
Abstract
A novel hyperbranched polysiloxane functionalized graphene oxide (HPBSi-GO) containing abundant primary amine, tertiary amine and hydroxyl groups was designed and successfully synthesized though efficient “grafting to” method. The as-synthesized HBPSi-GO was confirmed by FT-IR, XPS and AFM. Moreover, the HPBSi-GO was incorporated into epoxy (EP) resin matrix to fabricate HPBSi-GO/EP composites. The results showed that the HPBSi-GO has better dispersibility than that of unmodified GO in EP matrix. Interestingly, the mechanical and frictional properties of HPBSi-GO/EP composites were simultaneously superior to those of neat epoxy resin. AFM and TOM results revealed that the good properties mainly attributed to the unique inorganic–organic structure of HBPSi-GO, as well as the good interfacial adhesion between HBPSi-GO and EP matrix.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Epoxy resin (EP), as a kind of typical thermosetting polymer, is widely used in coatings, construction, electronic and other fields due to its good adhesive property, high chemical stability and excellent mold ability [1, 2]. However, the cured EP exhibits inherent brittleness resulting from its rigid structure, which hinders its potential applications in advanced fields such as automotive manufacturing, aerospace engineering and microelectronics industry [3,4,5]. Therefore, many materials (such as nano-SiO2 [6], nano-Al2O3 [7] and carbon nanomaterials [8, 9], etc.) have been introduced into EP resin to achieve excellent thermal and mechanical properties. Within those nanomaterials, graphene oxide (GO) has drawn great attention for its unique lamellar structure, enormous surface area and amounts of oxygen-containing groups. Tang et al. [10] prepared a series of GO/EP composites and found that GO could significantly enhance the toughness of the epoxy resin. Kausar et al. [11] found that GO could act as an ideal reinforcing material to enhance the thermal, mechanical and other properties of epoxy composites by summarizing the progress of GO modified epoxy resins. Nevertheless, the aggregation of GO caused by the Van der Waals force between GO sheets and the weak interfacial adhesion between GO and resin matrix weaken the enhancement effect between GO and EP [12]. Therefore, in the last few years, various kinds of functionalized GO have been fabricated by introducing small molecules and polymers [13,14,15,16,17,18] onto the graphene oxide sheets to improve various properties including the dispersity in composites and the interface bonding performance between fillers and matrix. Yuan et al. [13] attached γ-methacryloxypropyltrimethoxysilane (KH-570) onto GO sheets via the covalent bonding, and the novel fillers can effectively improve wear and corrosion resistance of polyimide matrix. Joseph et al. [14] synthesized the GO covalently grafted polyhedral oligomeric silsesquioxanes (POSS) for extremely low dielectric property in syndiotactic polystyrene. Furthermore, the functionalized GO with octa-aminophenyl polyhedral oligomeric silsesquioxanes (OapPOSS) [15] and diamine-functionalized GO with 4,4′-diaminodiphenyl sulfone (DDS) [16] were synthesized to enhance the EP composites, the results showed that the fillers have better dispersity on the entire composites, and the composites showed considerable improved thermal and mechanical properties compared to those of pristine EP. Therefore, the functionalized graphene oxide is widely used as reinforcing filler to enhance the mechanical and other properties of epoxy resin.
Hyperbranched polysiloxane (HBPSi), as a kind of inorganic–organic hybrid material, has aroused great interests in recent years because of its highly branched structure, low surface energy and high density of functional groups [19, 20]. Therefore, the functionalized GO with HBPSi has better dispersity in the composites and enhanced interfacial adhesion between nanoparticles and resin matrix. Yan’s group [21, 22] prepared GO functionalized by hyperbranched polysiloxane through hydrosilylation reaction, and incorporated it into cyanate ester (CE) and bismaleimide (BMI) to fabricate nanocomposites. It is found that the mechanical properties, thermal stability, moisture resistance and tribological performance of the nanocomposites are superior to those of neat CE and BMI resin. However, to our knowledge, no report on the EP composites modified by HBPSi-GO has been reported up to now. Recently, an innovative strategy to synthesize HBPSi [23] was proposed, which overcomes the significant shortcomings of conventional approaches: the catalysts for hydrosilylation reaction are precious and hydrolysis of organosiloxane results in gelation easily. Furthermore, Niu et al. [24] synthesized a novel hyperbranched polysiloxane contains epoxy groups and doped it into BMI resin to prepared HBPSi-EP/BMI composites. The results showed that the optimal loading of HBPSi-EP (8.0 wt%) could greatly improve the toughness and thermal performance of BMI composites, which exhibited the unexpected effect in thermosetting resins.
