
Abstract
In this work, we are evaluating the hydrogen adsorption capacity at 298 K of cobalt-doped MOF-5 using the Grand Canonical Monte Carlo method. We substitute eight, sixteen, and thirty-two zinc atoms of MOF-5 with cobalt atoms and we obtain Co8-MOF-5, Co16-MOF-5 and CoMOF-5 respectively. For each of these molecules, we determine the pore diameters, the surface, the pore volume, the isosteric heat, and the storage capacities of these doped MOFs. The results show that for Co8-MOF-5 and Co16-MOF-5, doping decreases the pore volume and increases the density. This will lead to an increase in volumetric capacity and a decrease in gravimetric capacity. However, we note a strong adsorbent-adsorbate attraction compared to undoped MOF. This is justified by a high excess capacity for materials with a small surface.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Storage remains the only major obstacle to the large-scale use of hydrogen in the energy sector today. Indeed, hydrogen is not a source of energy, but a vector, just like electricity. It is used to transport the energy produced by a primary source (petroleum, uranium). It is presented as a possible substitute for hydrocarbons and an efficient way to store renewable energies (wind, solar and hydro), main electricity over a long period. However, its application remains very expensive, because its storage is done by the bias two very energy-consuming methods. Compression under a pressure of 700 bar presents key problems in terms of material, design, and sealing; moreover, reaching the desired pressure requires more than 10% of the stored energy. The cryogenic method, on the other hand, causes large losses of energy during the liquefaction and evaporation processes [1, 2].
However, Metal-Organic Frameworks (MOFs) structures have received very important attention over the past two decades for applications in renewable energy and environmental science [3]. Indeed, the large number of combinations of organic linkers and metal connectors that can be used in principle makes it possible to design materials for a wide variety of potential applications [4,5,6]. These MOFs have the promise that by modifying building blocks, organic ligands or metallic nodes, i.e. the electronic structure, we can design an optimal material for various applications [7, 8]. Their characteristics, such as high porosity, large surface area, tunable structure, and modifiable functionality, make them very promising to be applied in gas storage and separation [9,10,11,12]. This is why the scientific community is placing great emphasis on the study of adsorption of hydrogen by these MOFs at room temperature and pressure to reduce the costs associated with storage. But at room temperature, their storage capacity becomes low, because of MOFs-H2 interactions which are very weak [3, 13, 14].
Different popular strategies to increase the adsorption capacity of hydrogen at room temperature are reported in the literature: increasing the length of organic linkers [15], incorporation post-synthesis of metallic species [16], post-synthesis anchoring of metals (Li or Cr, Co) in the aromatic rings of the linker [17], the improvement of the dipole moment of the group of metal oxides in MOFs [18, 19]. Other promising ways are to decorate the attraction of hydrogen by metals on their surfaces. Examples of hydrogen attracting metals are Li [17, 20], Na [21], K [21] Ca [22, 23], Sc [24] and Ti [25–27]. MOF having two metals in the structure is also presented as a likely solution [28, 29]. Indeed, the doping of different metals in the MOF nodes can generate defects in the MOF, which would improve their intrinsic properties and thus affect their gas absorption [30, 31].
MOFs can be used as semiconductors. But their applications in areas such as photovoltaics, detection, and photocatalysis [8, 32,33,34] are limited by their band gaps relatively wide. This is how Choi and al., 2009 [35] proposed a substitution of the Zinc molecules of MOF-5 to study the effect of doping on the band gap. In 2010, Botas et al., [36] Having noticed the high affinity of Co with H2, studied the adsorption capacity of Co-doped MOF-5. They noted that the adsorption of H2, CH4, and CO2 by MOF-5 systematically increases with the Co content, in particular at high pressure at 77 K. Yang et al., [37] In 2014 demonstrate that in addition to increasing the adsorption capacity, doping of MOF-5 with Co (II), can considerably improve the structural stability of MOF-5 with respect to humidity but also exhibits solvatochromic behavior to detect small solvent molecules. The substitution of the Zinc atoms of MOF-74 by Cobalt atoms also offered good performances in gas adsorption, Botas et al., [38] Show that the isosteric heat increased with the concentration of cobalt. Montes et al. [4] shows that the cobalt-based URJC-3 molecule has hydrogen storage performance not only better than activated carbon and purified single-walled carbon nanotubes but also exceeds absorption gravimetric of hydrogen reached by most MOFs, despite their moderate surfaces due to small pores [4].
