
Abstract
The present paper reports the density functional theory calculations of IR, NMR and UV–Visible, nonlinear optical (NLO) properties, highest occupied molecular orbital (HOMO)-lowest unoccupied molecular orbital (LUMO) energy band gap as measure of reactivity, molecular electrostatic potential surface (MESP), thermodynamic parameters and natural bond orbital (NBO) properties of the title molecule as well as its possible protein–ligand interactions using molecular docking approach. Theoretical NMR chemical shifts (1H and 13C) as well as the characteristics regions in IR active vibrations are in fair agreement with their experimental counterparts. UV–Visible absorption peak resulting from electronic transitions HOMO-5 → LUMO, HOMO-4 → LUMO observed at 324.27 nm is also in good agreement with its experimental value. The first order hyper polarizability of this molecule has been calculated to be 3182.124 × 10–33 esu which is 9.26 times to that of Urea indicating its suitability for nonlinear optical applications. The HOMO–LUMO energy band gap has been found to be reasonably small ≈ − 0.14596 a.u and MESP surface to be a chemically reactive site suited for drug activity. It is observed that the C11-atom of ring is more prone to the electrophilic attack. NBO analysis reveals that donor (O23) π → π *(C18-N22) acceptor interaction corresponds to the highest second order perturbation energy E(2) = 29.12 kcal/mole associated with electron delocalization. This biomolecule binds with 1GCN, 1HSG, 1X2J, 2NMO, 3I40 and 5C5S protein receptors showing that this molecule possesses multifunctional biological activity. Nonlinear optical and multifunctional biological activity reported in our study may be exploited for the novel applications of the heading molecule.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The plant-derived biomolecules are being used as natural medicine right from the ancient times and have become important due to their costless synthesis and noble properties. The quantum mechanical calculations on such molecules for tracing the molecular path way of their activity are possible at this time because the computational and theoretical chemistry methods are now available [1]. Vincosamide-N-Oxide having molecular formula C26H30N2O9 comprising of 67 atoms has already been reported to isolated from the fruits of Anthocephalus cadamba plant and its anti-lung cancer activity against cell lines H1299 have already been reported [2], however the high level quantum chemical analysis for elaborating the electronic structure properties to trace the molecular pathway towards its bioactivity and exploring its other novel applications are yet to be performed to the best of our knowledge. Due to the relatively large number of constituent atoms in the title molecule, the quantum chemical computation of all the properties implementing high level theory required for the accuracy of the results becomes less practical in terms of cost and computing time via general model chemistry. That is why depending upon the less computing time requirement, such properties have been computed by adopting DFT approach [3, 4] popular for the consistency of outcomes with the experimental results in the recent reports on the bio molecules [5,6,7]. The IR active vibrations, (1H &13C NMR) chemical shifts and UV–visible spectra have been computed using DFT and compared with their reported experimental counterparts in order to validate the adopted model chemistry at the optimized structure of the heading molecule. In order to decide the reactivity of this molecule, we have calculated the global and local reactivity descriptors both, using the same theory. Since NLO properties have been calculated at the same theoretical level because of its reported importance in various optoelectronics applications [8, 9]. The NBO analysis has been performed at the same level of density functional theory to predict the Lewis type NBO (donor)- Non-Lewis type NBO (acceptor) interaction associated with the largest delocalization. The biological activity of this natural molecule has been theoretically examined by using molecular docking approach which has been reported to be an important theoretical mode for the evaluation of the biological activity [10]. Since, the 1GCN [11], 1HSG [12] and (1X2J, 5C5S) [13, 14] protein receptors have been reported to be a target for an anti-diabetic, anti-immunodeficiency virus and anti-lung cancer agent along with G Protein-Coupled receptors are the novel structures for anti diabetic drug agent [15, 16] hence the binding of the titled molecule as drug agent with these protein receptors has been examined via the same molecular docking reported to be useful tool in computer-aided drug design due to the importance of shape-matching in drug-macromolecule interactions [17]. Docking of this molecule with the human insulin protein receptor (3I40) [18] and pancreatic cancer receptor (2NMO) [19] has also been performed to ascertain its anti diabetic mellitus activity and the nucleic acid residue at the binding sites have been located with the objective of visualizing its active layer.
The layer containing residue sites of binding in ligand–protein complex generated after completion of docking process has been viewed in the light of its MESP, chemical reactivity descriptors and NBO analysis. An attempt has been made to form the theoretical basis for studying its bioactivity using these quantum chemical analyses. The reports of the present study may be a good contribution in the area of computational chemistry of large natural biomolecule and applicable for evolving the principle of the spontaneous bioactivity contained in such molecules and developing the multifunctional natural drug agent.