Herein, a novel hyperbranched polysiloxane containing primary amine, tertiary amine and hydroxyl groups was prepared via transesterification through simple one-pot method. The HBPSi covalently-grafted graphene oxide (HPBSi-GO), which was synthesized by a simple “grafting to” method, was introduced into epoxy resin to fabricate HPBSi-GO/EP composites. The results demonstrated that the as-prepared nanoparticles can improve the mechanical and frictional properties of the composites. This is mainly ascribed to the excellent dispersity and the unique inorganic–organic structure of HBPSi-GO.
2 Experimental
2.1 Materials
Diglycidyl ether of bisphenol A (DGEBA)-type epoxy resin (E-51) with epoxy value of 0.48–0.54 mol/100 g was purchased from Guangzhou Suixin Chemical Co., Ltd., China. 4,4′-Diaminodiphenylsuifone (DDS, Shanghai Aladdin Biochemical Technology Co., Ltd., China) was used as curing agent. GO nanosheets were prepared from natural graphite flakes (500 mesh, Qingdao Hensen Graphite Co., Ltd., China) by a modified Hummers method [25, 26]. (3-Aminopropyl) triethoxysilane (APTES) was purchased from Jingzhou Jianghan Fine Chemical Co., Ltd., China. Dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd., China. Ethanol absolute and acetone were purchased from Guangdong Guanghua Sci-Tech Co., Ltd., China. All reagents were used as received without further purification.
2.2 Synthesis of HBPSi-GO
The schematic diagram of the synthesis of HBPSi-GO is shown in Fig. 1. Firstly, A novel hyperbranched polysiloxane (HBPSi) containing primary amine, tertiary amine and hydroxyl groups was synthesized by transesterification [23, 27, 28] of APTES with MDEA.

The schematic diagram of the synthesis of HBPSi-GO
Secondly, the HBPSi-GO was synthesized by introducing HBPSi into the GO sheets via amide formation between primary amines of HBPSi and carboxyl groups of GO. The detailed process is presented as follows: 0.3 g GO was dispersed in 130 mL DMF by ultrasonic treatment for 30 min. Successively, the obtained GO suspension, 1.0 g DCC and 0.2 g DMAP were added into a 250 mL three-necked flask, respectively, in which DCC and DMAP acted as dehydrating agent and catalyst. Subsequently, 1.0 g HBPSi was dissolved in 10 mL DMF, then the suspension was added into the flask. The mixtures reacted at 70 °C for 24 h under nitrogen atmosphere. Then the product was washed with ethanol absolute for three times and dried in a vacuum oven at 60 °C for 12 h. The structure of HBPSi-GO is shown in Fig. 1.
2.3 Fabrication of HBPSi-GO/EP composites
The HBPSi-GO/EP composites with different contents of fillers were fabricated using a casting method. Firstly, HBPSi-GO was dispersed into acetone by ultrasonic treatment for 15 min and then mixed with E-51 epoxy. The mixtures were heated for 20 min at 100 °C in oil bath with mechanical stirrer to remove the solvents. Next, the curing agent DDS was doped into the mixtures with a certain percentage. Subsequently, the mixtures were heated to 130 °C for 30 min with mechanical stirrer. Afterwards, the EP pre-polymer was poured into a pre-heated mold coated with release agent and put in a vacuum oven to remove the bubbles. Finally, the sample was cured at 170 °C for 4 h and 230 °C for 2 h, then the mold was cooled to room temperature and demolded to obtain the HBPSi-GO/EP composites.
