
Abstract
Phosphate geopolymers are part of chemically bonded phosphate cements obtained from an aluminosilicate and phosphate solution. Their structure consisting of phosphate bonds makes them suitable for use as refractory material. This study deals with the influence of phosphoric acid concentration (6, 8 and 10 mol/L) on the stability of volcanic ash-based phosphate geopolymers exposed to 100, 600 and 1000 °C. The results reveal that the onset of crystallization is about 600 °C with the formation of aluminum phosphate (V) and tridymite, then crystallization of iron (III) phosphate (V) and hematite at 1000 °C. The degree of crystallization of these phases increases with phosphoric acid concentration. The geopolymers obtained with 8 mol/L of phosphoric acid showed the best thermal stability at 1000 °C in terms of compressive strength change. The maximum thermal linear shrinkage recorded was 3%. The major phases of all geopolymers remain stable up to 1000 °C, after which the melting of phases happens.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Geopolymers are known as 3D network inorganic polymer obtained by chemical reaction of a solid precursor (aluminosilicate) and an alkaline or acidic medium [1]. Sometimes the alkaline part in powder form is directly mixed with the aluminosilicate and water used as liquid phase to make the binder. In that case it is called one-part geopolymer [2]. The geopolymer gained popularity because of the potential low CO2 emission and energy efficiency during its manufacturing compared to conventional cement [3, 4]. Moreover, the superior thermal stability of geopolymer compared to Portland cement has also increased the interest in that material for high-temperature applications [5]. However, this characteristic is closely related to the type of aluminosilicate used as well as the synthesis conditions [6,7,8]. For example, an addition of calcium sources to Metakaolin (MK) improves the thermal resistance of geopolymers in terms of compressive strength compared to solely MK-based geopolymers when exposed at 1000 °C [9]. It was reported a loss of 40% of initial strength after a cycle of heating–cooling of geopolymer from volcanic ash at 1000 °C [10]. When prepared in an alkaline medium, the phases of volcanic ash-based geopolymers are not stable beyond 1000 °C as they start melting contrary to MK-based ones. It was also shown that volcanic ash based-geopolymers obtained with potassium silicate as alkaline solution develops superior stability at elevated temperatures with thermal shrinkage less than 3% compared to the ones with sodium silicate [11]. In general, when exposed to elevated temperatures the physical properties of geopolymers obtained in alkaline solution are significantly deteriorated in the range of 750–900 oC [12, 13]. The latter constitutes the highest temperature range of application of geopolymer obtained in alkaline medium in the field of heat resistance materials. This is due to cations present in the alkaline solution which lower the temperature of glass transition, hence induce sintering with the decrease of the mechanical strength along with severe thermal shrinkage [8, 11]. Up to now, few studies deal with the phase stability or physical properties of geopolymers obtained in an acidic medium after exposure at elevated temperatures.
The phases of MK-based geopolymers obtained with phosphoric acid were reported to be thermally stable, with linear shrinkage of 5.3% after exposure at 1450 °C [14]. Also, phosphoric acid activation of synthetic Al2O3–2SiO2 powders showed extremely high thermal stability of phases with no sign of melting up to 1550 °C [15]. Recent work reported that the structural evolution of phases with temperature and their stability depend on the phosphate content of the matrix [16]. However, MK-based phosphate geopolymers have low thermal stability in terms of compressive strength evolution. Bewa et al. reported a decrease of more than 75% of the compressive strength development only after exposure at 200 °C [17]. While Celerier et al. [18] showed the complete destruction and/or extensive cracks apparition with a significant decrease of the compressive strength of MK-based phosphate geopolymer after heating to 1000 °C. These behaviours at elevated temperatures depend on the Al/P ratio of the matrix, the reactivity of the MK used and its wettability. Moreover, the high-water content of the MK-based phosphate geopolymer was also responsible of the destruction of the binding phase because of the dehydroxylation that happens after heating. Since MK particles have a high specific surface area, thus high-water demand and high liquid to solid mass ratio are required for reaching good workability.
The investigation of geopolymers from volcanic ash and phosphoric acid has never been reported yet. Since the improved thermal stability of phases of MK-phosphate geopolymer was due to the high stability of the phosphate bond, the low thermal stability of phases of volcanic ash-based geopolymer in alkaline solution could also be improved when synthesized in acidic medium. Moreover, volcanic ash has a lower specific surface area compared to MK and generally low liquid to solid mass ratio is required for achieving good workability [19]. This work aims to investigate the influence of phosphoric acid concentration on the thermal stability of volcanic ash-based phosphate geopolymers for high-temperature applications. These include the study of the transformation and transition of phases, the thermal shrinkage as well as porosity and compressive strength of geopolymers exposed to high temperatures. The thermal transformation and transition of phases were investigated by X-ray diffractometry (XRD), Fourier transformed infrared spectroscopy (FTIR), Thermogravimetry analysis coupled with Differential Scanning Calorimetry (TGA–DSC) and Dilatometry analysis. The porosity was determined using mercury intrusion porosimeter (MIP) and the microstructure by using scanning electron microscope with energy dispersive X-ray spectroscopy.