Motivated by the fact that MOF-5 is an excellent representative of porous materials and by ability of the known Co (II) ion to improve the stability and adsorption properties of materials, we examine in this paper the ability of MOF-5 doped with Co(II) ion to adsorb hydrogen at a temperature of 298 K by varying the pressure from 1 to 100 bar.
2 Method
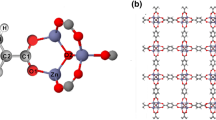
Figure 1 shows the four molecules studied. Fig. 1a shows the molecule MOF-5. Fig. 1b shows the Co8-MOF-5 molecule which has eight zinc atoms substituted by cobalt atoms. Fig. 1c shows the Co16-MOF-5 molecule in which sixteen zinc atoms are substituted by cobalt atoms. Fig. 1d shows the CoMOF-5 molecule in which all the zinc atoms of the MOF-5 molecule are substituted with cobalt atoms. The substituents are not located in the same position periodically but are randomly located with the same rate of substitution.

Representative structures for each cobalt substitution rate, where Co, Zn, C, O, and H are shown in green, violet, grey, red, and white, respectively. All atoms are drawn by a stick model except Co and Zn, which are drawn with a ball and stick model for emphasis. a MOF-5, b Co8-MOF-5, c Co16-MOF-5, d CoMOF-5.
The Grand Canonical Monte Carlo method consists in generating series of representative configurations of the system, linked together by arbitrarily chosen movements executed in a stochastic manner [39]. To ensure that each system is sampled in an equilibrium state, a total of 10,000 cycles are run for each simulation. The first half for equilibrium and the last half for production, which is sufficient to obtain consistent results [40]. Each cycle consists of N Monte Carlo motions, where N is the greatest number between 20 and the number of hydrogen molecules adsorbed in the elementary cell at the beginning of each point in the simulation. The three types of MC motions used, which are translation, rotation and complete re-insertion of hydrogen molecules, are considered with a probability equal to 1. The insertion of a new hydrogen molecule is done in a random position and orientation [41, 42]. To determine the hydrogen adsorption isotherms at 298 K between 1 and 100 bar in the proposed structures, hydrogen adsorption was calculated for seven pressure points, 1, 10, 20, 40, 60, 80 and 100 bar.
Having a periodically symmetrical structure, hydrogen adsorption can be simulated on a finite part as on the link or on the MOF-5 cluster. However, Mueller and Ceder show that such an approach can give misleading results [43]. Furthermore, since cobalt ions are randomly substituted in MOF-5, we simulate hydrogen uptake over the entire elementary cell.
In the simulation, the adsorbate and adsorbent molecules are considered rigid. In this case, the only interactions to be considered are intermolecular interactions between unbound atoms, Van der Waals interactions and electrostatic interactions [42]. These interactions are modelled by the Lennard-Jones (LJ) + Coulomb potentials as represented by equation (1).
Where ε is the depth of the potential well, σ is the finite distance at which the interparticle potential is zero, r is the distance between the particles. \({q}_{1}\) and \({q}_{2}\) Are the positive or negative magnitudes of the charges.
The hydrogen molecules have been modeled according to the Darkrim-Levesque model [44]. The LJ parameters for the MOF atoms were assigned according to the Universal Force Field (UFF) [45] and Dreiding [46]. A limit value of 12 Å was used for all LJ interactions, and Ewald summations were used to calculate Coulomb interactions. The Lorentz—Berthelot [47] mixing rules used to determine the LJ parameters for the interactions between atoms. The charges of the atoms were determined using load balancing with RASPA code.
The geometric area was calculated using a probe with a diameter of 2.958 Å equal to the diameter of the hydrogen atom. The void fraction, which is the void of a structure divided by the total volume, is measured using helium, because helium hardly adsorbs at room temperature. The pore diameters of the different structures are calculated from the largest sphere included; which is simply the greatest distance attributed to the Voronoi nodes [48]. Indeed, the algorithm iterates over all Voronoi nodes in a periodic unit cell of a structure and finds the node with the largest distance to a neighboring atom. The isosteric heat of adsorption is calculated as described in the work of Vlugt et al. [49]. Using a pressure of 0.5 bar assumed to be low load [50]. Indeed, at low load, the gas phase is assumed to be ideal [51], the heat is relatively constant and reflects the interactions between hydrogen and MOF, with a small contribution from hydrogen-hydrogen interactions [50].
Calculations of pore volume, largest cavity diameter, specific surface area, were calculated using Zoe ++ [52] while the helium fractions under vacuum, adsorption heats and hydrogen adsorption capacities were determined using RASPA-2.0 (David Dubbeldam et al. [53]).