2.4 Characterization
Fourier transform infrared (FT-IR) spectra of the samples were collected using a PerkinElmer-283B FTIR spectrometer (Perkin Elmer Inc., Waltham, USA) ranging from 4000 to 400 cm−1 with a resolution of 2 cm−1. The chemical change and elemental compositions of the samples surface were confirmed by X-ray photoelectron spectroscopy (XPS, Quantum 2000, ULVAC-PHI Inc., Japan), and Al Kα (1486.8 eV) was used as the X-ray source. Atomic force microscopy (AFM, Hitachi High-Tech Science Co., Japan) images of the GO and HBPSi-GO were observed with tapping mode to study the morphologies. X-ray diffraction (XRD) was carried out with Bruker D8 ADVANCE (Bruker AXS GmbH, Germany), using Cu Kα radiation (λ = 1542 nm). The XRD data were recorded from 5° to 85° with the scanning rate of 0.02° s−1. Raman spectra were obtained on a Raman microscope (Renishaw Invia Plus, UK) using a 633 nm argon ion laser. Thermogravimetric analyses (TGA) of GO and HBPSi-GO were investigated with a Q50 thermal analyzer (TA Instrument, USA) from 50 to 800 °C with a heating rate of 10 °C min−1 in argon atmosphere. Transmission optical microscopy (TOM, MM-8, Pu Dan Optical Instrument Co., Ltd., China) was used to verify the dispersion stability of GO and HBPSi-GO in epoxy matrix. Bending and impact tests were accomplished with a universal testing machine (CMT-6303, Shenzhen SANS Testing Machine Co., Ltd., China) according to the Chinese standard GB/T2567-2008. The dimensions of samples for tending and impact tests were (80 ± 0.2) × (15 ± 0.2) × (4 ± 0.2) mm3 and (80 ± 0.2) × (10 ± 0.2) × (4 ± 0.2) mm3, respectively, and each item has more than five effective samples. Scanning electron microscope (SEM) was carried out on a Hitachi S-570 instrument (Hitachi High-Tech Science Co., Japan) to observe the surface morphology of impact fracture, and the specimens were treated by spray-gold before testing.
3 Results and discussion
3.1 HBPSi-GO characterization
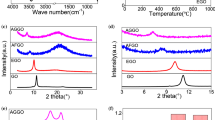
XPS spectra were obtained to illuminate chemical change and element composition on the surface of specimens. The full range spectra of GO and HBPSi-GO are presented in Fig. 2a. The peaks for the C 1s and O 1s are observed at 284.27 and 532.15 eV, respectively. However, as for the spectrum of HBPSi-GO, three new peaks appear at 101.80, 153.25 and 400.09 eV, attributing to Si 2p, Si 2s and N 1s, respectively. The N and Si elements were derived from HBPSi, demonstrating the successful synthesis of HBPSi-GO.

XPS spectra of a GO and HBPSi-GO; b C 1s of GO; c C 1s of HBPSi-GO, and d FT-IR spectra of GO and HBPSi-GO
Moreover, three characteristic peaks can be separated from the XPS spectrum of C 1s of GO, 284.8 (C=C bond), 287.0 (C–O bond) and 288.4 eV (C=O bond). As a comparison, five components can be found in the spectrum of C 1s of HBPSi-GO, which are located at 283.8 (C–Si bond), 284.8 (C=C bond), 285.7 (C–N bond), 286.4 (C–O bond) and 288.8 eV (C=O bond). The appearance peaks of C–Si and C–N bonds confirm that the HBPSi has been successfully grafted onto the GO surface.
The FTIR spectra of GO and HBPSi-GO are shown in Fig. 2d. The characteristic absorption peaks of GO can be seen at 3400 (–OH), 1724 (C=O), 1626 (C=C) and 1055 cm−1 (C–O). By contrast, the disappeared absorption peak at 1724 cm−1 is assigned to carboxyl group of GO. In addition, the new absorption peaks at 2934, 1119, 1649 and 1536 cm−1 are related to methyl groups, Si–O–C bond, C=O bond and N–H of secondary amide group, respectively, indicating that the HBPSi has been chemically grafted onto the surface of GO sheets.