2 Theoretical methods
The ground state structure of the title molecule was obtained first by applying B3LYP hybrid functional- 6–31 + G (d, p) basis set combination in DFT method [20, 21] and its frequency calculation at the same level of theory has been performed to ascertain the existence of true minimum at the potential surface visualized via gauss-view 05 program [22]. The 1H and 13C NMR chemical shifts have been calculated by using the most popular GIAO approach at the same level of theory. The UV–visible absorption spectrum and electronic transitions involved in it were analyzed using the known TD-DFT method at the same theoretical level. The effect of solvent (ethanol) is accounted by the reported polarizable continuum model of Tomasi and coworkers [23]. Theoretical calculations of HOMO, LUMO and MESP have been performed by using same DFT approach. Gaussian 09 program package [24] has been used for implementing the DFT model quantum chemistry calculations on the title molecule. In order to screen the biological activity of the title molecule, the protein receptors retrieved from Protein Data Bank (PDB) [25,26,27,28,29,30] has been allowed to interact with the title molecule as ligand using molecular docking approach implemented through the Auto dock 4.2 program package [31, 32].
3 Results and discussions
3.1 Geometry optimization, active modes of vibrations, IR spectra and single point energy
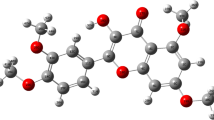
The optimized geometry of the titled molecule consisting of 67 atoms has been displayed in Fig. 1 and the total energy calculated at the single point of the potential surface named as the single point energy is ≈ −1,126,473.87 kcal/mol. The frequency calculation reveals 195 active fundamental modes of vibration which is in agreement with the reported formula of maximum (3 N-6) numbers of such modes of vibrations in a nonlinear molecule containing N atoms [33]. None of these frequencies are imaginary indicating the existence of true minimum at potential surface. The vibrational frequencies corresponding to these fundamental modes are assigned via gauss-view program. The calculated frequency of each fundamental mode has been scaled with a factor of 0.9648 because the hybrid functional B3LYP in DFT approach tends to overestimate the fundamental modes as reported by Merrick et al. [34]. The calculated and scaled frequencies along with their detailed vibrational assignments for each active mode of vibrations have been attached as supplementary material-1. The vibrations in the region 3094–3029 cm−1 occurring at the active modes of vibrations 188–181, are observed to be C-H stretching which in agreement with reported characteristic region 3100–3000 cm−1 [35] appeared due to the aromatic ring. The vibrational modes 132–95 shows the frequency range 1294–1003 cm−1 which correspond to C–H in plane bending frequencies appearing in line with the reported range 1300–1000 cm−1 [36]. The C–H out of plane bending vibrations have been reported to be appearing in the region 1000–750 cm−1[37] which are aligned with the theoretical vibrations occurred at the active modes 94–70 in the region 997–750 cm−1. The C–C stretching vibrations in titled compound are found in the region 1573–1305 cm−1 corresponding to the vibrational mode 161–133. Theoretically obtained C-H stretching due to the aromatic ring, O–H stretching due to the hydroxyl group and C = O vibrations due to the carbonyl group occurring at the region 3094–3029 cm−1, 3700–3649 cm−1 and 1730 cm−1 respectively in this molecule are in fairly good agreement with the corresponding experimental values at 3075–2851 cm−1, at 3424 cm−1 and 1612 cm−1 respectively reported in reference [2] which indicates the compatibility of the DFT optimization and frequency calculations within the theoretical constraints. The low region frequencies of other modes have also been obtained and the variation of the IR intensity with the calculated scaled frequency has been shown in Fig. 2.