3 Results
The surfaces of the four materials were determined to evaluate the influence of Co(II) doping on their texture. The values given in Table 1 show that the gravimetric surfaces are 3934,2640; 3178; 3898 m2.g−1 for MOF-5, Co8-MOF-5, Co16-MOF-5, and Co-MOF-5 respectively. It is noted that the surface area first decreases when 8 zinc atoms are substituted with cobalt, and then increases from 16 substituted zinc atoms. When we are at 100% of the substituted atoms, the surface area increases and becomes approximately equal to that of MOF-5. The decrease in the surface value of Co8-MOF-5 relative to MOF-5 was first understood as an effect of the variation in the size of the substituted metal ion in the cluster. Indeed, according to Botas et al., [38] the Co2 + ion is smaller than the Zn2 + ion. However, the increase in surface area observed for Co16-MOF-5 and CoMOF-5, rather allows us to understand that the variation of the gravimetric surfaces of Co8-MOF-5, Co16-MOF-5 and CoMOF-5 could not be influenced only by the size of the metal ion. Therefore, to better understand, the calculation of the pore diameters of these MOFs gives values which are 15.54, 13.33; 14.17 and 15.31 Å for MOF-5, Co8-MOF-5, Co16-MOF-5, and CoMOF-5 respectively.
In Fig. 2, it can be seen that the curves representing pore diameters and gravimetric surfaces as a function of the level of Co(II) doped in MOF-5 give similar appearances. It can therefore be understood that the Co(II) doping of MOF-5 has an effect on the length of the ligands. A result that is similar to that of Choi et al, who also showed that elementary cell lattice parameters varied as a function of doping. They determined by the DFT method that these elementary cell lattice parameters were 26.552, 23.430; 24.578 and 26.194 Å for MOF-5; Co8-MOF-5, Co16-MOF-5, and CoMOF-5 respectively [35]. The latter conclude that this is a combination of the effects of the cobalt ion radius and p orbital in organic linkers.

the curves representing the diameters of the pores and the gravimetric surfaces as a function of the level of Co (II) doped in the MOF-5
The gravimetric and volumetric adsorption capacities for MOF-5 shown in Table 2 are 1.35 wt% and 9.38 gH2.L−1at a pressure of 100 bar and a temperature of 298 K. In the recent works of El Kassaoui et al., [54] it has been shown that the connector consisting of the metal oxide cluster interconnected by the organic binding molecules becomes unstable when it stores 12 hydrogen molecules for the horizontal and inclined orientations and 10 molecules for the vertical. In the current work, we simulate adsorption on the whole elementary cell which consists of several connectors. Our results obtained for MOF-5 are similar to those obtained by Frost and Snurr [50] which are close to the results obtained experimentally by [55]. Thus, the doping of MOF-5 with Cobalt (II) should not influence its stability because, according to the work of Choi et al., the substitution of Zn by Co was more stable because in terms of energy this molecule was more exothermic [35]. Yang et al. [37] also demonstrated that doping MOF-5 with Co (II) dramatically improves the structural stability of MOF-5 with respect to humidity. So, as the difference between the adsorption capacities of doped MOFs and MOF-5 is not very high in the pressure range of this study, the adsorption of hydrogen in the MOF molecules doped with cobalt (II) should not affect their stability.
Fig. 3 shows the hydrogen adsorption isotherms of MOF-5, Co8-MOF-5, Co16-MOF-5, and CoMOF-5 over a pressure range of 1–100 bar at a temperature of 298 K. It is first observed that the gravimetric capacities of all the materials studied evolve as the pressure increases. However, it should be noted that the maximum capacities reached in our simulation range at 100 bar vary according to the Co (II) doping rate. In Table 2, we can see that these values are 1.35, 1, 1.13 and 1.4 wt% for MOF-5, Co8-MOF-5, Co16-MOF-5 and CoMOF-5 respectively.

Absolute gravimetric hydrogen adsorption in MOF-5, Co8-MOF-5, Co16-MOF-5 et le CoMOF-5 materials at 298 K.
In the literature, it has been established that hydrogen adsorption capacity is proportional to the heat of adsorption and the gravimetric surface area [56, 50]. Fig. 4 shows the evolution of the gravimetric surface, pore volume, heat of adsorption, and the gravimetric adsorption capacity from H2 at 100 bar with the number of substituted Co(II) atoms in MOF-5. The heat of adsorption and the gravimetric surfaces evolve in opposition during doping, and the adsorption capacity is proportional to the gravimetric surface and the pore volume. Fig. 5 shows that the curves representing the variation of the H2 adsorption capacity and the gravimetric surface merge from the doping of MOF-5 with 16 ions of Co(II) to total substitution. It can therefore be said that in the present case, the gravimetric hydrogen adsorption capacity of the Co(II)-doped MOF-5 is mainly influenced by the variation of the gravimetric surface.