AFM images were obtained to investigate the morphologies of GO and HBPSi-GO. From these images, the lamellar structure of GO and HBPSi-GO are clearly observed. Figure 3a shows the thickness of single-nanolayer GO after ultrasonication is 1.0–1.3 nm, which is consistent with the previous reports [29]. By contrast, the functionalized GO reveals an average thickness of 3.2–5.0 nm. The increscent thickness of the nanolayers demonstrates that the HBPSi functionalized GO has been successfully synthesized.

AFM images of a GO and b HBPSi-GO
The XRD patterns of GO and HBPSi-GO are shown in Fig. 4a. The characteristic peak of 2θ at 9.68° is related to the (001) plane of GO. And the d-spacing of (001) plane is calculated to 0.91 nm according to Bragg’s law, which is corresponded to the AFM result. In comparison, the (001) peak of the modified GO is invisible, at the same time, a new wide peak of 2θ appears at 21.2°, which shows that the HBPSi-GO has been stripped to single-layer structure and part of the GO have been reduced during the modification process. These XRD analysis indicate that the HBPSi-GO has an enhanced disordered structure after grafting.

a XRD spectra of GO and HBPSi-GO, and b Raman spectra of GO and HBPSi-GO
Raman spectroscopy is an effective tool for characterizing carbonaceous materials [15]. As shown in Fig. 4b, Raman spectra of GO and HBPSi-GO simultaneously exhibit two peaks near 1320 and 1590 cm−1, which are attributed to the D and G bonds of GO, respectively. A general rule is that higher disorder in carbon products leads to a boarder G bond, as well as a higher relative intensity ratio of ID/IG [30]. The ID/IG value of HBPSi-GO (1.17) is higher than that of GO (1.01), indicating that the HBPSi-GO has a higher degree of disorder.
TGA were conducted to investigate the thermal stability of GO and HBPSi-GO. As seen in Fig. 5, a slight weight loss of GO and HBPSi-GO below 100 °C is mainly due to the evaporation of adsorbed water. For GO, a major weight loss (47.7%) between 100 and 310 °C is mainly ascribed to the decomposition of oxygen-containing functional groups [31]. In contrast, the decomposition of hydroxyl and carboxyl groups in HBPSi-GO causes a lesser mass loss (21.9%) between 100 and 310 °C. When the temperature arrives to 800 °C, the HBPSi-GO shows the char residual weight of 54.2%, increased by 43.1% than that of unmodified GO (37.9%). The enhanced thermal resistance of HBPSi-GO is because that the existence of HBPSi can generate SiO2 covering layer at the high temperature, which can resist the decomposition process of HBPSi-GO effectively.

TGA curves of the GO and HBPSi-GO
3.2 HBPSi-GO/EP composites characterization
TOM was carried out to investigate the dispersion of nano-filler in resin matrix. Figure 6 shows the images of (a) 0.2 wt% HBPSi-GO/EP, (b) 0.6 wt% HBPSi-GO/EP, (c) 1.0 wt% HBPSi-GO/EP and (d) 1.0 wt% GO/EP composites. It is found that the HBPSi-GO can be well dispersed in epoxy composites, although parts of agglomeration of fillers still exist. As a comparison, in 1.0 wt% GO/EP composites, the GO sheets present serious agglomeration in epoxy matrix. The great dispersion of HBPSi-GO in epoxy composites is attributed to the exhibition of active functional groups on the fillers’ surface, which can actively interact with the epoxy matrix via chemical bonds.

TOM images of EP composites filled with a 0.2 wt% HBPSi-GO, b 0.6 wt% HBPSi-GO, c 1.0 wt% HBPSi-GO and d 1.0 wt% GO
It is generally believed that the mechanical strengths of composites are closely related to the good dispersibility of nano-fillers and interaction between fillers and polymeric matrix [32, 33]. As can be seen in Fig. 7, the impact and flexural strengths of the HBPSi-GO/EP composites increase continuously with the addition of HBPSi-GO. When the content of HBPSi-GO is 0.6 wt%, the impact and flexural strengths arrive to the maximum (132.2 MPa and 17.0 kJ/m2), increased by 23.9% and 56.0%, respectively, in comparison with those of the pristine epoxy resin (106.7 MPa and 10.9 kJ/m2). Those can attribute to the following causes: (1) the excellent dispersibility of HBPSi-GO avoids agglomeration in resin matrix, making filler evenly disperse in composites, which can prevent the excessive growth of micro-cracks. (2) The amounts of –NH2 and –OH groups on HBPSi-GO’s surface participate in the cross-linking reaction during curing process, thus improving the interface bonding performance between HBPSi-GO and epoxy resin, further promoting that the loading energy transfer from the EP matrix to the fillers. (3) The unique inorganic–organic structure of HBPSi-GO can enhance the compatibility between EP matrix and fillers. Based on the above consideration, the strengthening and toughening mechanisms of HBPSi-GO/EP composite is shown in Fig. 8. However, when the filler content exceeds 0.6 wt%, the mechanical strengths of HBPSi-GO/EP systems decrease with the increasing of HBPSi-GO, which can be attribute to the agglomeration of HBPSi-GO in epoxy matrix. Moreover, the high content addition of HBPSi-GO will hinder the curing reaction of epoxy resin, resulting in the low level cross-linking density of composites [34].