Optimized geometry of vincosamide-N-Oxide

IR Spectra of vincosamide-N-Oxide
3.2 NMR analysis
The theoretical 13C and 1H chemical shifts in NMR spectra are compared with the corresponding experimental values in order to get insight for the conformational changes large bio molecules [38]. The optimized structure of this molecule is used to calculate the 1H and 13C chemical shifts using B3LYP/6–31 + G (d, p) level of computation in DFT via GIAO approach depicted in Table 1. The GIAO approach is most popular formalism which shows a reasonable correlation with the experimental data within the theoretical constraints for fairly large systems as per the available literature [38,39,40,41,42,43]. The 1H chemical shift for 6H, 45H, 48H atoms are observed at triplet degeneracy, 5H, 13H, 14H, 35H, 65H, 39H atoms at double degeneracy and rest H-atoms at singlet degeneracy. 13C chemical shift of all the C-atoms are observed at singlet degeneracy. The chemical structure including the computationally assigned serial number of the atoms in the optimized geometry of the title molecule obtained via Gauss View 05 program, used for NMR chemical shift calculation has been displayed in Fig. 3.The good conformity between theoretical and experimental values of 13C and 1H NMR chemical shifts are observed which indicates that the conformation of the theoretically optimized geometry of the title molecule is congruent to that used in experimental characterization and is in line with our earlier report on other such molecules [44, 45].

Sketch diagram of the optimized geometry of Vincosamide-N-Oxide
3.3 UV–Visible spectrum analysis
The UV–Visible spectra incorporating the effect of solvent (ethanol) has been obtained using TD-DFT method because Briquet and Vercauteren reported that this is compatible for getting consistent theoretical results with the experiments [46] and shown in Fig. 4. We have analyzed the excited states by specifying the excitation energies, wavelengths and oscillator strengths listed in Table 2. The oscillator strength is the measure of how strongly the particular electronic transition is allowed in absorption. The maximum peak occurs at 324.27 nm at excitation energy 3.8235 eV corresponding to the orbital transitions H-5 → LUMO, H-4 → LUMO with oscillator strength 0.2244 which is largest amongst all and hence these transitions would be strongly allowed. The UV–Visible analysis predicts four electronic transitions involved in producing UV–visible absorption band out of which the maximum peak occurs at 324.27 nm leading to a good agreement with its experimental vale of 324 nm reported in reference [2]. The prediction of the electronic transitions, oscillator strengths and excitation energies corresponding to the absorption peak in UV spectrum, is an important advantage of the present study.

UV–Visible spectra of Vincosamide-N-Oxide
3.4 Nonlinear optical properties (NLO)
The energy of a nonlinear molecule (ξ) subjected by an weak and homogeneous electric field (E) is given by
where ξ0 is the energy of the unperturbed molecules, El is the field at the origin, μl, αlm and βlmn are the components of dipole moment, polarizability and first hyperpolarizability, respectively. The total static dipole moment μ, the mean polarizability α0, the anisotropy of the polarizability Δα and the mean first order hyperpolarizability β0, are defined as:
where,
The first order hyperpolarizability is a third rank tensor whose 27 components of the 3D matrix can be reduced to 10 components because of the Kleinman symmetry [47]. The calculated values of μ, α and β are listed in Table 3. Since the values of the polarizabilities (∆α) and the hyperpolarizabilities (βtot) of the GAUSSIAN 09 output are obtained in atomic units (a.u.), the calculated values have been converted into electrostatic units (e.s.u.) (for α; 1 a.u = 0.1482 × 10–24 e.s.u., for β; 1 a.u = 8.6393 × 10–33 e.s.u.). The calculated values of dipole moment (D) for the title compounds were found to be 8.8450D respectively, which are approximately 6.22 times than those of urea (D = 1.3732 D). Urea is one of the prototypical molecules used in the study of the NLO properties of molecular systems. The first hyperpolarizability of title molecules is approximately 9.26 times than those of urea (β of urea is 343.272 × 10–33 esu). This result indicates that Vincosamide-N-Oxide is also suited for nonlinear optical applications.
3.5 Chemical reactivity analysis
3.5.1 Global reactivity descriptors and thermodynamic parameters
In case of our molecule, HOMO = − 0.243a.u and LUMO = − 0.097.a.u. The band gap is −0.097–(−0.243) = 0.146a.u which is reasonably small indicating that this natural biomolecule is soft molecule as per Pearson who proposed the HOMO–LUMO energy band gap to be the measure of softness of molecule for chemical reaction [48] which explains the eventual charge transfer within the molecule. The ionization potential (I), electronic affinity (A), electronegativity (χ), chemical potential (µ), global hardness (η), global softness (S) and global electrophilicity index(ω) are the global reactivity descriptor explained in term of HOMO–LUMO energy band gap [49, 50] which are listed as supplementary material-2. The ability of electron transportation and excitation properties is qualitatively predicted through HOMO and LUMO [51, 52] wherein HOMO primarily acts as an electron donor and LUMO acts as an electron acceptor and tends to create chemical reactivity through electronic transition [53]. The HOMO and LUMO plots of the title compound have been shown in Fig. 5. The low HOMO–LUMO energy gap favors the high value of polarizability and hyper polarizbilty which are measure of NLO property [54].