2 Materials and methods
2.1 Materials
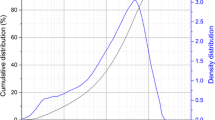
Volcanic ash (VA) was collected along “the Line of Cameroon” and ground in a ball mill to get a powder with particle size < 200 μm. The chemical analysis carried out by X-ray fluorescence is reported in Table 1. The particle size distribution carried out using a laser particle size analyzer (MASTERSIZER S, Malvern, UK) is depicted in Fig. 1. It shows a trimodal distribution with characteristic diameters as follows: d10 = 0.26 μm d50 = 3.7 μm and d90 = 57 μm. Orthophosphoric acid (H3PO4, 85wt%) was used to prepare the hardeners. The latter were obtained by diluting the as received orthophosphoric acid with water to get concentrations of phosphoric acid solution 6, 8 and 10 mol /L.

Particles size distribution of milled volcanic ash
2.2 Experimental methods
The geopolymer pastes were prepared by mixing in an automatic Hobart mixer for 3 min volcanic ash and phosphoric acid according to liquid to solid mass ratios of 0.32, 0.36 and 0.4 respectively. These different ratios were chosen according to the texture of each paste which allows easy pouring in the mold before losing any workability. The pastes obtained were poured in 40 mm cubic moulds, vibrated for 3 min and covered with a plastic foil to avoid cracks or pores due to rapid water evaporation. The specimens were kept at 27 ± 3 °C and 62%Relative humidity for 24 h before demolding, then stored in the same curing condition for 28d. Afterward, the geopolymer specimens were heated to a temperature of 100, 600 and 1000 °C at a heating rate of 5 °C/min in a programmable electrical furnace and kept at this temperature for 4 h. The analysis of the mineralogical composition of the geopolymers before and after exposure to elevated temperatures was carried out using X-ray diffractometry (Bruker X-ray diffractometer (D8 Discovery, US)). The device operates with Cu Kα1 radiation (λ = 1.54 Å), at 35 kV, 45 mA. The XRD patterns were recorded in the range of 15–70° 2θ. The changes in the chemical bonds of unheated and heated geopolymers were investigated by FTIR spectroscopy (Nicolet 5700 spectrometer). The analysis was carried out on KBr pellets. For each sample, 32 scans with a resolution of 4 cm−1 were recorded in the frequency range 4000–400 cm−1. The porosity and pore size distribution of unheated geopolymers were investigated by mercury intrusion porosimeter (Auto pore IV 9500 Micromeritics) It operates with the set-time equilibrium of 10 s and pressure ranges of 345 kPa and 228 MPa that allows to measure pore size range between 0.005 and 360 μm. The compressive strength of geopolymers before and after exposure to elevated temperatures was measured thanks to an automatic compressive test machine (Impact Test Equipment Limited) with a maximum load of 250 KN. The microstructure was carried out on unheated geopolymer using Zeiss Gemini SEM 500 NanoVP microscope (Oberkochen, Germany) coupled with an Energy-Dispersive X-ray spectroscopy (EDX) that operate at low-Vacuum mode with 15 kV acceleration voltage.
Thermal transformation and transition phases were monitored using Thermogravimetry analysis coupled with Differential Scanning Calorimetry (Linseis, Model STA PT1000). The equipment operated in the temperature ranges 20–1000 °C with a heating rate of 20 °C/min at an ambient atmosphere. An optical dilatometer (ESS MISURA HSM ODHT Model 1600/80) was used to assess changes of length of specimens shaped 5 × 5 × 15 mm3.