Effect of HBPSi-GO amounts on flexural strength and impact strength of EP resin

The strengthening and toughening mechanisms of HBPSi-GO/EP composite
To further investigate the toughening mechanism of HBPSi-GO, surface morphologies of the impact fracture for EP resin with different filler contents (pristine epoxy resin, 0.6 wt% HBPSi-GO/EP, 1.0 wt% HBPSi-GO/EP) were observed by SEM, as shown in Fig. 9. It can be observed that the fracture surface of neat epoxy is smooth and flat, corresponding to the features of brittle rupture (Fig. 9a). For epoxy composite with 0.6 wt% HBPSi-GO, the fracture surface becomes rougher, and more areas with the dimples can be observed in the image, presenting the typical characteristics of ductile fracture. When the loading of HBPSi-GO arrives to 1.0 wt%, the aggregation of HBPSi-GO can be seen in view region, which results in the stress concentration and impact failure.

SEM images of the fracture surfaces of the HBPSi-GO/EP systems with various contents of HBPSi-GO (a 0.0 wt%, b 0.6 wt%, c 1.0 wt%)
To explorer the frictional behavior of HBPSi-GO/EP composites, the tests were conducted with rotate speed of 200 rpm/min loading 196 N under dry sliding for 120 min. The coefficient of friction (COF) of neat EP resin and 0.6 wt% HBPSi-GO/EP composite are shown in Fig. 10. It can be seen that the COF of neat EP is unstable during all the test process, and the poor friction performance is attributed to the lack of stable transfer film on the surface of the steel counterpart ring. As a comparison, the COF curve of 0.6 wt% HBPSi-GO/EP composite exists a run-in stage and then keeps a steady state, and the average COF dips by 21.43% than that of pristine EP resin. The significant decrease of average COF of EP composites after adding HBPSi-GO is ascribed to the fact that a uniform and dense transfer film is formed on the surface of the steel counterpart ring, making the friction mainly occur between the composite and the transfer film. At the same time, the unique lamellar structure of GO, good dispersibility of HBPSi-GO as well as the good interfacial strength between fillers and resin matrix play important roles in decreasing friction coefficient.

The coefficient of friction (COF) of neat EP resin and 0.6 wt% HBPSi-GO/EP composite
4 Conclusion
A novel functionalized GO with HBPSi containing primary amine, tertiary amine and hydroxyl groups was synthesized by “grafting to” method, and HBPSi-GO exhibits good dispersibility and thermal stability. When the amount of HBPSi-GO is 0.6 wt%, the flexural and impact strengths of epoxy composites achieve the maximums of 132.2 MPa and 17.0 kJ/m2, increasing by 23.9% and 56.0% compared with that of pristine epoxy matrix, respectively. More importantly, in the same filler content, the frictional behavior of the HBPSi-GO composite is superior, at the same time, the frictional coefficient is lower and more stable than that of pure matrix, which are mainly attributed to the unique inorganic–organic structure of HBPSi-GO, as well as the good interfacial adhesion between HBPSi-GO and EP matrix. Hence, this work will provide a promising method to fabricate HBPSi-functionalized GO, improving the mechanical and frictional properties of epoxy resin.