Evolution of the gravimetric surface, the heat of adsorption, the pore volume and the absolute gravimetric adsorption capacity of H2 at 100 bar with the number of doping of Co (II) in MOF-5

Evolution of the gravimetric surface, of the pore volume and of the absolute gravimetric adsorption capacity of H2 at 100 bar with the number of Co (II) doping in the MOF-5
Figure 6 shows the volumetric hydrogen adsorption isotherms at 298 K for a pressure range from 1 to 100 bar. It can be seen that like gravimetric adsorption capacities, volumetric adsorption capacities vary as a function of pressure. The maximum capacities obtained at a pressure of 100 bars for the different MOFs are 9.38, 11.11; 10.09 and 9.93 gH2.L−1 respectively for MOF-5, Co8-MOF-5, Co16-MOF-5, and CoMOF-5. Thus, Co8-MOF-5 which had the smallest gravimetric capacity shows the largest volumetric capacity and CoMOF-5 has the smallest capacity. We understand that the factors influencing the gravimetric and volumetric adsorption capacities of our cobalt-doped materials are different. To better analyze these factors that influence the volumetric adsorption capacity, we have plotted in Fig. 7 a variation of the different parameters as a function of the doping rate.

Absolute volumetric hydrogen adsorption in MOF-5, Co8-MOF-5, Co16-MOF-5 et le CoMOF-5 materials at 298 K.

Variation of volumetric adsorption capacities, isosteric heat of adsorption, density, and gravimetric surface area with the number of Co (II) ions doped in the MOF-5 molecule.
Figure 7 shows the curves of the variations in volumetric adsorption capacity, isosteric heat of adsorption, density and gravimetric surface area with the number of doped Co(II) ions in the MOF-5 molecule. It can be seen that the gravimetric surface area varies in opposition to the volumetric adsorption capacity. But that the volumetric adsorption capacity of the doped MOFs is proportional to the isosteric heat of adsorption and to the density of these MOFs because they have curves that give similar aspects.
Figure 8 shows the variation in volumetric and gravimetric hydrogen adsorption capacities at 100 bar and 298 K with the number of doped Co (II) ions in the MOF-5 molecule. It can be stated that these two capacities vary in opposition to the number of cobalt ions doped in the MOF-5. The volumetric capacity is proportional to the isosteric heat of adsorption and to the density of the doped materials while the gravimetric capacity is proportional to the gravimetric surface

Variation of volumetric, Gravimetric and Excess hydrogen adsorption capacities at 100 bar and 298 K with the number of Co (II) ions doped in the MOF-5 molecule.
The excess adsorption capacity represents the capacity of the hydrogen that is bound to the surface of the material due to Van der Waals and Coulomb forces. Indeed, it is given by the ratio of the mass of hydrogen molecules that binds to materials by the sum of the masses of these hydrogen molecules and the mass of the materials. This capacity is the one that should best orient us on the forces of interactions during an adsorption process. From the isotherms shown in Fig. 9, we can easily see that Co8-MOF-5 binds more hydrogen molecules to its surface, unlike MOF-5 which has the smallest capacity. According to the work of Frost and Snurr, the adsorption capacity of excess hydrogen at high loads (high pressures) and at room temperature is well correlated with the surface and therefore the free volume [50], because, the Van der Waals interactions are very weak, the adsorption of hydrogen would be monolayer. However, it should be noted that it is the material with the smallest surface area that has the largest excess capacity. Co8-MOF-5 has small pores, which also have strong interactions with the hydrogen molecule. For this reason, Co8-MOF-5 has the highest isosteric heat of adsorption. We can therefore conclude that for cobalt-doped MOFs, the excess adsorption at high charge is well correlated with the isosteric heat of adsorption. Therefore, if the heat of adsorption is high, it can have on the surface of MOFs a multilayer hydrogen adsorption at room temperature.

Excess hydrogen adsorption in MOF-5, Co8-MOF-5, Co16-MOF-5 et le CoMOF-5 materials at 298 K.
