
Abstract
Different properties such as the structural, elastic, thermal, electronic, magnetic as well as the Curie temperature and the formation energy of the cubic perovskites PrXO3 compounds (X = V, Cr, Mn, and Fe) are studied by using the density functional theory based on the full-potential linearized augmented plane wave with local orbitals method with the generalized gradient approximation (GGA) for the exchange correlation potential as applied in WIEN2k code. The GGA + U approximation is also used to treat the f-states of Pr atoms and d-states of X atoms. We have also applied the analytical techniques for the structural parameters. The calculated structural parameters by both methods are in good agreement with experimental results. The calculated critical radii of the compounds exhibit ion conductivity as well as oxygen migration. The PrVO3, PrCrO3 and PrFeO3 compounds have a ductile nature, while PrMnO3 is brittle. The calculated electronic properties reveal the metallic nature for the studied compounds. Double cell optimizations, density of states as well as magnetic moment confirm that these compounds are ferromagnetic metals. The negative value of formation energy confirms the stability of these compounds. Large and small values of Curie temperature in these compounds show strong and weak interaction among the magnetic atoms, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Perovskites have great importance in industrial applications because of their commonly occurring in nature and interesting structural, elastic, electronic, magnetic, optical and high thermoelectric properties [1]. The unique characteristics in most of these compounds are high temperature superconductivity, electric and catalytic behavior [2, 3]. In microelectronics, the telecommunication, superconductivity, colossal magneto-resistance and ionic conductivity are the physical properties of interest among the perovskites [4]. On the basis of the electronic behavior most of the perovskites have half metallic character [5]. The half metallic behavior makes the potential material to speed up data with less energy consumption and also increase the circuit integration density. So, these types of materials are used in spintronic as well as technological field [6].
The transition metals oxides are much suitable to study the ferromagnetic response. Praseodymium (Pr) contains the electronic arrangement that is similar the other rare earth elements called as cerium (Ce) and lanthanum (La) [7,8,9,10]. In this regard, PrXO3 (X = V, Cr, Mn, and Fe) perovskite compounds have many applications in fuel cells, gas separation membranes and also in chemical reactors [11]. These compounds possess high magneto electric coupling [11]. The PrXO3 (X = V, Cr, Mn, and Fe) compounds have strong ferromagnetic order due to the strong magneto electric coupling and magneto electric effect arises in these compounds at ambient temperature. Due to its interesting magnetic properties these compounds have potential applications in random access memory (RAM) architectures etc. [12].
The PrVO3 was reported by Copie et al. [13] using high-resolution transmission electron microscopy studies. They concluded that PrVO3 exhibits an insulating character and low-temperature hard-ferromagnetic behavior below 80 K. Saber et al. [5] studied the structural, optical and thermoelectric properties of rare earth based PrVO3 perovskites by using the DFT calculations. The band structures have been analyzed and DOS confirmed the half metallic ferromagnetic behavior within this compound. Electrical conductivity, Seebeck coefficient and power factor were being used to elaborate the thermoelectric behavior which proved that PrVO3 compound is suitable for spintronic and thermoelectric applications. Wang et al. [14] used the powder samples of PrVO3 compound to explain the temperature dependent behavior at 20 K and 90 K. Different properties are observed such as the resistance changes due to the temperature which classified this compound as insulator. Some other properties such as the magnetic and the structural phase transition were also studied, which are the characteristics for the spin reorientation and magnetic ordering, respectively.
The PrCrO3 have been characterized by different experimental techniques [15] to check the magnetic response. Rezaiguia et al. [15] studied the electronic and magnetic properties of the cubic perovskites of PrCrO3 by using the first-principle calculations, where the electronic properties favor the complete half metallic character of PrCrO3 in GGA and GGA + U calculations, whereas the compound have an integer total magnetic moment. Murtaza et al. [5] reported the electronic, structural and magnetic properties of PrCrO3 compound by using DFT calculations with full potential linearized augmented plane wave plus local orbitals method. The half metallic ferromagnetism in this compound was confirmed by investigating the electronic band structure and density of states (DOS). Transports, structural and mechanical, and half-metallic ferromagnetic properties of PrMnO3 compound are investigated by Gupta et al. [16]. They investigated the ground state structure inclusive of elastic and transport properties of PrMnO3 perovskite oxide. Wei et al. [17] also studied theoretically the PrMnO3 compound by employing the DFT calculations and investigating the electronic structure and surface properties of this compound. Benstali et al. [15] investigated the structural, electronic and magnetic properties of PrFeO3 perovskite compound by using the first-principles study with GGA and GGA + U approximations: this study shows that the PrFeO3 compound has a metallic behavior in the framework of GGA and GGA + U potentials. Joshi et al. [18] studied visible light photo-catalytic activity of nano-crystalline PrFeO3 perovskite for hydrogen generation in ethanol–water system, by using sol–gel, template and combustion method. A nano-crystalline PrFeO3 perovskite in ortho-ferrite type was produced at 700 °C.