HOMO–LUMO plots for Vincosamide-N-Oxide
MESP surface is very useful to understand the potential sites for electrophilic (negative region) and nucleophilic (positive region) reactions [55, 56] which is well suited for recognition of one molecule by another through this potential, as in the case of drug- receptor interactions [57, 58] shown in Fig. 6.The values of the electrostatic potential at the surface are displayed by different colors in the order of red < orange < yellow < green < blue. The color code of these maps is in the range between −9.481 a.u. (deepest red) and 9.481 a.u. (deepest blue) in the titled compound, where blue indicates the most electropositive i.e. electron poor region and red indicates the most electronegative region, i.e. electron rich region. It is evident that the most electronegative region is located around 23O- atom which effectively acts as electron donor in molecule.

MESP surface for Vincosamide-N-Oxide l
By default, the program package used in the present study carries out thermo chemical analysis at 298.15 K temperature and 1 atmosphere of pressure. The thermodynamic parameters namely zero-point energy (ZPE), zero-point corrected vibrational energy (E), heat capacity at constant volume (CV), entropy (S), Gibb’s free energy (G) and enthalpy (H) have been obtained and given in supplementary material-2. The total energy of this molecular system is given by Etotal = E0 + Evibrational + Erotational + Etranslational, where E0 = Eelectronic + ZPE where zero-point energy correction (ZPE) to the electronic energy accounts for the effect of vibrations persisting even at 0 K in the molecule which has been estimated to be 342.11 kcal/Mol in case of title molecule. The final vibrational energy E, Enthalpy H = E + RT and Gibbs free energy G = H-TS are subject to the thermal correction which have been accounted in the results.
3.5.2 Local reactivity descriptors
The relative site selectivity of the atoms belonging to the active region (high layer) within the title molecule is determined by Fukui function \(\left( {f_{k}^{ \pm } } \right)\), local softness \((s_{k}^{ \pm }\)), and local electrophilicity \(\left( {\omega_{k}^{ \pm } } \right)\) which have been calculated as per reported equations [59,60,61,62] given below:
where q(N) is the charge on nth atom for neutral molecule while q(N + 1) and q(N − 1) are the same for its anionic and cationic chemical species, respectively. S and \(\omega\) are global softness and electrophilicity index respectively. The values of these descriptors are presented in Supplementary Maerial-3. If molecule is attacked by a soft reagent, it has tendency to react at the site where the value of local reactivity descriptor is largest and when attacked by hard reagent it tends to react at the site where the value of the same is smallest [63]. It is observed that the largest value of all local reactivity descriptors corresponds to 11C connected to the ring. Therefore, this site is favorable to the nucleophilic as well as electrophilic attack although, 11C site is more prone to the nucleophilic attack than electrophilic attack due to \(f_{k}^{ + }\) > \(f_{k}^{ - }\) as per the report of Morell et al. [64].
3.6 NBO analysis
NBO analysis helps in identifying individual bonds and the energies associated with loan -pair electrons which play an important role in the chemical process [65,66,67]. It provides the second order perturbative estimates of filled ‘donor’- ‘acceptor’ empty orbital (bond-antibond) interaction in NBO basis which leads to the loss of occupancy from the localized NBOs of idealized Lewis structure into Non-Lewis empty orbitals known as ‘delocalization correction to the zeroth-order natural Lewis structure’. For each donor NBO (i) and acceptor NBO (j), the stabilization energy E(2) associated with electron delocalization is computed [68, 69]. Second order perturbation theory analysis of the Fock matrix in NBO basis is listed in Table 4.
We observe that greatest stabilization energy of delocalization = 29.12 kcal/mol, is associated with the donor LP (O23) π → π *(C18-N22) acceptor interactions. We note that the atoms O23 has already been identified to be the sites of electrophilic attack in MESP also. The same atoms have been observed to be formed residue sites of interaction in ligand (title molecule)-protein complex.
