
Abstract
Eliminating dye mixtures in the aquatic environment has been of considerable interest due to the future sustainability of the environment, hence it needs to utilize photocatalytic material with synergistic influence aimed at efficient removal of these pollutants. Titania nanoparticles (NPs) dispersed on dealuminated clinoptilolite (TiO2–DCP) was synthesized via a sol–gel route. To determine the synergistic influence of as-prepared catalysts on enhanced degradation of dye mixtures (methyl orange—MO and methylene blue—MB), the physicochemical and electrochemical properties were investigated in detail. In comparison to TiO2, the prepared TiO2–DCP photocatalyst possessed excellent photocatalytic activity on the degradation of dye mixtures (83%—MO and 94%—MB) after 180 min of ultraviolet light irradiation. The partial aggregation reduction of titania NPs, improved absorption edge for more photons generations and efficient photogenerated charge carriers separation by DCP catalyst support are crucial factors for TiO2–DCP photocatalyst, synergistic influence on dye mixture removal in this study. The catalytic activity of TiO2–DCP could be reused six reaction times for the removal of the dye mixture with a promising potential for environmental remediation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The indiscriminate discharge of industrial wastewater into the water bodies is a major concern to human health, safety and also the ecosystem [1]. However, industrial wastewater from synthetic dyes is a serious pollutant that affects the ecosystem [2, 3]. These synthetic dyes are made up of aromatic structures with different functional groups attached to them, which makes them very difficult to eliminate in wastewater. The removal of synthetic dyes with aromatic bonds is very paramount before being discharged into the ecosystem [4]. Conventional approaches such as adsorption [5], sedimentation [6, 7], biological methods [8], chlorination, ion exchange [9] and membrane separation [10, 11] have been applied in the elimination of these synthetic dyes discharge. These approaches are limited by their conversion of treated synthetic dye effluents into innocuous substances that requires further purification treatments, while high cost and time consumption also hinder their large scale applications [12, 13]. Heterogeneous photocatalysis under advanced oxidation processes (AOPs) is a promising approach for synthetic dyes removal from water [14], with complete mineralization of pollutants at low cost and ambient temperature conditions [15,16,17,18,19].
Among the semiconductor materials, titanium dioxide (TiO2) is extensively employed as a photocatalyst for the degradation of toxic and recalcitrant pollutants in wastewater due to its chemical stability and nontoxicity [20, 21]. However, the efficiency of pure TiO2 for degradation of organic pollutants is hindered for practical applications due to its high aggregation tendency, the wide bandgap energy (3.2 eV), the fast recombination rate of photogenerated charge carriers, and post-separation from aqueous solution [22,23,24]. To enhance the efficiency of pure TiO2, many studies have been carried out with the incorporation of TiO2 with inert support materials, since heterogeneous catalysis is a surface-based process [25,26,27], that requires adsorption of pollutant by the support material towards the catalyst surface. Support materials utilised for TiO2 dispersion includes silica [28], clay [29], activated carbon [30] glass [31] and zeolites [32, 33]. Zeolites with three-dimensional (3D) framework structures aid the material transparency to light absorption, ability to participate in the electron transfer process, also their availability in natural form (clinoptilolite) and environmentally friendly makes them good support material for TiO2 dispersion [34,35,36]. However, dealumination is an effective approach for improving the physico-chemical properties of natural zeolites [22, 37]. Dealumination contributes largely to removal of amorphous Al-species in the zeolitic framework, thus improving the crystallinity and porosity due to Al elimination [38].
The degradation of dye pollutants employing photocatalyst material is very vital; however, most attention has been focused on a single dye component [39], in comparison to the removal of dye mixtures. As far as we know, the photocatalytic activity of the TiO2 dispersed on modified natural zeolite (clinoptilolite) on the degradation of dye mixtures [40] component has not been reported in the literature before. The purpose of this study is to investigate how the physicochemical and electrochemical properties of titanium dioxide dispersed on dealuminated clinoptilolite (TiO2–DCP) photocatalyst, synergistically influence the degradation of dye mixtures of methylene blue and methyl orange under UV light irradiation. Moreover, the reaction mechanism based on the prepared active photocatalyst properties is also discussed in this study.
2 Experimental
2.1 Materials
Methylene blue (MB, 99%) and methyl orange (MO, 98%) dyes (Fig. 1) were obtained from Sigma Aldrich. Hydrochloric acid (HCl, 36–38%), sodium hydroxide (NaOH), barium sulphate (BaSO4, 99%), Dimethylformamide (DMF, 98%) and sodium sulphate (Na2SO4) were from Merck Company, then used without further purification. Titanium-n-butoxide, ethanol, all solution was prepared using deionized water. The pH of the solution was adjusted with HCl or NaOH solution.