From the above it is clear that different physical properties were reported in the past on the structural, elastic, electronic and magnetic properties of these compounds but to the best of our knowledge no detail study is available on the thermal, elastic and magnetic properties of this important group of compounds which motivate us to perform these calculations in order to provide reference data for the experimentalist and to complete the existing theoretical works on these materials.
2 Method of calculation
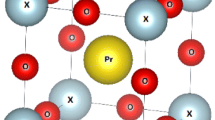
In the current study, the calculations were carried out by using the full-potential linearized augmented plane wave plus local orbitals (FP-LAPW + lo) method based on the density functional theory (DFT) as implemented in the WIEN2k code [19]. In the ideal cubic structure (space group \( Pm\bar{3}m \) (no. 221)) [20] of the perovskite PrXO3 (X = V, Cr, Mn, and Fe) compounds, the Pr, X and O atoms are located at (0, 0, 0), (0.5, 0.5, 0.5) and (0.5, 0.5, 0) sites, respectively. The X or transition metal atoms have their position at the body center of cube and bounded through six atoms of Pr (praseodymium) atoms. The unit cell of the PrVO3 compound as a prototype is shown in Figs. 1 and 2. All the calculations in the present work are performed with 3000 k-points for single cell optimization and 250 k-points for double cell optimization. The RMT× Kmax parameter is taken to be 7.00 for both GGA [21] and GGA + U schemes [22, 23]. The GGA plus U approximation is used to treat the 4f orbitals of Pr atom and 3d orbitals of transition metals. The U values are optimized in the range of 7.075 to 7.891 eV. Single cell optimization is taken to treat the mechanical, structural and thermal properties but the double cell optimization with SCF (self-consistent field) calculations are taken to treat the electronic and magnetic properties of these compounds.

Unit cell of PrVO3as a prototype

Double cell structure of PrVO3 as a prototype
3 Results and discussion
3.1 Structural properties
The ground state properties of the PrXO3 (X = V, Cr, Mn, Fe) compounds are determined through calculating the total energies versus different volumes around the equilibrium volume and then fitted to the Murnaghan’s equation of state [24]. The computed lattice constant (a0), bulk modulus (B0), its first pressure derivative (B′) and the ground state energy (E0) are presented in Table 1. Further the ionic radii are used to calculate the lattice constants by using the following empirical formula:
In above equations the factors α, β and γ are constants that have the values of 0.06741, 0.4905 and 1.2921, respectively. rPr is the ionic radius of Pr (1.13 Å), rO is the ionic radius of O (1.35 Å) and rX (X = V, Cr, Mn, and Fe) is the ionic radius of V, Cr, Mn and Fe which are about 0.640, 0.530, 0.800, and 0.645 Å, respectively. Table 1 show that the calculated lattice constants with GGA and analytical method are in close agreement with the experimental results as compared to other theoretical results, which shows the consistency of the current calculation in further explaining the electronic and magnetic properties. The values of the bulk modulus B0 that are obtained by optimizations are also listed in Table 1 for all the studied PrXO3 perovskite compounds. It is observed that B0 increases in going from V to Mn atoms and then decreases to Fe while V0 and E0 decreases from V to Fe atoms. Therefore, PrMnO3 compound is harder and less compressible than the other compounds in the group. Binding energy gives the information about the stability of a given materials and are estimated by the comparison of ionic radii. From above discussions, the ionic radius of Mn atom is greater than V, Cr, and Fe atom which indicates that binding energy of PrMnO3 compound is smaller than the other compounds in the group. The larger binding energy of PrCrO3 compound predicts that this material is more stable than the other compounds. One more structural parameter is the bond length, which have their importance in the symmetry of the perovskite structure. The calculated bond length values are also listed in Table 1. The bond length is used to calculate the tolerance factor (t) by using following expression:
In the above expression, X is V, Cr, Mn or Fe atoms. The calculated tolerance factors for the PrXO3 compounds are also quoted in Table 1. From Table 1 it is concluded that the calculated tolerance factor have a close agreement with the calculated values in references [25, 26]. The critical value of tolerance factor for cubic perovskites is between 0.93 and 1.02. Through Table 1, the calculated values of the tolerance factor lies in this range, which confirms the cubic structure of PrXO3 compounds. Other structural parameter is the critical radius (rC) which has their importance in the activation energy of oxygen migration. It can be calculated by using following empirical formula [26]:
Vacancy mechanism is responsible for oxygen migration as vacancy moves in a crystal from one B site cation to other A site cation, where this migration affects the bulk transport properties. We can remark from the calculated critical radii (see Table 1) that the PrCrO3 compound has greater migration energy than other compounds in the group.