3.7 Screening of bioactivity
Vincosamide-N-Oxide biomolecule as ligand binds with 1GCN, 1HSG, 1X2J, 2NMO, 3I40 and 5C5S protein receptors screened through molecular docking approach. The calculated free energy of binding and intermolecular energy of each protein–ligand interaction has been depicted in Table 5. The free energy of binding of each protein–ligand interaction is significantly negative which reveals that the labeled molecule can be explored as potential inhibitor against the diabetic mellitus, HIV, Lung Cancer and Pancreatic Cancer. The nucleic acid residue site of binding is ASP-9 in 1GCN binding complex located around 67O atom of the heading molecule. The same are LYS-20, APS-25 & GLY-27 in 1HSG complex located around O23, O53 & O67-atoms respectively, APS-389 & GLU-449 in 1X2J complex located around O23 & O67-atoms, GLU-184 & BGC-501 in 2NMO complex located around O23 & O67-atoms, TYR-14 & LEU-13 in 3I40 complex located around O23 & O67-atom and ASN-3 located around N22-atom, HIS-232 & LEU-233 in 5C5S complex located around O23 & O67-atoms. In this way, the binding has been observed to be mediated through the O-atoms of the title molecule at all the residue sites. The molecular surface of the complex in each protein–ligand interaction has been show in Figs. 7, 8, 9, 10, 11 and 12. On the basis of the biological activity evaluated in the present study, the labeled molecule may be explored as multifunctional drug candidate.

Molecular surface of the complex produced in Vincosamide-N-Oxide -1GCN binding

Molecular surface of the complex produced in Vincosamide-N-Oxide -1HSG binding

Molecular surface of the complex produced in Vincosamide-N-Oxide -1X2J binding

Molecular surface of the complex produced in Vincosamide-N-Oxide -2NMO binding

Molecular surface of the complex produced in Vincosamide-N-Oxide -3I40 binding

Molecular surface of the complex produced in Vincosamide-N-Oxide -5C5S binding
4 Conclusion
The density functional theory at DFT-B3LYP/6–31 + G (d,p) level is compatible for geometry optimization without introducing the conformational changes evident from the fair agreement between theoretical and experimental spectral characteristics (IR, NMR and UV–Visible). HOMO, LUMO, MESP, NLO properties, local reactivity descriptors and NBO properties computed applying the same level of theory on the title molecule are significant characteristics for structural description of this molecule. Reasonably low HOMO–LUMO energy band gap makes this biomolecule soft for the chemical reaction which may be one of the reasons of its inherent bioactivity. The MESP surface of the title molecule is suited for the drug activity wherein electrophilic sites are located around the 23O atom which is the epicenter of its biological activity. The first order hyperpolarizability of title molecules is approximately 9.26 times greater than that of the prototype molecule Urea indicates that this biomlecule can be explored for nonlinear optical applications. The reactive sites identified through local reactivity descriptors match well with those identified through molecular docking of the title molecule with protein receptor’s 1GCN, 1HSG, 1X2J, 2NMO, 3I40 and 5C5S. The NBO analysis also predicts the largest electron delocalization is associated with donor (O23) π → π *(C18-N22) acceptor interaction which indicates that these atoms are the suitable active sites at surface of the title molecule and that is why the nucleic acid residue sites of ligand–protein complex are located around these atoms. The present study finds the title molecule not only biologically active for drug applications but also optically active for nonlinear optical applications. The findings of the present investigations infer that apart from its reported activity against the lung Cancer Cell Line H1299; it possesses the biological activity against the several types of lung cancer, HIV and diabetic mellitus.