Chemical structure of a methyl orange and b methylene blue
2.2 Synthesis and characterization of catalysts
The synthesis of dealuminated clinoptilolite (DCP) and titanium dioxide on dealuminated clinoptilolite (TiO2–DCP) has been described elsewhere in our previous studies [41]. Dealuminated clinoptilolite (DCP) was prepared via the ion exchange method, while TiO2–DCP was prepared via a sol–gel method. Natural Clinoptilolite (CP) obtained from the Pratley Perlite mining company (South Africa) was crushed in an agate mortar and sieved to less than 100 µm particle size. The micro sieved CP particle was subjected to dealumination, by adding 20 g into 1 M NH4Cl stirred solution at 90 °C for 24 h to achieve the desired ion-exchange process. The resultant mixture was filtered, washed several times with distilled water until the supernatant pH was neutral. The treated CP sample was dried at 150 °C overnight, then calcined in the muffle oven at 550 °C for 8 h required for activation of the dealuminated Clinoptilolite (DCP). For TiO2–DCP synthesis, 1 g of the prepared DCP powder was dispersed in 20 mL aqueous solution of ethanol. A mixture of 10 mL of titanium-n-butoxide in 50 mL ethanol and water solution (1:1) was added drop wise to the stirred solution of DCP mixture, which was further stirred for 3 h at 25 °C. The product was separated, thoroughly rinsed with distilled water, dried at 120 °C overnight and calcined at 550 °C for 4 h.
The powder X-ray diffraction pattern of the TiO2–DCP and TiO2 was examined using Bruker X-ray diffractometer with Cukα source in a 2θ range of 10 to 80º. The morphology and composition of the TiO2–DCP were analyzed using scanning electron microscopy equipped with EDAX system for Energy Dispersive Spectroscopic analysis. The bond vibrations of TiO2–DCP and DCP samples were analyzed on Fourier transform infrared (FTIR) spectrophotometer machine (Perkin Elmer spectrum 400) at the wavelength range of 600–4000 cm−1. Thermogravimetric analysis (TGA) was carried out using Perkin Elmer STA 6000 thermal simultaneous analyzer at a heating rate of 10 °C/min from 30 to 900 °C within 19.8 ml/mins under nitrogen purge stream. The diffuse reflectance spectra of TiO2–DCP and TiO2-samples were recorded using a UV–visible spectrophotometer (PerkinElmer, Lambda 950, USA) in the wavelength range of 200–900 nm. The photocurrent measurements were conducted using Biologic SP 240 potentiostat workstation using a standard three-electrode cell, comprising of a platinum wire as a counter electrode, Ag/AgCl (saturated KCl) as a reference electrode and a (WE). The working electrode was prepared by dispersing the prepared material (5 mg) in a 0.2 mL DMF solution, then 20 µL was drop cast on the WE and 0.2 M Na2SO4 used as the electrolyte solution. First, cyclic voltammetry (CV) was recorded from −0.80 V to 0.20 V in 0.2 M Na2SO4 at a scan rate of 50 mV s−1 to estimate the electrochemical behavior of the prepared photocatalysts. Electrochemical impedance spectroscopy (EIS) was carried out at the open-circuit potential and recorded over a frequency range of 100 kHz to 40 mHz with an ac amplitude of 5 mV.
2.3 Photocatalytic degradation procedure
The photocatalytic activities of the TiO2–DCP, TiO2, and DCP were conducted in an ACE photochemical reactor (Sigma-Aldrich, Z567523) of 1.0 L capacity, 10 cm diameter, 15 cm height, and the light source was a mercury arc lamp (450 W), placed in a 5 cm diameter quartz tube. The wavelength range of the mercury arc lamp through the quartz tube is within the range of 300–600 nm. A fixed volume of 500 mL dye mixtures of MO and MB solution of 40 ppm was used as initial concentration. The dye mixtures solution with the prepared photocatalysts (0.1 g) were magnetically stirred for 60 min in the dark to achieve adsorption–desorption equilibrium, then the light is switch on. The degradation of the dye mixtures was carried out for 180 min under the ultraviolet (UV) light irradiation. 4 mL of the solution was withdrawn from the reactor after designated minutes of irradiation time, centrifuge, filter and the concentration decrease of dye mixtures (MO and MB) was determined using UV–Vis spectrophotometer (Perkin Elmer Lamba 25) at 465 and 665 nm respectively. All analysis were done in triplicates, and the mean values with standard errors were reported. The degradation efficiency (%R) of the photocatalyst on dye mixtures removal was determined with the expression below:
where Co is dye mixtures initial concentration at the beginning of light illumination and Ct concentration at certain reaction time t.
3 Results and discussion
3.1 Characterization
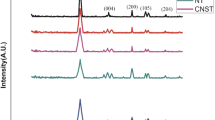
In the XRD pattern, the diffraction planes of DCP in the TiO2–DCP photocatalyst in Fig. 2 are crystalline exhibiting reflections at 2θ values at 13.3°, 17.3°, 23.1°, 30.2°, 32.0° corresponds to diffraction card peak (JCPDS Card No. 025-1349). The diffraction card (JCPDS Card No. 021-1272) of TiO2 anatase peak at 2θ = 25.6°, 38.1°, 48.1°, 53.9°, 55.1°, 62.9° and 75.2°, while the card data (JCPDS Card No. 021-1276) of TiO2 rutile peaks [42] is at 2θ = 27.4° and 44.1° for TiO2–DCP and TiO2 samples in Fig. 2. The average crystallite sizes for the TiO2 and TiO2–DCP were calculated according to the Scherrer equation [43] on the peak at 25.6° of anatase TiO2. According to the equation, TiO2 and TiO2–DCP photocatalyst has a crystallite size of 11.6 and 22.6 nm respectively. The heterojunction between TiO2 and DCP affirm more adhesion and aggregation of TiO2 nanoparticles.

