokrzycki L (2016) Remarkably enhanced density and specific activity of active sites in Al-rich Cu-, Fe-and Co-beta zeolites for selective catalytic reduction of NOx. Appl Catal B 189:65. https://doi.org/10.1016/j.apcatb.2016.02.020
Tang B, Dai WL, Li LD (2014) A procedure for the preparation of Ti-Beta zeolites for catalytic epoxidation with hydrogen peroxide. Green Chem 16:2281. https://doi.org/10.1039/c3gc42534g
Maschmeyer T, Rey F, Thomas JM (1995) Mono-and bifunctional heterogeneous catalytic transformation of terpenes and terpenoids. Nature 378:159. https://doi.org/10.1038/378159a0
Kessler H, Patarin J, Darie CS (1994) The opportunities of the fluoride route in the synthesis of microporous materials. Stud Surf Sci Catal 85:75. https://doi.org/10.1002/chin.199514339
Naraki Y, Ariga K, Sano T (2016) Synthesis of Fe-based BEA zeolites in fluoride media and their catalytic performance in the NH3-SCR of NOx. Adv Porous Mater 2:125. https://doi.org/10.1166/apm.2016.1110
Luo YY, Li MR, Chen XH (2020) Microporous Mesoporous Mater. https://doi.org/10.1016/j.micromeso.2019.109675
Xie B, Song J, Ren L (2008) Fast synthesis of hierarchical nanosized pure Si-Beta zeolite via a steam-assisted conversion method. Chem Mater 20:4533. https://doi.org/10.1021/cm801167e
Lipsch JMJG, Schuit GCA (1969) The CoO-MoO3-Al2O3 catalyst: II. The structure of the catalyst. J Catal 15:174. https://doi.org/10.1016/0021-9517(69)90021-9
Xiao FS, Zheng S, Xu R (1998) Dispersion of inorganic salts into zeolites and their pore modification. J Catal 176:474. https://doi.org/10.1006/jcat.1998.2054
Larlus O, Valtchev VP (2005) Control of the morphology of all-silica BEA-type zeolite synthesized in basic media. Chem Mater 17:881–886. https://doi.org/10.1021/cm048799r
Ren LM, Wu QM, Xiao FS (2012) Solvent-free synthesis of zeolites from solid raw materials. J Am Chem Soc 37:15173. https://doi.org/10.1021/ja3044954
Xu R, Pang W, Yu J (2007) Chemistry of zeolite and related porous materials. Wiley, Singapore. https://doi.org/10.1002/9780470822371
Karin M, Bilge Y (2011) One-step synthesis of hierarchical zeolite beta via network formation of uniform nanocrystals. J Am Chem Soc 133:5284. https://doi.org/10.1021/ja108698s
Wang BR, Lin M (2018) Silica beta synthesized under alkaline conditions. Microporous Mesoporous Mater 266:43. https://doi.org/10.1016/j.micromeso.2018.02.049
Chen XW, Mao JJ, Tang Y (2014) Fast synthesis of nanosized zeolite beta from a low-seeded, low-templated dry gel with a seeding-steam-assisted conversion method. J Mater Chem A 2:1247–1251. https://doi.org/10.1039/C3TA14235C
Moller K, Yilmaz B, Bein T (2011) One-step synthesis of hierarchical zeolite beta via network formation of uniform nanocrystals. J Am Chem Soc 133:5284. https://doi.org/10.1021/ja108698s
Luo YY, Zhu YD, Chen XH (2020) Fast synthesis of hierarchical Al-free Ti-BEA plate-like nanocrystals from low-templated dry gel via a steam-assisted conversion method. Green Chem 22:1681. https://doi.org/10.1039/c9gc03869h
Nogier JP, Millot Y, Man PP (2009) Probing the incorporation of Ti(IV) into the BEA zeolite framework by XRD, FTIR, NMR, and DR UV–jp810722bis. J Phys Chem C 12:4885. https://doi.org/10.1021/jp8099829
Nogier J, Millot Y, Man Pascal P (2009) Nature, environment and quantification of titanium species in TiSiBEA zeolites investigated by XRD, NMR, DR UV–Vis and XPS. Nature 130:588. https://doi.org/10.1007/s10562-009-9960-8
Bordiga S, Buzzoni R (1996) Structure and reactivity of framework and extra-framework iron in Fe-silicalite as investigated by spectroscopic and physicochemical methods. J Catal 158:486. https://doi.org/10.1006/jcat.1996.0048
Pérez-Ramírez J, Groen JC, Brückner A (2005) Evolution of isomorphously substituted iron zeolites during activation: comparison of Fe-beta and Fe-ZSM-5. J Catal 232:318. https://doi.org/10.1016/j.jcat.2005.03.018
Zhang H, Chu L, Xiao FS (2013) One-pot synthesis of Fe-Beta zeolite by an organotemplate-free and seed-directed route. J Mater Chem A 1:3254. https://doi.org/10.1039/c3ta01238g
Chupin C, van Veen AC, Konduru M (2006) Identity and location of active species for NO reduction by CH4 over Co-ZSM-5. J Catal 241:103. https://doi.org/10.1016/j.jcat.2006.04.025
Guczi László, Bazin Dominique (1999) Structure and selectivity of metal catalysts: revisiting bimetallic zeolite systems. Appl Catal A 188:163. https://doi.org/10.1016/S0926-860X(99)00214-8
Price GL, Kanazirev V (1991) Ga2O3/HZSM-5 propane aromatization catalysts: formation of active centers via solid-state reaction. ChemInform. https://doi.org/10.1002/chin.199105087
Price GL, Kanazirev V (1990) Ga2O3/HZSM-5 propane aromatization catalysts: formation of active centers via solid-state reaction. J Catal 126:267. https://doi.org/10.1016/0021-9517(90)90065-R
Inagaki S, Matsukata M (2007) Changes of intermediate-range structure in the course of crystallization of zeolite beta. Microporous Mesoporous Mater 101:50. https://doi.org/10.1016/j.micromeso.2006.10.039
Ogura M, Nakata S, Matsukata M (2001) Effect of NH+4 exchange on hydrophobicity and catalytic properties of Al-free Ti–Si–Beta zeolite. J Catal 199:41. https://doi.org/10.1006/jcat.2000.3156
Inagaki S, Matsukata M (2007) Elucidation of medium-range structure in a dry gel-forming* BEA-type zeolite. J Phys Chem C 28:10285. https://doi.org/10.1021/jp0668044
Ge P, Luo YL, Chen XH (2018) Synthesis of ERB-1 by a steam-environment crystallization method and further application in the post-synthesis of Ti-MWW zeolite. Appl Catal A Gen 564:218. https://doi.org/10.1016/j.apcata.2018.07.015
Acknowledgements
The authors acknowledge the support from the National Natural Science Foundation of China (21571036).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
†Electronic Supplementary information (ESI) available: chemicals involved in the M-BEA synthesis, characterization description, schematic diagram of simple experimental process, XRD pattern and nitrogen test of Co-BEA sample obtained without any seeds, O1s XPS spectra of Fe-BEA samples and UV–Vis spectra of Ga-BEA samples.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Luo, Y., Chen, X. & Lv, X. One-step synthesis of [Al,F]-free hierarchical M-BEA nanocrystals. SN Appl. Sci. 2, 1144 (2020). https://doi.org/10.1007/s42452-020-2930-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-2930-x