In these last three figures, we represent the isosteric heat of hydrogen adsorption at 298 K as a function of the different hydrogen adsorption capacities of the four MOFs studied, obtained over a pressure range of 1–100 bars. We can observe that doping MOF-5 with cobalt increases the isosteric heat of hydrogen absorption for Co8-MOF-5 and Co16-MOF-5. However, Fig. 10a reveals that this increase in isosteric heat does not influence the absolute gravimetric capacity of hydrogen adsorption of these two MOFs. But for CoMOF-5 which has characteristics such as pore volume, pore diameter and a surface similar to MOF-5, we notice that it has a slightly higher absorption capacity. It can be concluded that at room temperature, hydrogen adsorption is monolayer and therefore more bound to the gravimetric surface. For this reason, the increase of isosteric heat does not influence the gravimetric adsorption capacities of Co8-MOF-5 and CO16-MOF-5.

a Correlation of the isosteric heat of adsorption with the total gravimetric hydrogen uptake for the four MOFs at 298 K and for pressures ranging from 1 to 100 bars. b Correlation of the isosteric heat of adsorption with total volumetric H2 Uptake for the four MOFs at 298 K and for pressures ranging from 1 to 100 bars. c Correlation of iosteric heat of absorption with excess adsorption for the four MOFs at 298 K and for pressures ranging from 1 to 100 bar.
Figure 10b shows that the volumetric hydrogen adsorption capacity at room temperature is higher for the MOF having the greatest isosteric heat. It can, therefore, be said that doping MOF-5 with cobalt improves the volumetric adsorption capacity of this MOF.
The excess hydrogen absorption capacity, as shown in Fig. 10c, depends on the isosteric heat of absorption. Other previous work had shown that this capacity could be influenced by the surface available to fix the hydrogen molecules and the interactions. It is observed here that the MOFs which have weak surfaces are those which fix the most hydrogen. This reveals that the interactions produced during doping with cobalt are more important.
The differences between the adsorbed quantities of the various materials are relatively small. However, the methodology used in this work gives good numerical accuracy. Indeed, the method used here was also used in our recent work [57], and had provided results that are in line with the results found in the literature and closed to experimental results [50, 55]. As the error bars observed on the pressures of the simulation boxes are quite important in molecular simulation [53], we have determined them, and it should be noted that in Figs. 3, 6, and 9, these results are accurate because the error bars do not impinge on the differences between the adsorption capacities of the four MOFs studied. So there is indeed a variation in the adsorption capacities during doping. In the same way, we can see in the last three Fig 10a, b, c that there is also a variation in the isosteric heat of adsorption during doping.
4 Conclusion
At the end of this work in which we studied the adsorption of hydrogen at room temperature in MOF-5 doped with cobalt (II) by the Grand Canonical Monte Carlo method, we demonstrated that the doping increases for Co8-MOF-5 and Co16-MOF-5 the isosteric heat of adsorption and the density of these MOFs, but decreases the gravimetric surface area of these MOFs. The reduction of the surface area of these two materials during doping reduces their gravimetric hydrogen adsorption capacity. For these cobalt-doped MOFs, it can be seen that the gravimetric and volumetric hydrogen adsorption capacities are closely related to the gravimetric surface area and isosteric heat of adsorption respectively. It should also be noted that between CoMOF-5 and MOF-5 which have approximately the same surface area and isosteric heat, the cobalt doping improves the gravimetric and volumetric hydrogen adsorption capacities very slightly by 0.05 wt% and 0.55 gH2.L−1at 298 k and 100 bar respectively. It can be concluded that partial doping of MOFs with cobalt can effectively improve the adsorption of hydrogen at room temperature if the doped molecules retain their surface.