References
Young DC (2001) Computational chemistry. Wiley, New York
Mishra DP, Khan MA, Yadav DK, Rawat AK, Singh RK, Ahamad T, Hussain MK, Saquib M, Khan MF (2018) Monoterpene indole Alkaloids from Anthocephalus cadamba fruits exhibiting anticancer activity in human lung cancer cell line H1299. Chem Sel 3:8468–8472. https://doi.org/10.1002/slct.201801475
Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140(4A):A1133. https://doi.org/10.1103/PhysRev.140.A1133
Parr RG, Yang W (1994) International series of monographs on chemistry. University Press, Oxford
Taşal E, Sıdır İ, Gulseven Y, Oğretir C, Onkol T (2009) Vibrational spectra and molecular structure of 3-(piperidine-1-yl-methyl)-1,3-benzoxazol-2(3H)-one molecule by density functional theory and Hartree-Fock calculations. J Mol Struct 923:141–152. https://doi.org/10.1016/j.molstruc.2009.02.017
Srivastava AK, Pandey AK, Jain S, Misra N (2015) FT-IR spectroscopy, intra-molecular C–H–O interactions, HOMO, LUMO, MESP analysis and biological activity of two natural products, triclisine and rufescine: FT and QTAIM approaches. Spectrochim Acta 136:682–689
Zhongqiang L, Zhao-XuChen BJ (2019) Density functional theory studies on the structures and vibrational spectroscopic characteristics of nickel, copper and zinc naphthalocyanines. Spectrochimica Acta Part A 217:8–17. https://doi.org/10.1016/j.saa.2019.03.029
Andraud C, Brotin T, Garcia C, Pelle F, Goldner P, Bigot B, Collet A (1994) Theoretical and experimental investigations of the nonlinear optical properties of vanillin, polyenovanillin, and bisvanillin derivatives. J Am Chem Soc 116:2094–2102. https://doi.org/10.1021/ja00084a055
Geskin VM, Lambert C, Bredas JL (2003) Origin of high second- and third-order nonlinear optical response in ammonio/borato diphenylpolyene zwitterions: the remarkable role of polarized aromatic groups. J Am Chem Soc 125:15651–15658. https://doi.org/10.1021/ja035862p
Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, Olson AJ (2010) Virtual screening with AutoDock: theory and practice. Expert Opin Drug Discovery 5:597–607. https://doi.org/10.1517/17460441.2010.484460
Sasaki K, Dockerill S, Adamiak DA, Tickle IJ, Blundell T (1975) X-ray analysis of glucagon and its relationship to receptor binding. Nature 257:5529. https://doi.org/10.1038/257751a0
Chen Z, Li Y, Chen E, Hall DL, Darke PL, Culberson C, Shafer JA, Kuo LC (1994) Crystal structure at 1.9-A resolution of human immunodeficiency virus (HIV) II protease complexed with L-735,524, an orally bioavailable inhibitor of the HIV proteases. J Biol Chem 269:26344–26348
Padmanabhan B, Tong KI, NakamuraY OT, Scharlock M (2006) Structural basis for the defects of human lung cancer somatic mutations in the repression activity of Keap1 on Nrf2. RCSB PDB. https://doi.org/10.2210/pdb1X2J/pdb
Yi FS, Ren JQ, Feng W (2015) Crystal structure of human myosin 9b RhoGAP domain at 2.2 angstrom. RCSB PDB
Oh DY, Oefsky JM (2016) G protien-coupled receptors as targets for anti-diabetic therapeutics. Nat Rev Drug Discov 5:161–172. https://doi.org/10.1038/nrd.2015.4
Sasaki K, Dockerill S, Adamiak DA, Tickle IJ, Blundell T (1975) X-ray analysis of glucagon and its relationship to receptor binding. Nature 257:751–757. https://doi.org/10.1038/257751a0
Hawkins PC, Skillman AG, Nicholls A (2007) Comparison of shape-matching and docking as virtual screening tools. J Med Chem 50:74–82. https://doi.org/10.1021/jm0603365
TimofeevV I, Chuprov-Netochin RN, SamiginaV R, Bezuglov VV, Miroshnikov KA, Kuranova IP (2010) X-ray investigation of gene-engineered human insulin crystallized from a solution containing polysialic acid. Acta Crystallogr Sect F Struct Biol Cryst Commun. 66(3):259–263. https://doi.org/10.1107/S1744309110000461
Coppin L, Vincent A, Frénois F, Duchêne B, Lahdaoui F (2017) Galectin-3 is a non-classic RNA binding protein that stabilizes the mucin MUC4 mRNA in the cytoplasm of cancer cells. Sci Rep 7:43927. https://doi.org/10.1038/srep43927
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652. https://doi.org/10.1063/1.464913
Lee C, Yang W, Parr RG (1988) Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789. https://doi.org/10.1103/PhysRevB.37.785
Dennington R, Keith T, Millam J (2009) Gauss view, version 5. Semichem Inc., Shawnee Mission KS