3 Results and discussion
3.1 Influence of phosphoric acid concentration on mineralogy at 27 and 1000°C
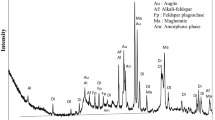
The phase compositions of raw volcanic ash and geopolymers before and after exposure to 1000 °C are depicted in Figs. 2 and 3 respectively. The main minerals identified in volcanic ash using X’Pert High Score Plus software are as follows: augite (PDF# 98-005-6926); albite high (PDF# 98-010-0499); jadeite-diopside (PDF# 98-006-9709); forsterite, ferroan (PDF# 98-003-9795) and magnetite (PDF# 98-008-5807). In the unheated geopolymers, no new crystalline phases were identified (Fig. 2), but a significant decrease of peaks intensity of major minerals is visible. This behaviour is like what happens with volcanic ash-based geopolymers obtained in alkaline solution [20, 21]. The intensities of the peaks of augite (Al0.1Ca0.83Fe0.34Mg0.77Na0.02O6Si1.92Ti0.02) at 29.80° (2.99 Å), 35.53° (2.52 Å) and 35.74° (2.51 Å) respectively decrease by 38, 27 and 28% respectively after reaction of volcanic ash with 6 mol/L acid solution. Also, intensities of the main peaks of albite (NaAlSi3O8) at 2θ = 27.73° (3.21 Å), 23.57° (3.77 Å) and 21.87° (4.05 Å) respectively are reduced by 26, 22 and 17% respectively. These data show that augite is more dissolved than albite. This is likely due to the preferential dissolution of augite (Pyroxene group) in low pH (below 3–4) in comparison with albite (Feldspar group) [22, 23]. However, the extent of dissolution of those minerals is not proportional to the acid concentration, though it remains low in volcanic ash. A tentative explanation of this behaviour might be the rapid dissolution of amorphous phase and fast setting as observed during mixing, which would have delayed the further dissolution of unreacted particles with time. Nevertheless, a deep investigation to understand the dissolution behaviour of volcanic ash in an acidic medium is requires and this will be carried out in our upcoming studies.

X-ray patterns of volcanic ash and resulting geopolymers at 27 °C

X-ray patterns of geopolymer after heating at 1000 °C
The X-ray patterns of heated geopolymers at 1000 °C (Fig. 3) show several changes in the phase compositions compared to unheated ones. The main diffraction peaks of minerals like Jadeite-Diopside (PDF# 98-006-9709); Forsterite, ferroan (PDF# 98-003-9795) and Magnetite (PDF# 98-008-5807) are no longer identifiable, which means that they were transformed into other phases. It can also be seen the significant decrease of intensities of peaks at 2θ = 29.80° (2.99 Å) and 27.74° (3.21 Å) corresponding to the main peak of augite and albite respectively with acid concentration. This happens along with the change in the profile of the broad peak at 2θ = 35.74 (2.51 Å) on the X-ray pattern of volcanic ash which becomes narrow. The extent of the decrease of the main peak intensity of these major minerals shows that in addition to acid concentration, the stability of augite is more sensitive to temperature than albite. The new crystalline phases are aluminum phosphate (V) (PDF# 98-028-0307), tridymite (PDF# 98-009-4090), hematite (PDF# 98-008-8418) and iron (III) phosphate (V) (PDF# 98-009-8063). Their main peaks are as follows: 2θ = 20.46° (4.33 Å), 21.52° (4.12 Å) and 23.15° (3.84 Å) for aluminium phosphate (V); 2θ = 20.32° (4.36 Å), 21.62° (4.10 Å) and 23.08° (3.85 Å) for tridymite); 2θ = 25.68° (3.46 Å), 37.63° (2.38 Å) and 20.33° (4.36 Å) for iron(III) phosphate(V); 2θ = 33.25° (2.69 Å), 35.72° (2.51 Å) and 54.23° (1.70 Å) for hematite. Iron phosphate minerals have a similar structure like the SiO2-based minerals [24]. It can be noticed an increase of these main peak intensities with the increase of phosphoric acid concentration. The formation of aluminum phosphate(V) and silica-based minerals such as tridymite are common in phosphate geopolymers treated at 1000 °C [15, 17, 25,26,27]. The formation of these two minerals is due to the crystallization of amorphous AlPO4-SiO2 solid solution constituting the binding phase of the phosphoric acid-based geopolymer [27]. Thus, the increase of the amount of these minerals demonstrates the extent of dissolution of reactive phases that happens with the increase of phosphoric acid concentration. Moreover, the formation of hematite and iron phosphate is new in high-temperature heating of phosphate geopolymers. Their presence is the proof that in addition to AlPO4-SiO2 solid solution as binder in phosphate geopolymer there is also FePO4 in the case of ferro-aluminosilicate materials like volcanic ash. Moreover, similarly to aluminum phosphate and tridymite, the presence of hematite and iron phosphate comes from the partial breakdown of the Fe–O–P bonds of the amorphous phase of the geopolymer binder.
Structural bond changes in geopolymer with varying phosphoric acid concentration before and after exposure to elevated temperatures are highlighted in Figs. 4 and 5 respectively. The vibration band appearing at 1650 and 3420 cm−1 in all IR spectra are respectively characteristic of bending and stretching vibrations of H–O–H and H–O bonds of water molecules. In Fig. 4, the band on VA at 1020 cm−1 is characteristic of stretching vibrations of Si–O bonds of silicate chain. With 6 mol/L of phosphoric acid used as a hardener, that band has shifted by about 52 cm−1 to a higher value after geopolymerization. It further shifted by 11 cm−1 when the acid concentration rises respectively to 8 and 10 mol/L. Such change is also noticeable on the band at 750 cm−1 of volcanic ash, which is characteristic of stretching vibration of Si–O bonds of the silicate network, that shifted to 792 cm−1 on geopolymer. This observation indicates that there is a formation of new bonds as a result of reaction occurring [21, 28]. Such a phenomenon can be attributed to the formation of the polymeric structure of geopolymer binder which structural unit includes connection among tetrahedra [AlO4]5−, [FeO4]5−and [PO4]5−. The changes observed in that main vibration band also ascertain that increasing the concentration of phosphoric acid improves the extent of the dissolution of reactive phases as stated by XRD patterns. The bands appearing at 630, 578 and 541 cm−1 are respectively related to the various vibrating modes of the Si–O bonds in the silicate chains of volcanic ash [29].