References
Bossel U, Eliasson B (2019) Energy and the Hydrogen Economy US DOE, EERE. Available online: https://www.afdc.energy.gov/pdfs/hyd_economy_bossel_eliasson.pdf
Von Helmolt R, Eberle U (2007) Fuel cell vehicles: status 2007. J Power Sour 165(2):833–843. https://doi.org/10.1016/j.jpowsour.2006.12.073
Zhang H, Nai J, Yu L, Lou XW (2017) Metal-organic-framework-based materials as platforms for renewable energy and environmental applications. Joule 1(1):77–107. https://doi.org/10.1016/j.joule.2017.08.008
Montes H, Leo P, Orcajo G, Rodríguez-Diéguez A, Choquesillo-Lazarte D, Martos C, Botas J, Martínez F, Calleja G (2019) Novel versatile cobalt azobenzene-based metal−organic framework as hydrogen adsorbent. Chem Phys Chem. https://doi.org/10.1002/cphc.201801151
Furukawa H, Cordova KE, O’Keeffe M, Yaghi OM (2013) The Chemistry and applications of metal-organic frameworks. Science 341(6149):1230444–1230444. https://doi.org/10.1126/science.1230444
D’Alessandro DM, Smit B, Long JR (2010) Carbon dioxide capture: prospects for new materials. Angewandte Chemie Int Ed 49(35):6058–6082. https://doi.org/10.1002/anie.201000431
Zhou H-C, Long JR, Yaghi OM (2012) Introduction to metal-organic frameworks. Chem Rev 112(2):673–674. https://doi.org/10.1021/cr300014x
Maria A, Christopher I, Fatmah M, Berend S, Olga A (2019) Metal substitution as the method of modifying electronic structure of metal−organic frameworks. J American Chem Soc 141(15):6271–6278. https://doi.org/10.1021/jacs.8b13667
Lee K, Howe JD, Lin L-C, Smit B, Neaton JB (2015) Small-molecule adsorption in open-site metal-organic frameworks: a systematic density functional theory study for rational design. Chem Mater 27(3):668–678. https://doi.org/10.1021/cm502760q
Li H, Wang K, Sun Y, Lollar CT, Li J, Zhou H-C (2018) Recent advances in gas storage and separation using metal–organic frameworks. Mater Today 21(2):108–121. https://doi.org/10.1016/j.mattod.2017.07.006
Planas N, Dzubak AL, Poloni R, Lin L-C, McManus A, McDonald TM, Neaton BJ, Long JR, Smit B, Gagliardi L (2013) The mechanism of carbon dioxide adsorption in an alkylamine-functionalized metal-organic framework. J American Chem Soc 135(20):7402–7405. https://doi.org/10.1021/ja4004766
Witman M, Ling S, Anderson S, Tong L, Stylianou KC, Slater B, Smit B, Haranczyk M (2016) In silico design and screening of hypothetical MOF-74 analogs and their experimental synthesis. Chem Sci 7(9):6263–6272. https://doi.org/10.1039/c6sc01477a
Brangier E, Vivian R, Bornet C (2019) Méthodes d’ergonomie prospective appliquées à l’identification de besoins pour des systèmes d’énergie à base d’hydrogène: étude exploratoire. Psychologie Française. https://doi.org/10.1016/j.psfr.2019.02.002
Suh MP, Park HJ, Prasad TK, Lim D-W (2011) Hydrogen storage in metal-organic frameworks. Chem Rev 112(2):782–835. https://doi.org/10.1021/cr200274s
Rosi NL, Eddaoudi M, Kim J, O’Keeffe M, Yaghi OM (2002) Advances in the chemistry of metal–organic frameworks. CrystEngComm 4(68):401–404. https://doi.org/10.1039/b203193k
Li Y, Yang RT (2006) Significantly enhanced hydrogen storage in metal−organic frameworks via spillover. J American Chem Soc 128(3):726–727. https://doi.org/10.1021/ja056831s
Blomqvist A, Araujo CM, Srepusharawoot P, Ahuja R (2007) Li-decorated metal organic framework 5: a route to achieving a suitable hydrogen storage medium. Proc Natl Acad of Sci 104(51):20173–20176. https://doi.org/10.1073/pnas.0708603104
Srepusharawoot P, Araújo CM, Blomqvist A, Scheicher RH, Ahuja R (2008) A comparative investigation of H2 adsorption strength in Cd- and Zn-based metal organic framework-5. J Chem Phys 129(16):164104. https://doi.org/10.1063/1.2997377
Wongprakarn S, Prasongkit J, Srepusharawoot P (2014) Hydrogen adsorption of Be-, Zn-, and Cd-zeolitic imidazolate framework-23: a comparative study. Jpn J Appl Phys 53(8S3):8NK03
Srepusharawoot P, Swatsitang E, Amornkitbamrung V, Pinsook U, Ahuja R (2013) Hydrogen adsorption of Li functionalized covalent organic framework-366: an ab initio study. Int J Hydrog Energy 38(33):14276–14280. https://doi.org/10.1016/j.ijhydene.2013.08.102