References
Shi Y, Wang G (2016) The novel silicon-containing epoxy/PEPA phosphate flame retardant for transparent intumescent fire resistant coating. Appl Surf Sci 385:453–463
Ligoda-Chmiel J, Śliwa RE, Potoczek M (2017) Flammability and acoustic absorption of alumina foam/tri-functional epoxy resin composites manufactured by the infiltration process. Compos Part B Eng 112:196–202
Raimondo M, Guadagno L, Speranza V, Bonnaud L, Dubois P, Lafdi K (2018) Multifunctional graphene/POSS epoxy resin tailored for aircraft lightning strike protection. Compos Part B Eng 140:44–56
Su Z, Wang H, Tian K, Huang W, Guo Y, He J, Tian X (2018) Multifunctional anisotropic flexible cycloaliphatic epoxy resin nanocomposites reinforced by aligned graphite flake with non-covalent biomimetic functionalization. Compos Part A Appl Sci Manuf 109:472–480
Wang Y, Liu W, Qiu Y, Wei Y (2018) A one-component, fast-cure, and economical epoxy resin system suitable for liquid molding of automotive composite parts. Materials 11(5):685–696
Xiao C, Tan Y, Yang X, Xu T, Wang L, Qi Z (2018) Mechanical properties and strengthening mechanism of epoxy resin reinforced with nano-SiO2 particles and multi-walled carbon nanotubes. Chem Phys Lett 695:34–43
Duan Z, He H, Liang W, Wang Z, He L, Zhang X (2018) Tensile, quasistatic and dynamic fracture properties of nano-Al2O3-modified epoxy resin. Materials 11:905–921
Gojny F, Wichmann M, Kopke U, Fiedler B, Schulte K (2004) Carbon nanotube-reinforced epoxy-compo sites: enhanced stiffness and fracture toughness at low nanotube content. Compos Sci Technol 64(15):2363–2371
Yang S, Lin W, Huang Y, Tien H, Wang J, Ma C, Li S, Wang Y (2011) Synergetic effects of graphene platelets and carbon nanotubes on the mechanical and thermal properties of epoxy composites. Carbon 49(3):793–803
Tang J, Zhou H, Liang Y, Shi X, Yang X, Zhang J (2014) Properties of graphene oxide/epoxy resin composites. J Nanomater 2014:1–5
Kausar A, Rafique I, Anwar Z, Muhammad B (2016) Perspectives of epoxy/graphene oxide composite: significant features and technical applications. Polym Plast Technol 55(7):704–722
Tang L, Wan Y, Yan D, Pei Y, Zhao L, Li Y, Wu L, Jiang J, Lai G (2013) The effect of graphene dispersion on the mechanical properties of graphene/epoxy composites. Carbon 60:16–27
Yuan R, Ju P, Wu Y, Ji L, Li H, Chen L, Zhou H, Chen J (2019) Silane-grafted graphene oxide improves wear and corrosion resistance of polyimide matrix: molecular dynamics simulation and experimental analysis. J Mater Sci 54(16):11069–11083
Joseph A, Nagendra B, Surendran K, Gowd E (2019) Sustainable in situ approach to covalently functionalize graphene oxide with POSS molecules possessing extremely low dielectric behavior. Langmuir 35:4672–4681
Wang X, Song L, Yang H, Xing W, Kandola B, Hu Y (2012) Simultaneous reduction and surface functionalization of graphene oxide with POSS for reducing fire hazards in epoxy composites. J Mater Chem 22(41):2237–2243
Yu J, Jung J, Choi Y, Choi J, Yu J, Lee J, You N, Goh M (2016) Enhancement of the crosslink density, glass transition temperature, and strength of epoxy resin by using functionalized graphene oxide co-curing agents. Polym Chem UK 7(1):36–43
Wan Y, Tang L, Gong L, Yan D, Li Y, Wu L, Jiang J, Lai G (2014) Grafting of epoxy chains onto graphene oxide for epoxy composites with improved mechanical and thermal properties. Carbon 69:467–480
Shi M, Lin T, Hu Y, Peng J, Li J, Zhai M (2020) Functionalization of graphene oxide by radiation grafting polyhedral oligomeric silsesquioxane with improved thermal stability and hydrophilicity. J Mater Sci 55(4):1489–1498