XRD patterns of TiO2–DCP and TiO2
The EDAX spectra (Fig. 3a, b) obtained for the TiO2–DCP catalyst indicates that TiO2 nanoparticles are distributed on the HCP surface, with the partial aggregation of spherical titania nanoparticles. The observed Ti and O peaks were consistent with the formation of TiO2 nanoparticles on the DCP, while peaks of Al and Si represent the DCP. The EDAX spectra confirm the dispersion of titania within the DCP matrix, which restrained agglomeration of TiO2 partially and will contribute to the catalyst higher activity on dye mixtures removal.

EDX spectra of TiO2–DCP
The FTIR spectra of TiO2–DCP and DCP are shown in Fig. 4a, the zeolitic water stretching vibration at 3400 cm−1 was broader with the TiO2–DCP sample as a result of dispersion of titania on the DCP surface [44]. The distorted OH bending of CP is pronounced with TiO2–DCP at 1620–1640 cm−1 which indicated the co-existence and assemblage of TiO2 within the DCP matrix. The sharp absorption peak at 1070 cm−1 is attributed to an asymmetrical stretching vibration band of Si(Al)-O in tetrahedral Si(Al)O4 in DCP [45]. The stretching vibrations of Ti–O–Si and Ti–O–Al appeared at 750 cm−1 were evident for both samples. The slight peak change at 3400 and 1620 cm−1 with TiO2–DCP photocatalyst (due to more surface hydroxyl groups) evidence a firm interaction between the TiO2 and DCP [45,46,47].

a FTIR spectra plot of TiO2–DCP and DCP; b thermal analysis of TiO2–DCP
The thermogravimetric analysis (TGA) pattern of TiO2–DCP is shown in Fig. 4b. The TGA result is coherent with the literature [48] and weight loss occurs at 3 different temperatures, which are attributed to exchangeable cations present in the zeolite and also the presence of volatile organic species. The first major weight loss occurred at 30–200 °C is due to desorption of physisorbed water and hydroxyl species from the HCP cavities [49, 50]. At this weight loss for TiO2–DCP, the decomposition of titanium hydroxide to form TiO2 could be another factor. The weight loss at 200–500 °C is ascribed to exchangeable cations within the HCP and third weight loss at 500–800 °C is ascribed to water present in the silanol nest and the phase transformation of the photocatalyst.
The optical properties of TiO2, and TiO2–DCP shown in Fig. 5a. The TiO2 catalyst has its absorption edge less than 410 nm in the UV-DRS spectra, while the TiO2–DCP sample has an absorption edge at about 420 nm. The incorporation of DCP in the TiO2–DCP show improved light-harvesting ability in the optical spectrum, which is due to their 3D framework structure. The estimated bandgap energy (Ebg) of TiO2 and TiO2–DCP is calculated from the following equation below:
where Ebg is the bandgap energy in eV and λ is the wavelength in nanometres. The bandgap (Ebg) values of TiO2 and TiO2–DCP are 3.08 eV, and 2.94 eV respectively. The shift to the lower energy for TiO2–DCP photocatalyst is very advantageous for more absorption of photons in enhanced degradation of the dye mixtures.