Barone V, Cossi M (1997) A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J Chem Phys 107:3210–3221. https://doi.org/10.1063/1.474671
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr. J A, Peralta J E, Ogliaro F, Bearpark M, Heyd J J, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowskiand J, Fox DJ (2009) Gaussian 09,Revision B.01,Gaussian Inc. J Comput Chem, 30: 2785
https://www.rcsb.org/structure/1HSG Accessed 11 May 2018
https://www.rcsb.org/structure/1GCN Accessed 10 Aug 2018
https://www.rcsb.org/structure/1X2J Accessed 22 Jan 2019
https://www.rcsb.org/structure/2NMO Accessed 17 Dec 2018
https://www.rcsb.org/structure/3I40 Accessed 03 Dec 2018
https://www.rcsb.org/structure/5C5S Accessed 19 Jan 2019
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem 30:2785–2791. https://doi.org/10.1002/jcc.21256
Forli S, Olson AJ (2012) A force field with discrete displaceable waters and desolvations entropy for hydrated ligand Docking. J Med Chem 55:623–638. https://doi.org/10.1021/jm2005145
Silversten RM, Bassler GC (1991) Spectrometric identification of organic compound. Wiley, London
Merrick JP, Morran D, Radom L (2007) An evaluation of harmonic vibrational frequency scale factors. J Phys Chem A 111:11683. https://doi.org/10.1021/jp073974n
Silverstein RM, Webster FX (1998) Spectrometric identification of organic compounds, 6th edn. Wiley, New York
Arjunan V, Govindaraya ST, Ravindran P, Mohan S (2014) Exploring the structure-activity relations of N-carbethoxyphthalimide by combining FTIR, FT-Raman and NMR spectroscopy with DFT electronic structure method, Spectrochim Acta Part A 120:473–488. https://doi.org/10.1016/j.saa.2013.10.025
Sundaraganesan N, Ilakiamani S, Joshua BD (2007) FT-Raman and FT-IR spectra, ab initio and density functional studies of 2-amino-4,5-difluorobenzoic acid, Spectrochim Acta Part A 67:287–297. https://doi.org/10.1016/j.saa.2006.07.016
Schlick T (2010) Molecular modeling and simulation: an interdisciplinary guide, vol. 21, 2nd edn. Springer, Berlin. https://doi.org/10.1007/978-1-4419-6351-21
London F (1937) Théorie quantique des courants interatomiques dans les combinaisons aromatiques. J Phys Rhodium 8:397–409. https://doi.org/10.1051/jphysrad:01937008010039700
Mcweeny R (1962) Perturbation theory for the Fock-Dirac density matrix. Phys Rev 126:1028. https://doi.org/10.1103/PhysRev.126.1028
Ditchfield R (1974) Self-consistent perturbation theory of diamagnetism. Mol Phys 27:789–807. https://doi.org/10.1080/00268977400100711
Dodds JL, Mcweeny R, Sadlej AJ (1977) Self-consistent perturbation theory. Mol Phys 37:1779–1791. https://doi.org/10.1080/00268977700102961
Wolinski K, Hilton JF, Pulay P (1990) Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J Am Chem Soc 112:8251–8260. https://doi.org/10.1021/ja00179a005
Mishra AK, Tewari SP (2019) Structure, spectra and bioactivity of pentyl ester of chlorogenic acid: DFT study. Emerg Mater Res 8:651–662. https://doi.org/10.1680/jemmr.18.00129
Mishra AK, Tewari SP (2019) 7-Hydroxy-6-methoxy-coumarian to be a multifunctional bioactive natural molecule: a theoretical study. Mater Today: Proc 15:400–408. https://doi.org/10.1016/j.matpr.2019.04.100
Briquet L, Vercauteren DP, Andre JM, Perpete EA, Jacquemin D (2007) On the geometries and UV/Vis spectra of substituted trans-azobenzenes. Chem Phys Lett 435:257–262. https://doi.org/10.1016/j.cplett.2006.12.065
Kleinman DA (1962) Nonlinear dielectric polarization in optical media. Phys Rev 126:1977. https://doi.org/10.1103/PhysRev.126.1977
Pearson RG (1986) Absolute electronegativity and hardness correlated with molecular orbital theory. Proc Natl Acad Sci USA 83:8440–8441. https://doi.org/10.1073/pnas.83.22.8440
Pearson RG (1989) Absolute electronegativity and hardness: applications to organic chemistry. J Org Chem 54:1423–1430. https://doi.org/10.1021/jo00267a034
Padmanabhan J, Parthasarathi R, Subramanian V, Chattaraj PK (2007) Electrophilicity-based charge transfer descriptor. J Phys Chem A 111:1358. https://doi.org/10.1021/jp0649549
Belletete M, Morin JF, Leclerc M, Durocher G (2005) A theoretical, spectroscopic, and photophysical study of 2–7-carbazolenevinylene-based conjugated derivatives. J Phys Chem 109:6953–6959. https://doi.org/10.1021/jp051349h
Zhenminga D, Heping S, Yufang L, Dianshenga L, BoL L (2011) Experimental and theoretical study of 10-methoxy-2-phenylbenzo[h]quinoline. Spectrochim Acta Part A 78:1143–1148. https://doi.org/10.1016/j.saa.2010.12.067