Zhou C, Liang G, Gu A (2016) Thermally resistant unsaturated polyester resin with low dielectric loss based on special benzyl alcohol terminated hyperbranched polysiloxane for producing high efficiency motors using vacuum pressure impregnation technique. RSC Adv 6(8):6672–6678
Lian R, Lei X, Chen Y, Zhang Q (2019) Hyperbranched-polysiloxane-based hyperbranched polyimide films with low dielectric permittivity and high mechanical and thermal properties. J Appl Polym Sci 136(31):47771–47778
Zhang M, Yan H, Gong C, Li T (2014) Hyperbranched polysiloxane functionalized graphene oxide for dicyclopentadiene bisphenol dicyanate ester nanocomposites with high performance. Express Polym Lett 8(6):413–424
Yan H, Li S, Jia Y, Ma X (2015) Hyperbranched polysiloxane grafted graphene for improved tribological performance of bismaleimide composites. RSC Adv 5:12578–12582
Niu S, Yan H (2015) Novel hyperbranched polysiloxanes containing acetoacetyl groups synthesized through transesterification reaction. Macromol Rapid Commun 36(8):739–743
Niu S, Yan H, Li S, Tang C, Chen Z, Zhi X, Xu P (2016) A multifunctional silicon-containing hyperbranched epoxy: controlled synthesis, toughening bismaleimide and fluorescent properties. J Mater Chem C 4(28):6881–6893
Hummers W, Offeman R (1958) Preparation of graphitic oxide. J Am Chem Soc 80(6):1339
Marcano D, Kosynkin D, Berlin J, Sinitskii A, Sun Z, Slesarev A, Alemany L, Lu W, Tour J (2010) Improved synthesis of graphene oxide. ACS Nano 4(8):4806–4814
Niu S, Yan H, Chen Z, Yuan L, Liu T, Liu C (2016) Water-soluble blue fluorescence-emitting hyperbranched polysiloxanes simultaneously containing hydroxyl and primary amine groups. Macromol Rapid Commun 37(2):136–142
Bai L, Yan H, Bai T, Feng Y, Zhao Y, Ji Y, Feng W, Lu T, Nie Y (2019) High fluorescent hyperbranched polysiloxane containing beta-cyclodextrin for cell imaging and drug delivery. Biomacromol 20(11):4230–4240
Xu J, Xu M, Wu J, Wu H, Zhang W, Li Y (2015) Graphene oxide immobilized with ionic liquids: facile preparation and efficient catalysis for solvent-free cycloaddition of CO2 to propylene carbonate. RSC Adv 5(88):72361–72368
Kudin K, Ozbas B, Schniepp H, Prud’Homme R, Aksay I, Car R (2008) Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett 8(1):36–41
Xu Z, Gao C (2010) In situ polymerization approach to graphene-reinforced nylon-6 composites. Macromolecules 43(16):6716–6723
Chen Z, Yan H, Guo L, Li L, Yang P, Liu B (2019) A novel polyamide-type cyclophosphazene functionalized rGO/WS2 nanosheets for bismaleimide resin with enhanced mechanical and tribological properties. Compos Part A Appl Sci Manuf 121:18–27
Meng X, Wang M, Yang L, Ye H, Cong C, Dong Y, Zhou Q (2017) Effect of amino-functionalized graphene oxide on the mechanical and thermal properties of polyoxymethylene. Ind Eng Chem Res 37(4):15038–15048
Ryu S, Sin J, Shanmugharaj A (2014) Study on the effect of hexamethylene diamine functionalized graphene oxide on the curing kinetics of epoxy nanocomposites. Eur Polym J 52:88–97
Acknowledgements
This work was supported by the National Natural Science Foundation (21875188), the Natural Science Basic Research Plan in Shaanxi Province of China (2018JM2024) and the Key Research and Development Program of Shaanxi (2019ZDLGY04-08).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Guo, L., Yan, H., Chen, Z. et al. Graphene oxide grafted by hyperbranched polysiloxane to enhance mechanical and frictional properties of epoxy resin. SN Appl. Sci. 2, 473 (2020). https://doi.org/10.1007/s42452-020-2277-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-2277-3