3.2 Elastic properties
The elastic constants (Cij) are used to determine the effect on material by applied stress. These constants have significant role in finding information about the stiffness and stability of material. For the calculation of stiffness and stability of the given materials, numerical first-principle calculation method developed by Charpin and integrated in the WIEN2k code [25] was adopted. This code is used to compute the components of the stress for small strains. The calculated values of C11, C12, and C44 are shown in Table 2. The requirement of mechanical stability in cubic crystals leads to the following restrictions on the elastic constants [27]:
The above criteria for the given materials are satisfied, indicating that the compounds of interest are mechanically stable. Elastic anisotropy factor (A) is an elastic parameter that gives information about the elastic wave velocity in the crystal. This factor has their importance in engineering science, and it is extremely related to persuade micro-cracks in materials. Anisotropy factor in cubic perovskite crystals are calculated from the following relation:
For completely isotropic materials, the anisotropy factor A takes the value of zero and the deviation from zero is a measure of the degree of elastic anisotropy of the crystal [28]. The calculated values of the anisotropic factor are about 1.344, 1.605, 1.660, and 2.684 for PrVO3, PrCrO3, PrMnO3, and PrFeO3 compounds, respectively. They are all different than zero, suggesting that these compounds are anisotropic in nature, thus the PrFeO3 compound is characterized by a pronounced anisotropy. Furthermore, we have also computed the Kleinman parameter (ζ), which describes the relative positions of the cation and anion sub-lattices under volume-conserving strain distortions for which positions are not fixed by symmetry using the following relation [28, 29]:
It is known that a low value of ζ implies that there is a large resistance against bond bending or bond-angle distortion and vice versa. The computed values of ζ are tabulated in Table 2. One can remark that the PrMnO3 compound has more resistance against bond bending as compared to the other studied compounds. Another factor is the Poisson ratio (υ) which is the ratio between contraction perpendiculars to the applied load to the extension in the direction of applied load [30]. Poisson’s ratio gives the information about the hardness and stiffness of material [31]. From Table 2, it is noted that the PrMnO3 compound is stiffer than the other PrVO3, PrCrO3, and PrFeO3 compounds. The obtained values of υ are between 0 and 0.5, suggesting that all PrXO3 (X = V, Cr, Mn, and Fe) compounds show a larger lateral expansion. Mechanical properties such as ductility and brittleness of material can be explained from some proposed relationship. Pugh [32] has proposed a simple relationship that links empirically the plastic properties of materials with their elastic moduli by B/G factor. The critical value which separates ductile and brittle materials is around 1.75; if B/G ratio is strictly superior to 1.75, the material behaves in a ductile manner; otherwise, the material behaves a brittle manner. The values for Pugh’s criterion B/G ratio are equal to 5.99, 7.33, 1.41 and 1.88 for PrVO3, PrCrO3, PrMnO3 and PrFeO3 compounds, respectively. These values are greater than the critical value (1.75) except for the case of PrMnO3 compound. Therefore, the PrVO3, PrCrO3, and PrFeO3 compounds are classified as ductile materials; whereas, the PrMnO3 compound is brittle. The ductility/brittleness characteristic in materials can also be discussed via calculating the Cauchy’s pressure which it is defined as the difference between the two elastic constants (C12-C44). If the Cauchy’s pressure is positive (negative), the material is expected to be ductile (brittle). A quick look of Table 2 confirms the ductile nature of PrVO3, PrCrO3 and PrFeO3 compounds and the brittle behavior for PrMnO3 compound. Furthermore, Frantsevich et al. [33] relate the ductility/brittleness behavior of material to the Poisson’s ratio (υ). For brittle materials, Poisson’s ratio must be less than 1/3; otherwise, the material behaves in a ductile manner, where the above statement confirms on the compounds of interest about the ductility/brittleness behaviors. Other mechanical parameters, namely Young’s modulus (E), shear modulus (G), Lamé’s coefficients (µ) and (λ), and Poisson’s modulus (σ) which are the important of elastic moduli for applications, they can be derived from the elastic constants, by using the following standard relations [34,35,36]:
Physically, the shear modulus (G) and Lamé’s constant (μ) are equal and give information about shear stiffness of materials while the Lamé constant (λ) is related to the compressibility of the material. The calculated values of these elastic moduli are listed in Tables 2 and 3. Bonding and stiffness of materials is also indicated by Poisson’s ratio. The systems with interatomic interactions have values of σ are close to 0.25 [37], for covalent and metallic materials the value of σ is between 0.1 and 0.33. The calculated values of Poisson’s ratio are gathered in Table 3, clearly indicate that all the PrXO3(X = V, Cr, Mn, and Fe) compounds are effected with central contributions, suggesting the ionic character of these materials.