IR spectra of volcanic ash and resulting geopolymers at 27 °C

IR spectra of geopolymer after heating at 1000 °C
Figure 5. shows the IR spectra of geopolymer with varying acid concentration after heating at1000oC. After heating geopolymer, there is a significant decrease in the intensity of bands at 3420 and 1650 cm−1 which are characteristic of vibration modes of water molecules. Compared to the IR spectra of geopolymer cured at 27 °C (Fig. 4.) it can also be observed a shift of the wavenumber of the main band characteristic of Si–O vibration (1072–1083 cm−1) to a higher value (1112 cm−1) and the apparition of new bands at 1029 and 1064 cm−1 respectively. Other changes are the disappearance of the vibration band at 541 cm−1 and the appearance of the band at 713 cm−1 after heating. Similar changes were observed in previous work [16, 17]. In this study, these changes are attributed to the crystallization of tridymite and phosphate minerals as shown in XRD patterns of geopolymer after heating at 1000 °C.
3.2 Influence of heating temperature on the mineralogy of phosphate geopolymer obtained with 10 mol/L acid concentration
Figure 6 shows the X-ray patterns of volcanic ash-based geopolymers obtained with 10 mol/L of phosphoric acid after exposure to various temperatures. As it can be seen, the crystalline phases initially present in unheated geopolymers remain stable up to 600 °C, where the onset of crystallisation is observed. The latter phenomenon is however characterized by the formation of new minerals, aluminum phosphate (V) (PDF# 98-028-0307) and tridymite (PDF# 98-009-4090). Although the initial minerals remain up to 600 °C, their peak intensities somewhat diminish with increasing temperature. This is more prominent at 1000 °C where other new minerals (hematite and iron (III) phosphate (V)) are formed along with the disappearance of some of the initial ones (jadeite-diopside; forsterite, ferroan and magnetite). There is also a significant increase of the peak intensities of aluminum phosphate (V) and tridymite at 1000 °C. It arises from these results that, the stability of minerals of the studied system is temperature sensitive and the onset of crystallization is at around 600 °C.

X-ray patterns of geopolymer obtained with 10 mol/L acid concentration after heating at various
3.3 Physical characteristics and microstructure
The IR spectra of geopolymers obtained with 10 mol/L of phosphoric acid and heated at various temperatures are shown in (Fig. 7). The effect of heating is highlighted by the main changes between 600 and 1000 °C. The latter include the shift of the main vibration band from 1093 to 1122 cm−1, the disappearance of the bands at 541 cm−1 and the appearance of the bands at 713, 1029 and 1064 cm−1. These results confirm the fact that the onset of crystallisation of phosphate geopolymers is around 600 °C.

IR spectra of geopolymer obtained with 10 mol/L acid concentration after heating at various temperatures
3.3.1 Visual aspect
After maintaining geopolymers at various temperatures, changes in colour and the formation of cracks on the surface of some specimens were observed (Fig. 8). The specimens obtained with 6 mol/L of phosphoric acid show no sign of surface deterioration when exposed to 100 °C, whilst other specimens show some cracks. The latter seems to be more prominent and larger on the specimen obtained with 10 mol/L of phosphoric acid than the one obtained with the concentration of 8 mol/L. The presence of these cracks can be due to the evaporation of free water present in the pores network or to the dehydroxylation of geopolymer binder. Thus, the extent of cracks demonstrates that the geopolymer obtained with 10 mol/L of phosphoric acid undergoes more damages at 100 °C compared to other specimens. At 600 °C, the geopolymer specimens obtained with 6 and 8 mol/L of phosphoric acid display minor microcracks and there are changes of initial colours from black to light and dark brown. When heated to 1000 °C all the specimens do not show any sign of external damage except the colour which is now light red and becomes dark with increasing phosphoric acid concentration. Based on a correlation between the change of colours and the mineral evolution with temperature, it can be concluded that the change of colour observed at 600 °C results from the crystallisation of aluminium phosphate(V) and tridymite, whereas those observed at 1000 °C corresponds to the crystallisation of iron (III) phosphate and hematite.