Lewis DF, Loannides C, Parke DV (1994) Interaction of a series of nitriles with the alcohol-inducible isoform of P450: computer analysis of structure-activity relationships. Xenobiotica 24:401–408. https://doi.org/10.3109/00498259409043243
Agren H, Valtras O, Koch H, Jorgensen P, Helgaker T (1993) Direct atomic orbital based self-consistent-field calculations of nonlinear molecular properties: application to the frequency dependent hyperpolarizability of paranitroaniline. J Chem Phys 98:6417–6423. https://doi.org/10.1063/1.465099
Scrocco E, Tomasi J (1978) Electronic molecular structure, reactivity and intermolecular forces: an euristic interpretation by means of electrostatic molecular potentials. Adv Quantum Chem 11:115–193. https://doi.org/10.1016/S0065-3276(08)60236-1
Luque FJ, Lopez JM, Orozco M (2000) Perspective on “electrostatic interactions of a solute with a continuum: a direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor Chem Acc 103:343–345. https://doi.org/10.1007/978-3-662-10421-7
Scrocco E, Tomasi J (1973) Topics in current chemistry. 7: 95. https://www.springer.com/series/128
Li Y, Liu Y, Wang H, Xiong X, Wei P, Li F (2013) Synthesis, crystal structure, vibration spectral, and DFT studies of 4-aminoantipyrine and its derivatives. Molecules 18:877–893. https://doi.org/10.3390/molecules18010877
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516. https://doi.org/10.1021/ja00364a005
Parr RG, Szentpaly LV, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922–1924. https://doi.org/10.1021/ja983494x
Chattaraj PK, Lee H, Parr RG (1991) Principle of maximum hardness. J Am Chem Soc 113:1854–1855. https://doi.org/10.1021/ja00005a072
Chattaraj PK, Giri S (2007) Stability, reactivity, and aromaticity of compounds of a multivalent superatom. J Phys Chem A 111:11116–11121. https://doi.org/10.1021/jp0760758
Sponer J, Hobza P (1996) DNA base amino groups and their role in molecular interactions: Ab initio and preliminary density functional theory calculations. Int J Quantum Chem 57:959–970. https://doi.org/10.1002/(SICI)1097-461X(1996)57:5%3c959:AID-QUA16%3e3.0.CO;2-S
Morell C, Grand A, Labbe AT (2005) New dual descriptor for chemical reactivity. J Phys Chem A 109:205–212. https://doi.org/10.1021/jp046577a
Pophristic V, Goodman L, Guchhait N (1997) Role of lone-pairs in internal rotation barriers. J Phy Chem A 101:4290–4297. https://doi.org/10.1021/jp971020z
Guo D, Goodman L (1996) Nature of barrier forces in acetaldehyde. J Phys Chem 100:12540–12545. https://doi.org/10.1021/jp960182c
Weinhold F (2001) A new twist on molecular shape. Nature 411:539–541. https://doi.org/10.1038/35079225
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor view point. Chem Rev 88:899–926. https://doi.org/10.1021/cr00088a005
Liu CG, Su ZM, Guan XH, Muhammad S (2011) Redox and photoisomerization switching the second-order nonlinear optical properties of a tetrathiafulvalene derivative across six states: a DFT study. J Phys Chem C 115:23946–23954. https://doi.org/10.1021/jp2049958
Acknowledgements
AKM is thankful to the UGC, New Delhi for providing the minor research project on computational study on the bioactive natural compounds. Thanks are due to Dr. D. P. Mishra, Research fellow CDRI, Lucknow, India for the immense help and discussions regarding bioactive natural products. Authors are thankful to Prof. Neeraj Misra and Professor R.K. Shukla, Department of Physics, University of Lucknow for allowing us to access their computational facility. We are grateful to the Bioinformatics Resources & Applications Facility (BRAF), C-DAC Pune, India for permitting us to access its supercomputing facility for performing quantum chemistry calculations. We extend gratitude to Prof. Arthur J. Olson, department of Molecular Biology, Scripps Research Institute, La Jolla, USA for providing us the Auto Dock 4.2 Program for performing molecular docking study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mishra, A.K., Tewari, S.P. Density functional theory calculations of spectral, NLO, reactivity, NBO properties and docking study of Vincosamide-N-Oxide active against lung cancer cell lines H1299. SN Appl. Sci. 2, 1021 (2020). https://doi.org/10.1007/s42452-020-2842-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-2842-9