3.3 Thermal properties
One of the most important parameter is the Debye temperature (θD) which is used to find the thermal features of different materials. The Debye temperature is used to differentiate between the high (the low) temperature states in solid materials. The higher θD suggests a large thermal conductivity and melting temperature associated with materials. When the temperature is low, the excitation arises slowly from audible sensations. Here we have used standard methods with elastic constant data to calculate Debye temperature (θD). Debye temperature is efficiently calculated by using νm (average sound velocity) in the following expression:
where h, k, NA, n, M, ρ, and vm, are respectively Planck’s constant, Boltzman’s constant, Avogadro’s number, the number of atoms per formula unit, the molecular mass per formula unit, the density, and the average sound velocity obtained from the following relation of vm [38]:
where vl and vt are the longitudinal and transversal elastic wave velocities in an isotropic material, respectively; they can be obtained by using the shear modulus (G) and the bulk modulus (B) from Navier’s equation [39]:
We can be seen from Table 3 that the PrMnO3 compound has a higher thermal conductivity as compared to other compounds in the same group. As far as we study, there are no experimental and theoretical data available in the literature to be compared with our present calculations; so, in coming future experimental works test our calculated results.
3.4 Electronic properties
To know whether these compounds are metals, insulators or semiconductors, the study of the band structures is very important. Every material has different electronic properties due to its unique band structures. The spin polarized electronic band structures of the compounds are calculated by using WC-GGA and GGA + U potentials. The calculated band structure of all PrXO3(X = V, Cr, Mn, and Fe) cubic perovskites in both spin-up and spin-down channels along the high directions in the first Brillouin zone are depicted in Figs. 3 and 4 for WC-GGA and GGA + U approximations, respectively. Our results reveal that these compounds have a metallic nature with mixed valence and conduction bands (no energy band gap). To study the contribution of different states of the ions in the electronic band structures, we have computed the total density of states (TDOS) and partial density of states (PDOS) of the studied PrXO3(X = V, Cr, Mn, and Fe) cubic perovskites which are shown in Figs. 5 and 6 for GGA and GGA + U parameterizations, respectively. In the case of PrVO3 compound (see Fig. 6), participation of each state such as “s”, “p”, “d” and “f” states of Pr, V and O atoms are shown in majority-spin (spin up) and minority-spin (spin down). In spin-up case, the “Pr-d and Pr-f” states have maximum participation in conduction band while the “V-s and V-d” states have maximum participation in the valence band for spin-down, “Pr-d, Pr-f and V-s” states have maximum participation in conduction band, “V-s” states crossing the Fermi level and “V-d” have maximum participation in the valence band which is an indication for a metallic behavior of PrVO3 compound demonstrated by the GGA + U approximation. In the case of PrCrO3 compound, similarly all states are shown for both spin-up and spin-down directions. In spin-up orientation, the “Cr-s” states have a little participation in conduction band and an maximum participation in the valence band, the “Cr-d” states have a participation only in the valence band, “Pr-f and Cr-s” states in spin-up channel and “Cr-s” in spin-down channel crossing the Fermi level to make the metallic character of the material. For the PrMnO3 compound and in case of spin-up channel, the “Pr-f” states and “Mn-s” states lie at Fermi level in the spin-up and spin-down directions to make the material in metallic feature and the “Mn-d” states have almost the same participation in majority-spin and minority-spin cases for both conduction and valence bands. For the PrFeO3 system, the “Pr-f and Mn-s” states cross the Fermi level in spin-up and spin-down channels to make the material metallic and the “Pr-d” states have the same participation in spin-up and spin-down channels of both valence and conduction bands. Figure 6 shows that for all the studied compounds, the Pr-4f states cross the Fermi level in spin-up case, while they have a maximum contribution in valence band for spin-down case. From Figs. 5 and 6, the densities of Pr-4f states and of V-3d, Cr-3d, Mn-3d, and Fe-3d in all PrXO3 cubic perovskites show that these states are responsible for the metallic behavior within these compounds. The X-3d states are expended and cross the Fermi level in band structures due to each material shows the metallic behavior. It is also seen from the Fig. 6 that Pr-4f and V-3d, Cr-3d, Mn-3d, and Fe-3d states are shifted toward the valence band in GGA + U approximation, which indicate that GGA + U approximation is more appropriate for an accurate description of the system with 4f and 5f electrons.