The physical appearance of geopolymers before and after exposure to various temperatures
3.3.2 Porosity
The total porosity and pore size distribution of unheated geopolymers are shown in Figs. 9 and 10 respectively. Total porosity (Fig. 9) of geopolymers obtained with 6 or 8 mol/L of phosphoric acid are 22 and 21vol% respectively. The porosity significantly decreases to 10.7vol% when the acid concentration rises to 10 mol/L, which means that the latter has fewer pores compared to the formers. However, it is clearly shown in Fig. 10a that the specimen obtained with 10 mol/L of phosphoric acid exhibits larger pore diameters than those obtained with 6 and 8 mol/L respectively. The range of pore size (Fig. 10b)is 0.01–5 µm, 0.01–1 µm and 0.027–5 µm for specimens with 6, 8 and 10 mol/L of phosphoric acid respectively. Within these pore diameter ranges, the geopolymers obtained with 6 and 10 mol/L of phosphoric acid have a unimodal pore diameter distribution with maxima at 0.09 and 0.55 µm respectively. Whereas specimens obtained with 8 mol/L of phosphoric acid show a bimodal distribution with maxima at 0.04 and 0.06 µm respectively. The effect of pore size distribution on the thermal stability of geopolymers will be well discussed in the next section.

Total porosity of geopolymers

Pores size distribution of geopolymers: (a) Cumulative pores volume, (b) Differential volume of intruded mercury
3.3.3 Compressive strength
Figure 11 depicts the 28d compressive strength evolution of geopolymer before and after heating at various temperatures for 4 h. At room temperature, compressive strength increases with acid concentration. After heating at 100 °C, the strength of geopolymer obtained with 6 mol/L of phosphoric acid increases by about 84% (49 MPa) while that at the other temperatures decreases. For geopolymers obtained with 8 and 10 mol/L of phosphoric acid, the decrease of compressive strength is 14.7% (43 MPa) and 17% (60 MPa) respectively. This trend correlates well with the observation made on (Fig. 8) where major cracks were observed only on geopolymers obtained respectively with 8 and 10 mol/L of phosphoric acid. These observations allow surmising that although high dissolution of reactive phase using of 6 mol/L of phosphoric acid was observed in XRD patterns, the reaction of volcanic ash with 6 mol/L acid concentration remains low. Thus, when exposed at 100 °C, the heat supplied rather improves the geopolymerization leading to a more condensed and stronger polymer chain. This seems to be the contrary to geopolymerization with 8 and 10 mol/L acid concentration. In these latter cases, the reactions that take place are faster and lead directly to the formation of strong geopolymer structures at room temperature. Thus, the heat supplied has rather triggered the dehydroxylation of the binding phase, leading to the formation of cracks, hence weakening of the structure. The strength of geopolymer obtained with 10 mol/L of phosphoric acid continues to decrease till the temperature 1000 °C (26.5 MPa), whereas the strength of geopolymer obtained with 8 mol/L of phosphoric acid (56.5 MPa) remains high compared to the one before heating. This difference of behaviour might be due to the pores size distribution of the geopolymers. In fact, the geopolymer obtained with 8 mol/L of phosphoric has high number of pores with various diameters, which allows easy evaporation of water during the dehydroxylation stage of the binder with less impact on compressive strength.

Compressive strength evolution of geopolymers with temperature
3.3.4 Scanning electron microscopy-EDX
The microstructures of unheated geopolymers are shown in Fig. 12. The micrographs are characterized by the presence of several cracks that might come from the evaporation of water during the geopolymerization. That process is fostered by the heat released during the reaction, as when volcanic ash is mixed with the phosphoric acid an exothermic reaction occurs. It was reported that in phosphate cement the temperature of the fresh paste can reach up to 100 °C depending on the mix design [30]. The high temperature reached is due to the presence of metal oxides CaO and MgO in volcanic ash. All the samples are composed of a dense phase characteristic of the binder. Whereas the sample obtained with 6 mol/L shows the presence of unreacted particles translating the low formation of reaction products.

Micrographs of unheated geopolymer samples
3.4 Phases and dimensional stability
3.4.1 Mass loss and phase stability
The monitoring of the mass change, the phases transformation and transition of the geopolymer with temperature were carried out by TGA–DSC (Fig. 13). The TGA curves are characterized by a single mass loss of 13–14 wt% which starts nearly at room temperature and ends at around 600 °C. That mass loss is accompanied by an endothermic peak at 86 °C. These changes correspond to the removal of free water contained in the pores network and the dehydroxylation of polymer chains of the geopolymer binder [31, 32]. Between 600 and 1000 °C, there is not any significant mass change. The DSC curves show respectively a succession of three small exothermic peaks at 634–657 °C, 690 and 820 °C, and an endothermic peak at 805 °C. The first one (634–657 °C) corresponds to the crystallization of ferrous/ferric phosphate minerals from the amorphous phase. In the crystallization process of iron phosphate glasses, that first peak is generally followed by a second exothermic peak at 820 °C related to the crystallization of ferric phosphate minerals (FePO4) [33, 34]. The latter obviously confirms the crystallization of iron (III) phosphate identified only in the XRD patterns of geopolymer maintained at 1000 °C. The endothermic peak at 805 °C corresponds to the decomposition of remaining amorphous iron phosphate FePO4 to crystallized Fe2O3 [33]. This confirms the crystallization of hematite observed in XRD patterns. Noteworthy that during the phase transition of iron phosphate there is sometimes a competition between decomposition FePO4 into Fe2O3 and the crystallization of new form of FePO4 [35]. The temperature of the apparition of the first exothermic peak (634–657 °C) shifts to lower value with increasing phosphoric acid concentration. This is due to the increase of the content of Fe2+/Fe3+ ions in the bulk composition [33]. The exothermic peak appearing at 690 °C corresponds to the crystallization of aluminum phosphate and /or tridymite [27].