Srepusharawoot P, Blomqvist A, Araújo CM, Scheicher RH, Ahuja R (2011) Hydrogen binding in alkali-decorated iso-reticular metal organic framework-16 based on Zn, Mg, and Ca. Int J Hydrog Energy 36(1):555–562. https://doi.org/10.1016/j.ijhydene.2010.10.035
Sorokin PB, Lee H, Antipina LY, Singh AK, Yakobson BI (2011) Calcium-decorated carbyne networks as hydrogen storage media. Nano Lett 11(7):2660–2665. https://doi.org/10.1021/nl200721v
Lee H, Ihm J, Cohen ML, Louie SG (2010) Calcium-decorated graphene-based nanostructures for hydrogen storage. Nano Lett 10(3):793–798. https://doi.org/10.1021/nl902822s
Dixit M, Adit Maark T, Ghatak K, Ahuja R, Pal S (2012) Scandium-decorated mof-5 as potential candidates for room-temperature hydrogen storage: a solution for the clustering problem in MOFs. J Phys Chem C 116(33):17336–17342. https://doi.org/10.1021/jp302852h
Srinivasu K, Ghosh SK (2012) Transition metal decorated porphyrin-like porous fullerene: promising materials for molecular hydrogen adsorption. J Phys Chem C 116(48):25184–25189. https://doi.org/10.1021/jp3047517
Samolia M, Kumar TJD (2014) Hydrogen sorption efficiency of titanium-functionalized mg–bn framework. J Phys Chem C 118(20):10859–10866. https://doi.org/10.1021/jp501722z
Mashoff T, Takamura M, Tanabe S, Hibino H, Beltram F, Heun S (2013) Hydrogen storage with titanium-functionalized graphene. Appl Phys Letters 103(1):013903. https://doi.org/10.1063/1.4812830
Masoomi MY, Morsali A, Dhakshinamoorthy A, García H (2019) Mixed-metal mofs: unique opportunities in metal-organic framework functionality and design. Angewandte Chemie Int Ed. https://doi.org/10.1002/anie.201902229
Song X, Oh M, Lah MS (2013) Hybrid bimetallic metal-organic frameworks: modulation of the framework stability and ultralarge CO2 uptake capacity. Inorg Chem 52(19):10869–10876. https://doi.org/10.1021/ic400844v
Sara A, Parviz D, Hannes D, François-Xavier C, Henk V, Pascal V, Karen L (2019) (2018) Mixed-metal metal organic frameworks. Chem Soc Rev 48:2535–2565. https://doi.org/10.1039/c8cs00337h
Bitzer J, Kleist W (2018) Synthesis strategies and structural arrangements of isoreticular mixed-component metal-organic frameworks. Chem - A Eur J. https://doi.org/10.1002/chem.201803887
Hendon CH, Rieth AJ, Korzyński MD, Dincă M (2017) Grand challenges and future opportunities for metal-organic frameworks. ACS Cent Sci 3(6):554–563. https://doi.org/10.1021/acscentsci.7b00197
Wang W, Xu X, Zhou W, Shao Z (2017) Recent progress in metal-organic frameworks for applications in electrocatalytic and photocatalytic water splitting. Adv Sci 4(4):1600371. https://doi.org/10.1002/advs.201600371
Chen Y, Wang D, Deng X, Li Z (2017) Metal–organic frameworks (MOFs) for photocatalytic CO2 reduction. Catal Sci Technol 7(21):4893–4904. https://doi.org/10.1039/c7cy01653k
Choi JH, Choi YJ, Lee JW, Shin WH, Kang JK (2009) Tunability of electronic band gaps from semiconducting to metallic states via tailoring Zn ions in MOFs with Co ions. Phys Chem Chem Phys 11(4):628–631. https://doi.org/10.1039/b816668d
Botas JA, Calleja G, Sánchez-Sánchez M, Orcajo MG (2010) Cobalt doping of the MOF-5 Framework and its effect on gas-adsorption properties. Langmuir 26(8):5300–5303. https://doi.org/10.1021/la100423a
Yang J-M, Liu Q, Sun W-Y (2014) Co(II)-doped MOF-5 nano/microcrystals: solvatochromic behaviour, sensing solvent molecules and gas sorption property. J Solid State Chem 218:50–55. https://doi.org/10.1016/j.jssc.2014.06.004
Botas JA, Calleja G, Sánchez-Sánchez M, Orcajo MG (2011) Effect of Zn/Co ratio in MOF-74 type materials containing exposed metal sites on their hydrogen adsorption behaviour and on their band gap energy. Int J Hydrog Energy 36(17):10834–10844. https://doi.org/10.1016/j.ijhydene.2011.05.187
Frenkel D, Smit B (2002) "Understanding molecular simulation: from algorithms to applications, 2nd edn. Academic Press, San Diego