Spin polarized ferromagnetic electronic band structure of PrXO3 (X = V, Cr, Mn, Fe) with GGA method

Spin-polarized ferromagnetic electronic band structure of PrXO3 (X = V, Cr, Mn, Fe) with GGA + U potential

Total and PDOS of PrXO3 (X = V, Cr, Mn, Fe) with GGA method

Total and PDOS of PrXO3 (X = V, Cr, Mn, Fe) with GGA + U method
Electron density of spin for the studied PrXO3 (X = V, Cr, Mn, and Fe) compounds are calculated by employing the GGA + U scheme, where their results are presented in Figs. 7 and 8 for (100) and (110) planes, respectively. It shows the bonding nature between Pr and transition metal (X) and between oxygen and transition metal. For PrVO3 compound, the bonding between Pr and V atoms is completely ionic in both cases of spin-up and spin-down. Through the plane of (110) for spin-up and spin-down cases, the bonding between V and O atoms is metallic, whereas it has an ionic nature between Pr and O atoms. In PrCrO3 compound, the plane (100) shows an ionic bounding between Pr and Cr atoms in majority and minority spins, also it has a small polarization in spin-down case between Pr and Cr atoms, while the bounding between O and Cr atoms is covalent in the plane (110) for spin-up and-down directions. In PrMnO3 system, the spin-up plane (100) has a bonding between Pr and Mn atoms with small polarization while the spin-down bounding between Pr and Mn atoms is ionic. In PrFeO3, the spin-polarized bonding between Pr and Fe atoms is ionic for (100) plane, while the (110) plane display completely a metallic bonding between Fe and O atoms. The covalent bounding arises between X and O atoms is explained as: the oxygen valance shell (2p) needs 2 electrons to complete their outer most shell and X transition metals have + 2 or + 3 oxidation and shared with each other, which is the result of covalent bounding between these atoms.

Electron density of spin-polarized perovskites PrXO3 (X = V, Cr, Mn, Fe) in (100) plane

Electron density of spin-polarized perovskites PrXO3 (X = V, Cr, Mn, Fe) in (110) plane

Calculated total energy versus unit-cell volume of the cubic PrVO3 and PrCrO3 compounds in paramagnetic (PM), ferromagnetic (FM), and anti-ferromagnetic (AFM) phases

Calculated total energy versus unit-cell volume of the cubic PrMnO3 and PrFeO3 compounds in paramagnetic (PM), ferromagnetic (FM), and anti-ferromagnetic (AFM) phases
3.5 Magnetic properties
Magnetic properties are those properties which are caused by spin and orbital motion of electrons in an atom. Different materials have different properties such as the ferromagnetic and anti-ferromagnetic properties; these types of properties are due to unpaired electrons, which produce two types of exchanges, the double exchange as well as the super exchange [40]. In some cases, the ferromagnetism is due to the double exchange and the super exchange while the anti-ferromagnetism is due to the super exchange interaction. The double-exchange mechanism is a type of a magnetic exchange that may arise between ions in different oxidation states. The double-exchange interaction occurs between two atoms in which one atom have extra-electron compare to the other, while the super-exchange is a ferromagnetic or anti-ferromagnetic alignment which occurs between two atoms with the same number of electrons. Another difference between double-exchange and super-exchange is that in double-exchange electrons are delocalized while in super-exchange electrons are localized, these results are in the material displaying magnetic exchange coupling as well as metallic conductivity.
This information was first proposed by Zener [41]; he predicts electron exchange ways between the two species and gives important suggestions about the ferromagnetic and anti-ferromagnetic materials. He also gives the idea of indirect exchanges between rare earths through oxygen (X–O–X). Magnetic properties of these compounds are also studied by optimizing the double cell structure in paramagnetic, ferromagnetic and in anti-ferromagnetic phases, as shown in Fig. 2, by using Birch–Murnaghan’s equation of state [42]. The stable state is that which have the lowest ground state energy. Table 4 shows that all the compounds have the lowest energy in ferromagnetic phase; therefore, the ferromagnetic phase is most favorable in energy. Figures 9 and 10 show the optimizing plots for the ground state energies of these compounds.
Further investigation on the magnetic properties of PrXO3 compounds with calculated total, local and interstitial magnetic moments that are listed in Table 5. The local magnetic moment of V, Cr, Mn and Fe atoms in all PrXO3 systems are obtained through the GGA framework about 0.74348, 0.62925, 2.995635 and 2.67892, whereas those of the GGA + U approximation are about 1.09453, 2.112055, 3.012345 and 2.23419, respectively. The larger value of magnetic moment of Mn indicates that the magnetic behavior of PrMnO3 is stronger than that of PrVO3, PrCrO3 and PrFeO3 compounds. For all the all PrXO3 compounds, the positive values of magnetic moment is shown in Pr atom and in interstitial part, confirming that they are parallel to the magnetic moment of transition element (TM), and the negative values of the magnetic moment of oxygen in PrVO3 compound resulted that they are anti-parallel to V atom, in the goal to reduce the net magnetic moment. It is also shown in Table 5 that the total magnetic moment of all PrVO3, PrCrO3, PrMnO3 and PrFeO3 compounds is not integral (see Table 5). These calculated non integers values of total magnetic moment confirm that these compounds are ferromagnetic metals, as reported in References [43, 44] for other materials.