TGA (solid lines) and DSC (dotted lines) curves of geopolymers with varying phosphoric acid concentrations
3.4.2 Dimensional stability
The dilatometry curves (Fig. 14a) show that up to 1000 °C, thermal linear shrinkage of geopolymers are 1.8, 2.9 and 3.1% respectively. These shrinkages are mainly observed in two temperature ranges (110–140 °C and 630–665 oC). It is worth pointing out that beyond 1000 °C the higher sintering of volcanic ash particles induces the beginning of melting of mineral phases [10, 11]. This can explain the higher shrinkage beyond 1000 °C. Hence, this shrinkage was not included in the total shrinkage calculation because the material melts and does not withstand. The first shrinkage occurs between 25 to 600 °C and reaches the maximum at 110–140 °C. This shrinkage is of about 1.6, 2.3 and 2.4% for geopolymer specimens obtained respectively with 6, 8 and 10 mol/L of phosphoric acid. The increase of the first shrinkage with phosphoric acid concentration correlates with the extent of cracks as observed on the surface of specimens after exposure at 100 °C (Fig. 8). This shrinkage is linked to the mass loss recorded in TGA and which corresponds to the evaporation of water. The second shrinkage begins at around 600 °C and ends before 1000 °C, and is about 0.2, 0.6 and 0.7% for geopolymer specimens obtained respectively with 6, 8 and 10 mol/L of phosphoric acid. This second shrinkage, which does not correspond to any mass loss, is due to the crystallization of ferrous/ferric phosphate minerals. The shift of the maximum temperature related to this second shrinkage (Fig. 14b) agrees with the similar phenomenon observed in the DSC curves (Fig. 13), and which was ascribed to the increase of the amount of Fe2+/ Fe3+ ions in the bulk composition. The latter arising from the increased dissolution of the reactive phase of volcanic ash with the increase of phosphoric acid concentration. Most interesting in the differential curve of the percentage of expansion beyond 1000 °C is the delay of the temperature at which maximum shrinkage due to melting occurs (Fig. 14b). Such behaviour means that the increase of the dosage of phosphate bonds in the geopolymer binder prevents the early melting of phases.

Dimension change with temperature (a) % of expansion; (b) differential of the % of the expansion
4 Conclusion
The influence of phosphoric acid concentration on the thermal stability of volcanic ash-based phosphate geopolymers was reported. It appears that changes occurring in mineralogical phases are highly sensitive to temperature and phosphoric acid concentration. The major phases including geopolymer binder remain stable up to 1000 °C and beyond the latter temperature, the melting of geopolymers becomes predominant. The geopolymer binder is based on ferro-silico-aluminophosphate (AlPO4-SiO2-FePO4) chains. The total porosity of unheated geopolymers decreases from 22 to 10% with increasing phosphoric acid concentration. The heterogeneity of pore diameter distribution enhances the thermal stability of geopolymers in terms of strength loss and thermal shrinkage. The maximum strength loss was 63% and the highest thermal shrinkage 3% after heating geopolymers up to 1000 °C. The geopolymers produced with a phosphoric acid concentration of 8 mol/L demonstrated superior thermal stability. Thus, phosphate geopolymer is suitable for high-temperature applications up to 1000 °C.