Mert H, Deniz CU, Baykasoglu C (2020) Monte Carlo simulations of hydrogen adsorption in fullerene pillared graphene nanocomposites. Mol Simul. https://doi.org/10.1080/08927022.2020.1758696
Allen MP, Tildesley DJ (1987) Computer simulation of liquids. Clarendon Press Oxford, UK
Getman RB, Bae Y-S, Wilmer CE, Snurr RQ (2011) Review and analysis of molecular simulations of methane, hydrogen, and acetylene storage in metal-organic frameworks. Chem Rev 112(2):703–723. https://doi.org/10.1021/cr200217c
Mueller T, Ceder G (2005) A density functional theory study of hydrogen adsorption in MOF-5. J Phys Chem B 109(38):17974–17983. https://doi.org/10.1021/jp051202q
Darkrim F, Levesque D (1998) Monte carlo simulations of hydrogen adsorption in single-walled carbon nanotubes. J Chem Phys 109(12):4981–4984. https://doi.org/10.1063/1.477109
Rappe AK, Casewit CJ, Colwell KS, Goddard WA, Skiff WM (1992) UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J American Chem Soc 114(25):10024–10035. https://doi.org/10.1021/ja00051a040
Mayo SL, Olafson BD, Goddard WA (1990) DREIDING: a generic force field for molecular simulations. J Phys Chem 94(26):8897–8909. https://doi.org/10.1021/j100389a010
Boda D, Henderson D (2008) The effects of deviations from Lorentz-Berthelot rules on the properties of a simple mixture. Mol Phys 106(20):2367–2370. https://doi.org/10.1080/00268970802471137
Willems TF, Rycroft CH, Kazi M, Meza JC, Haranczyk M (2012) Algorithms and tools for high-throughput geometry-based analysis of crystalline porous materials. Microporous and Mesoporous Mater 149(1):134–141. https://doi.org/10.1016/j.micromeso.2011.08.020
Vlugt TJH, García-Pérez E, Dubbeldam D, Ban S, Calero S (2008) Computing the heat of adsorption using molecular simulations: the effect of strong coulombic interactions. J Chem Theory and Comput 4(7):1107–1118. https://doi.org/10.1021/ct700342k
Frost H, Snurr RQ (2007) Design requirements for metal-organic frameworks as hydrogen storage materials. J Phys Chem C 111(50):18794–18803. https://doi.org/10.1021/jp076657p
Snurr RQ, Bell AT, Theodorou DN (1993) Prediction of adsorption of aromatic hydrocarbons in silicalite from grand canonical monte carlo simulations with biased insertions. J Phys Chem 97(51):13742–13752. https://doi.org/10.1021/j100153a051
Martin RL, Haranczyk M (2014) construction and characterization of structure models of crystalline porous polymers. Cryst Growth Des 14(5):2431–2440. https://doi.org/10.1021/cg500158c
Dubbeldam D, Calero S, Ellis DE, Snurr RQ (2016) RASPA: molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol Simul 42(2):81–101. https://doi.org/10.1080/08927022.2015.1010082
El Kassaoui M, Lakhal M, Abdellaoui M, Benyoussef A, El Kenz A, Loulidi M (2020) Modeling hydrogen adsorption in the metal organic framework (MOF-5, connector): Zn4O(C8H4O4)3. Int J Hydrog Energy. https://doi.org/10.1016/j.ijhydene.2020.03.168
Panella, B., Hirscher, M., Roth, S (2005) Hydrogen adsorption in different carbon nanostructures. Carbon 43(10):2209–2214. https://doi.org/10.1016/j.carbon.2005.03.037
Firlej L, Pfeifer P, Kuchta B (2013) Understanding Universal Adsorption Limits for Hydrogen Storage in Nano Porous Systems. Adv Mater 25(41):5971–5974. https://doi.org/10.1002/adma.201303023
Assoualaye G, Djongyang N (2020) Influence of pore size and isosteric heat of adsorption of some metal–organic frameworks on the volumetric and gravimetric adsorption capacities of hydrogen at room temperature. Polym Bull. https://doi.org/10.1007/s00289-020-03350-w
Acknowledgement
The authors are thankful to the developers of the RASPA particularly Dr Randall Snurr and Zeo ++ software with which we made simulations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest in regard with this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Assoualaye, G., Tom, A. & Djongyang, N. Monte carlo study of hydrogen adsorption by MOF-5 doped with cobalt at ambient temperature and pressure. SN Appl. Sci. 2, 1815 (2020). https://doi.org/10.1007/s42452-020-03627-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-03627-9