3.6 Formation energy
The formation energy (Ef) is an important factor to test the stability of solid materials. Formation energy (Ef) is defined as the energy which is required to dissociate the links between different atoms of the solids. Therefore formation energy for the studied compounds PrXO3 at the zero temperature is calculated; where the negative sign of Ef reveals the stronger bonding which relates the atoms and their favorable allowing stability of the crystal [48].
The formation energy is also defined as the difference between total energy of the compound (E0) and the sum of pure energies which constitute the compound, taken at their stable structural phase. Additionally, the formation energy (Ef) of PrXO3 (X = V, Cr, Mn, and Fe) perovskite compounds is calculated by using the following formula [49, 50]:
where Ef is the system total energy, and EPr, EX, and E0 are the individual energies of Pr, X, and O sites, respectively. The calculated negative values of Ef as tabulated in Table 6 reveal that all the compounds PrXO3 compounds are thermodynamically stable.
3.7 Curie temperature
Curie temperature is that temperature at which certain magnetic materials have a sharp change in their magnetic properties. At low temperature magnetic dipole are align, but above the Curie point alignments becomes random. The Curie temperature of PrXO3 (X = V, Cr, Mn, and Fe) compounds is calculated as tabulated in Table 7. Table 7 lists the total energy in both ferromagnetic state (EFM) and anti-ferromagnetic state (EAFM), and Curie temperature (with both methods) for bulk PrVO3, PrCrO3, PrMnO3, and PrFeO3 perovskite compounds, by using GGA and GGA + U approximations. Curie temperature is calculated by using method 1 and method 2 as reported in References [51, 52].
In method 1, we can calculate the Curie temperature by using the following relation:
In the above equation KB is the Boltzmann constant and Jij is called exchange interaction which is defined as:
where EFM and EAFM are the total energy at ferromagnetic and anti-ferromagnetic states, respectively.
In method 2, the Curie temperature (TC) is also calculated by using the following expression [53].
Table 7 shows the Curie temperature and total energies at ferromagnetic and anti-ferromagnetic states. Large values of TC show strong interaction while smaller value of TC shows weak interaction among the magnetic atoms.
4 Conclusions
In this work, we have performed detailed investigation on the structural, thermal, elastic, electronic and magnetic properties of lanthanide oxides based on the cubic perovskite-type PrXO3 (X = V, Cr, Mn, and Fe) compounds by using the first-principles FP-APW + lo method within GGA and GGA + U frameworks. The computed lattice constants are consistent with the experimental results. It is found that the PrVO3, PrCrO3 and PrFeO3 compounds are ductile in nature whereas the PrMnO3 compound is brittle. The compounds of interest are metallic in nature, where the metallic behavior of these systems is due to Pr-4f and X-3d states. It is also concluded that these compounds are stable in ferromagnetic phase. The larger value of Curie temperature in PrMnO3 compound reveals that this compound has a strong interaction among the magnetic atoms as compared to other compounds in the group. The negative values of the formation energy of these compounds show that they are thermodynamically stable.