References
Davidovits J (2015) Geopolymer chemistry and applications, 4th edn. Institut Géopolymère, Saint-Quentin
Ke X, Bernal SA, Ye N, Provis JL, Yang J (2015) One-part geopolymers based on thermally treated red mud/NaOH blends. J Am Ceram Soc 98:5–11. https://doi.org/10.1111/jace.13231
Ouellet-Plamondon C, Habert G (2015) Life cycle assessment (LCA) and innovative applications of alkali-activated cements and concretes. In: Handbook of alkali-activated cements, mortars and concretes 2015, pp 663–686
Davidovits J (2015) False values on CO2 emission for geopolymer cement/concrete published. Sci Pap Geopolymer Inst Libr TechPap 24:1–9
Rivera OG, Long WR, Weiss CA, Moser RD, Williams BA, Torres-Cancel K, Gore ER, Allison PG (2016) Effect of elevated temperature on alkali-activated geopolymeric binders compared to portland cement-based binders. Cem Concr Res 90:43–51. https://doi.org/10.1016/j.cemconres.2016.09.013
Yadollahi MM, Benli A, Demirboğa R (2015) Effects of elevated temperature on pumice based geopolymer composites. Plast Rubber Compos 44:226–237. https://doi.org/10.1179/1743289815Y.0000000020
Tchakouté HK, Rüscher CH, Kong S, Kamseu E, Leonelli C (2016) Thermal behavior of metakaolin-based geopolymer cements using sodium waterglass from rice husk ash and waste glass as alternative activators. Waste Biomass Valoriz. https://doi.org/10.1007/s12649-016-9653-7
Elimbi A, Tchakoute HK, Kondoh M, Manga JD, Dika Manga J (2014) Thermal behavior and characteristics of fired geopolymers produced from local Cameroonian metakaolin. Ceram Int 40:4515–4520. https://doi.org/10.1016/j.ceramint.2013.08.126
Bernal SA, Rodríguez ED, Mejía De Gutiérrez R, Gordillo M, Provis JL (2011) Mechanical and thermal characterisation of geopolymers based on silicate-activated metakaolin/slag blends. J Mater Sci 46:5477–5486. https://doi.org/10.1007/s10853-011-5490-z
Lemougna PN, MacKenzie KJD, Melo UFC (2011) Synthesis and thermal properties of inorganic polymers (geopolymers) for structural and refractory applications from volcanic ash. Ceram Int 37:3011–3018. https://doi.org/10.1016/j.ceramint.2011.05.002
Lemougna PN, Chinje Melo UF, Delplancke MP, Rahier H (2013) Influence of the activating solution composition on the stability and thermo-mechanical properties of inorganic polymers (geopolymers) from volcanic ash. Constr Build Mater 48:278–286. https://doi.org/10.1016/j.conbuildmat.2013.06.089
Kong DLY, Sanjayan JG, Sagoe-Crentsil K (2007) Comparative performance of geopolymers made with metakaolin and fly ash after exposure to elevated temperatures. Cem Concr Res 37:1583–1589. https://doi.org/10.1016/j.cemconres.2007.08.021
Kong DLY, Sanjayan JG (2010) Effect of elevated temperatures on geopolymer paste, mortar and concrete. Cem Concr Res 40:334–339. https://doi.org/10.1016/j.cemconres.2009.10.017
Le-ping L, Xue-min C, Shu-heng Q, Jun-li Y, Lin Z (2010) Preparation of phosphoric acid-based porous geopolymers. Appl Clay Sci 50:600–603. https://doi.org/10.1016/j.clay.2010.10.004
Le-ping L, Xue-min C, Yan H, Si-dong L, Si-yu G (2012) The phase evolution of phosphoric acid-based geopolymers at elevated temperatures. Mater Lett 66:10–12. https://doi.org/10.1016/j.matlet.2011.08.043
Khabbouchi M, Hosni K, Mezni M, Zanelli C, Doggy M, Dondi M, Srasra E (2017) Interaction of metakaolin-phosphoric acid and their structural evolution at high temperature. Appl Clay Sci 146:510–516. https://doi.org/10.1016/j.clay.2017.07.006
Nobouassia Bewa C, Tchakouté HK, Fotio D, Rüscher CH, Kamseu E, Leonelli C (2018) Water resistance and thermal behavior of metakaolin-phosphate-based geopolymer cements. J Asian Ceram Soc 6:271–283. https://doi.org/10.1080/21870764.2018.1507660
Celerier H, Jouin J, Tessier-Doyen N, Rossignol S (2018) Influence of various metakaolin raw materials on the water and fire resistance of geopolymers prepared in phosphoric acid. J Non Cryst Solids 500:493–501. https://doi.org/10.1016/j.jnoncrysol.2018.09.005
Djobo JNY, Tchadjié LN, Tchakoute HK, Kenne BBD, Elimbi A, Njopwouo D (2014) Synthesis of geopolymer composites from a mixture of volcanic scoria and metakaolin. J Asian Ceram Soc 2:387–398. https://doi.org/10.1016/j.jascer.2014.08.003
Lemougna PN, Chinje Melo UF, Delplancke MP, Rahier H (2014) Influence of the chemical and mineralogical composition on the reactivity of volcanic ashes during alkali activation. Ceram Int 40:811–820. https://doi.org/10.1016/j.ceramint.2013.06.072