References
Ullah H, Naeem S, Murtaza G, Khenata R, Khalid MN (2013) First principle study of CsSrM3 (M = F, Cl). Phys B 414:91–96
Ullah H, Murtaza G, Khenata R, Muhammad S, Reshak AH, Wong KM, Omran SB, Ahmed ZA (2014) Structural, chemical bonding, electronic and magnetic properties of KMF3 (M = Mn, Fe Co, Ni) compounds. Comput Mater Sci 85:402–408
Ullah H, Murtaza G, Khenata R, Mohammad S, Naeem S, Khalid MN, Manzar A (2013) Structural, elastic, electronic and optical properties of CsMCl3 (M=Zn, Cd). Phys B 420:15–23
Guzik A, Talik E, Pajaczkowska A, Turczynski S, Kusz J (2014) Magnetic properties of manganese doped PrAlO3 monocrystalline fibres. Mater Sci Poland 32:633–640
Sabir B, Murtaza G, Mahmood Q, Ahmad R, Bhamu KC (2017) First principles investigations of electronics, magnetic, and thermoelectric properties of rare earth based PrYO 3 (Y=Cr, V) perovskites. Current Appl Phys 17:1539–1546
Chattopadhyay S, Nath TK (2011) Electrical and magnetoelectronic properties of La0.7Sr0.3MnO3/SiO2/p-Si heterostructure for spintronics application. Current Appl Phys 11:1153–1158
Banach JG, Temmerman WM (2004) Delocalization and charge disproportionation in La(1−x)SrxMnO3. Phys Rev B 69:054427
Cheong SW, Mostovoy M (2007) Multiferroics: a magnetic twist for ferroelectricity. Nat Mater 6:13–20
Gschneidner KA, Eyring L (1979) Handbook on physics and chemistry of rare earth, vol 26. North-Holland, Amsterdam
Ren Y, Nugroho AA, Menovsky AA, Strempfer J, Rutt U, Iga F, Takabatake T, Kimball CW (2003) Orbital-ordering-induced phase transition in LaVO3 and CeVO3. Phys Rev B 67:014107
Shpanchenko RV, Chernaya VV, Tsirlin AA, Chizhov PS, Sklovsky DE, Antipov EV, Khlybov EP, Pomjakushin V (2004) Synthesis, structure, and properties of new perovskite pbVO3. Chem Mater 16:3267–3273
Wang F, Zhang J, Yuan P, Yan Q, Zhang P (2000) Magnetic and transport properties of vanadate PrVO3. J Phys Condens Matter 12:3037–3040
Raccah PM, Goodenough JB (1967) First-order localized-electron ⇆ collective-electron transition in LaCoO3. Phys Rev B 155:932
McDannald A, Kuna L, Seehra MS, Jain M (2015) Magnetic exchange interactions of rare-earth-substituted DyCrO3 bulk powders. Phys Rev B 91:224415
Rezaiguia M, Benstaali W, Abbad A, Bentata S, Bouhafs B (2017) GGA + U study of electronic and magnetic properties of Pr(Fe/Cr)O3 cubic perovskites. J Supercond Novel Magn 30:2581–2590
Khandy SA, Gupta DC (2016) Investigation of the transport, structural and mechanical properties of half-metallic REMnO3 (RE = Ce and Pr) ferromagnets. RSC Adv 6:97641–97649
Zhou Y, Lü Z, Wei B, Wang Z, Zhu X (2015) Electronic structure and surface properties of PrMnO3 (001): a density functional theory study. Solid State Commun 201:31–35
Tijare SN, Bakrdjieva S, Subrt J, Joshi MV, Rayalu SS, Hishita S, Labhsetwar N (2014) Synthesis and visible light photocatalytic activity of nanocrystalline PrFeO3 perovskite for hydrogen generation in ethanol-water system. J Chem Sci 126:517–525
Blaha P, Schwarz K, Sorantin P, Trickey SK (1990) Full-potential, linearized augmented plane wave programs for crystalline systems. Comput Phys Commun 59:399–415
Howard CJ, Kennedy BJ, Chakoumakos BC (2000) Neutron powder diffraction study of rhombohedral rare-earth aluminates and the rhombohedral to cubic phase transition. J Phys Condens Matter 12:349
Anisimov VI, Gunnarsson O (1991) Density-functional calculation of effective Coulomb interactions in metals. Phys Rev B 43:7570
Anisimov VI, Zaanen J, Andersen OK (1991) Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys Rev B 44:943
Anisimov VI, Solovyev IV, Korotin MA, Czyzyk MT, Sawatzky GA (1993) Density-functional theory and NiO photoemission spectra. Phys Rev B 48:16929
Monir ME, Ullah H, Baltach H, Ashiq MG, Khenata R (2017) Mechanical and magneto-electronic properties of half-metallic ferromagnetism in Ti-doped ZnSe and CdSe alloys: Ab initio study. J Magn Magn Mater 442:107–117
Kumar A, Verma AS, Bhardwaj SR (2008) Prediction of formability in Perovskite-type oxides. Open Appl Phys J 1:11–19
Xu N, Zhao H, Zhou X, Wei W, Lu X, Ding W, Li F (2010) Dependence of critical radius of the cubic perovskite ABO3 oxides on the radius of A- and B-site cations. Int J Hydrogen Energy 35:7295–7301
Bouhemadou A (2008) Theoretical study of the structural, elastic and electronic properties of the GeX 2O4 (X = Mg, Zn, Cd) compounds under pressure. Modelling Simul Mater Sci Eng 16:055007
Kanoun MB, Merad AE, Cibert J, Aourag H, Merad G (2004) Properties of strained zinc-blende GaN: first-principles study. J Alloys Compd 366:86–93
Merad AE, Aourag H, Khalifa B, Mathieu C, Merad G (2001) Predictions of the bonding properties in Cd 1 − xZn xTe. Superlattice Micro-Struct 30:241–251
Fu H, Li D, Peng F, Gao T, Cheng X (2008) Ab initio calculations of elastic constants and thermodynamic properties of NiAl under high pressures. Comput Mater Sci 44:774–778
Ullah H, Murtaza G, Khenata R, Mohammad S, Manzar A, Omran SB, Ullah A, Muzammil M (2015) Mechanical, electronic and magnetic properties of Sm-based perovskite-type oxides SmMO3 (M = V, Fe and Co): an ab initio study. Indian J Phys 89:1133–1142
Pugh SF (1954) XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos Mag 45:823–843
Frantsevich IN, Voronov FF, Bokuta SA (1983) Elastic constants and elastic moduli of metals and insulators handbook. Naukova Dumka, Kiev, p 60
Voigt W (1928) Lehrbuch der Kristallphysik. Teubner, Leipzig
Reuss A, Angew Z (1929) Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. Math Mech 9:49–58
Nye JF (1957) Physical properties of crystals. Clarendon, Oxford
Ledbetter HM (1983) Materials at low temperatures, ed. by R.P. Reed, A.F. Clark. American Society for Metals, Metals Park
Anderson OL (1963) A simplified method for calculating the debye temperature from elastic constants. J Phys Chem Solids 24:909–917
Barsoum MW, El-Raghi T, Porter WD, Wang H, Chakraborty S (2000) Thermal properties of Nb2SnC. J Appl Phys 88:6313
Blundell S (2001) Magnetism in condensed matter, 1st edn. Oxford University Press), New York
Zener C (1951) Interaction between the d-shells in the transition metals II ferromagnetic compounds of manganese with perovskite structure. Phys Rev 82:403
Blaha P, Schwartz K, Luitz J (2001) WIEN2k, ISBN:3-9501031-1-2. Wien2k, Vienna University of Technology, 2002, improved and updated Unix version of the original copyrighted Wien-code
Hebert S, Pralong V, Pelloquin D, Maignan A (2007) Hexagonal perovskite cobaltites: unconventional magnetism at low temperature. J Magn Magn Mater 316:394–399
Longo JM, Racah PM, Goodenough JB (1968) Magnetic properties of SrRuO3 and CaRuO3. J Appl Phys 39:1327
Verma AS, Jindal VK (2012) Bulk modulus of cubic perovskites. J Alloy Compd 541:210–214
Bouadjemi B, Bentata S, Abbad A, Benstaali W, Bouhafs B (2013) Half-metallic ferromagnetism in PrMnO3 perovskite from first principles calculations. Solid State Commun 168:6–10
Birch F (1947) Finite Elastic Strain of Cubic Crystals. Phys Rev 71:809
Yakoubi A, Baraka O, Bouhafs B (2012) Structural and electronic properties of the Laves phase based on rare earth type BaM2 (M = Rh, Pd, Pt). Results Phys 2:58–65
Zeng ZH, Calle-Vallejo F, Mogensen MB, Rossmeisl J (2013) Generalized trends in the formation energies of perovskite oxides. Phys Chem Chem Phys 15:7526–7533
Rai DP, Shankar A, Ghimire MP, Khenata R, Thapa RK (2015) Study of the enhanced electronic and thermoelectric (TE) properties of ZrxHf1−x−yTayNiSn: a first principles study. RSC Adv 5:95353–95359
Kubler J, Fecher GH, Felser C (2007) Understanding the trend in the Curie temperatures of Co2-based Heusler compounds: Ab initio calculations. Phys Rev B 76:024414
Wurmehl S, Fecher GH, Kandpal HC, Ksenofontov V, Fesler C, Lin HJ, Morais J (2005) Geometric, electronic, and magnetic structure of Co2FeSi: Curie temperature and magnetic moment measurements and calculations. Phys Rev B 72:184434
Candan A, Ugur G, Charifi Z, Baaziz H, Ellialtioglu MR (2013) Electronic structure and vibrational properties in cobalt-based full-Heusler compounds: a first principle study of Co2MnX (X = Si, Ge, Al, Ga). J Alloy Compd 560:215–222
Acknowledgements
This work is supported by Mustapha Stambouli University of Mascara.
Funding
This study was funded by the University Training Research Projects (PRFU) of the National Scientific Research of Algeria (Grant Number B00L02UN290120180001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
El Amine Monir, M., Dahou, F.Z. Structural, thermal, elastic, electronic and magnetic properties of cubic lanthanide based perovskites type oxides PrXO3 (X = V, Cr, Mn, Fe): insights from ab initio study. SN Appl. Sci. 2, 465 (2020). https://doi.org/10.1007/s42452-020-2223-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-2223-4