Djobo JNY, Elimbi A, Tchakouté HK, Kumar S (2016) Reactivity of volcanic ash in alkaline medium, microstructural and strength characteristics of resulting geopolymers under different synthesis conditions. J Mater Sci 51:10301–10317. https://doi.org/10.1007/s10853-016-0257-1
Chen Y, Brantley SL (1997) Temperature- and pH-dependence of albite dissolution rate at acid pH. Chem Geol 135:275–290. https://doi.org/10.1016/S0009-2541(96)00126-X
McAdam AC, Zolotov MY, Sharp TG, Leshin LA (2008) Preferential low-pH dissolution of pyroxene in plagioclase-pyroxene mixtures: implications for martian surface materials. Icarus 196:90–96. https://doi.org/10.1016/j.icarus.2008.01.008
Reale P, Scrosati B, Delacourt C, Wurm C, Morcrette M, Masquelier C (2003) Synthesis and thermal behavior of crystalline hydrated iron(III) phosphates of interest as positive electrodes in li batteries. Chem Mater 15:5051–5058. https://doi.org/10.1021/cm031107z
Khan MI, Min TK, Azizli K, Sufian S, Ullah H, Man Z (2015) Effective removal of methylene blue from water using phosphoric acid based geopolymers: synthesis, characterizations and adsorption studies. RSC Adv. 5:61410–61420. https://doi.org/10.1039/c5ra08255b
Guo CM, Wang KT, Liu MY, Li XH, Cui XM (2016) Preparation and characterization of acid-based geopolymer using metakaolin and disused polishing liquid. Ceram Int 42:9287–9291. https://doi.org/10.1016/j.ceramint.2016.02.073
Lassinantti Gualtieri M, Romagnoli M, Gualtieri AF (2015) Preparation of phosphoric acid-based geopolymer foams using limestone as pore forming agent—thermal properties by in situ XRPD and Rietveld refinements. J Eur Ceram Soc 35(2015):3167–3178. https://doi.org/10.1016/j.jeurceramsoc.2015.04.030
Djobo JNY, Elimbi A, Tchakouté HK, Kumar S (2016) Mechanical activation of volcanic ash for geopolymer synthesis: effect on reaction kinetics, gel characteristics, physical and mechanical properties. RSC Adv 6:39106–39117. https://doi.org/10.1039/c6ra03667h
Basile-Doelsch I, Amundson R, Stone WEE, Masiello CA, Bottero JY, Colin F, Masin F, Borschneck D, Meunier JD (2005) Mineralogical control of organic carbon dynamics in a volcanic ash soil on La Réunion. Eur J Soil Sci 56:689–703. https://doi.org/10.1111/j.1365-2389.2005.00703.x
Liu Y, Qin Z, Chen B (2020) Influence of low-grade bauxite on the properties of magnesium phosphate cement. Constr Build Mater 242:118052. https://doi.org/10.1016/j.conbuildmat.2020.118052
Lassinantti Gualtieri M, Romagnoli M, Pollastri S, Gualtieri AF (2015) Inorganic polymers from laterite using activation with phosphoric acid and alkaline sodium silicate solution: mechanical and microstructural properties. Cem Concr Res 67:259–270. https://doi.org/10.1016/j.cemconres.2014.08.010
Wang YS, Provis JL, Dai JG (2018) Role of soluble aluminum species in the activating solution for synthesis of silico-aluminophosphate geopolymers. Cem Concr Compos 93:186–195. https://doi.org/10.1016/j.cemconcomp.2018.07.011
Ray CS, Fang X, Karabulut M, Marasinghe GK, Day DE (1999) Effect of melting temperature and time on iron valence and crystallization of iron phosphate glasses. J Non Cryst Solids 249:1–16. https://doi.org/10.1016/S0022-3093(99)00304-X
Oliver CJRG, Lovey FC, Caneiro A, Russo DO, Rodríguez DS, Soldera F, Fiscina JE, Rincón JM (2010) Crystallization of iron phosphate glasses containing up to 19 wt.% UO2.67. J Non Cryst Solids 356:2986–2993. https://doi.org/10.1016/j.jnoncrysol.2010.06.034
Aliouane N, Badeche T, Gagou Y, Nigrelli E, Saint-Gregoire P (2000) Synthesis and phase transitions of iron phosphate. Ferroelectrics 241:255–262. https://doi.org/10.1080/00150190008224999
Acknowledgements
Open Access funding provided by Projekt DEAL. This work was supported by the Alexander von Humboldt Foundation through the Georg Foster Postdoctoral research fellowship (CMR—1201499—GF-P). The authors are also grateful to Dr Elie Kamseu and Mr. Sylvain Tome for assistance in materials characterisation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Djobo, J.N.Y., Elimbi, A. & Stephan, D. Phase and dimensional stability of volcanic ash-based phosphate inorganic polymers at elevated temperatures. SN Appl. Sci. 2, 828 (2020). https://doi.org/10.1007/s42452-020-2616-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-2616-4