a UV–Vis absorption spectra as-prepared samples and b plot of (αhv)0.5 versus hν (photon energy) for TiO2–DCP and TiO2
PL analysis confirms the charge carrier separation for TiO2 and TiO2–DCP sample catalysts (Fig. 5b). The emission intensity of TiO2–DCP was lower than that of the TiO2 catalyst, indicating that the addition of DCP effectively restrained the recombination rate of photo-induced charge carriers in the TiO2–DCP catalyst. This results in more free reactive species formation for enhanced activity.
The CV of the TiO2 showed the lowest redox current intensity in Fig. 6a. When TiO2 NPs were immobilized on the DCP (TiO2–DCP), the redox current intensity was more enhanced as compared to the TiO2, which means higher catalytic activity and better stability for the TiO2–DCP catalyst. Overall, DCP presence in the composite (TiO2–DCP) facilitates efficient photogenerated charge carriers transfer from the TiO2 conduction band (CB), which invariably accelerates the formation of reactive oxidative species for enhanced photocatalytic activity on dye mixtures removal. The EIS Nyquist plot for TiO2 and TiO2–DCP sample are shown in Fig. 6b. The TiO2–DCP sample has a small arc radius as compared to TiO2 in the EIS plot (Fig. 6b), highlighting a small resistance charge for electron transfer and prevents recombination with holes at the composite interface.

a Cyclic voltammetry and b electrochemical impedance spectroscopy of the as-prepared samples (TiO2–DCP and TiO2)
3.2 Photocatalytic degradation of the dye mixtures
The photocatalytic activities of TiO2–DCP, TiO2, and DCP were evaluated on the degradation of dye mixtures solution under UV light irradiation. The degradation of the dye mixtures with no photocatalyst showed poor efficiency between 27 and 30% after 180 min UV irradiation, highlighting the significant presence of a catalyst required for enhanced degradation. However, TiO2–DCP showed enhanced degradation efficiency on the dye mixtures up to 83% (MO) and 94% (MB) than TiO2, DCP and photolysis alone. The high degradation efficiency of TiO2–DCP is attributed to abundance of surface hydroxy groups in reducing charge carrier seperation [51] (From Fig. 4a analysis), DCP support in reducing the bandgap of TiO2, thus enhancing absorption properties for more generation of reactive oxidative species. The DCP catalyst support (Fig. 6a), also restrain the recombination rate of photoinduced charge carriers, which in turn produces reactive oxidative species for enhanced degradation of the dye mixtures removal.
Figure 7b shows the UV absorption spectra of the dye mixtures solution at different UV light irradiation times with the TiO2–DCP photocatalyst. As depicted in Fig. 7b, the characteristic absorption peak of MO and MB in the dye mixtures significantly stand out at 465 and 665 nm respectively. The MB dye in this study degrades faster than MO with TiO2–DCP under UV light radiation (Fig. 7b), which is similar to other works degrading the dye mixtures at lower concentrations [52, 53]. The absorbance peak height for individual dye in the solution mixture reduced gradually for the UV irradiation time, highlighting the significant breakdown of the aromatic structure of the dye mixtures that occurred during the degradation process. Also, Table 1 highlights the comparison of the performance of TiO2–DCP with various photocatalysts [40, 52, 54,55,56] developed in recent years for the removal of dye mixtures of MO and MB. The catalytic performance of TiO2–DCP is much higher than that of Bi6O6(OH)3 and ZnO photocatalysts as presented in Table 1, while comparable or improved in performance with other photocatalyst as well.

a Photodegradation efficiency of prepared samples on dye mixtures solution; b absorption spectra decrease of the dye mixtures solution with degradation time in the presence of TiO2–DCP. Error bars represent standard deviations of triplicate tests
The plot of − In (Ct/Co) versus the irradiation time for the degradation of dye mixtures in the presence of prepared samples is shown in Fig. 8a. The first-order kinetic model as derived from the Langmuir–Hinshelwood kinetic equation was applied in this study to quantify the degradation efficiency of the samples using the expression below:

a Kinetic fit; b rate constant and half-life values of the prepared samples on the degradation of dye mixtures solution
The rate constant (k) was obtained from the regression analysis of the linear curve (Fig. 8a) in the plot and Fig. 8b shows the calculated values. The calculated rate constant of TiO2–DCP was 0.0093 and 0.0148 min−1 for MO and MB, which was 4.21 and 2.68, 4.31 and 3.40, 7.40 and 4.97 times higher than TiO2, HCP and photolysis alone respectively. The fast reaction rate of TiO2–DCP photocatalyst highlights its excellent removal of the dye mixtures.
Aside from the excellent photocatalytic efficiency of TiO2–DCP, the reusability of the photocatalyst was also studied for other potential applications in the removal of pollutants. The reusability of TiO2–DCP on the degradation of the dye mixtures was carried out for six consecutive cycles. After each cycle, the solution containing the dye mixtures and catalyst were recovered by centrifugation process and then washed with deionized water 3 times. The washed catalyst is then dried at 110 °C overnight and applied for the next cycle. Figure 9a shows the degradation efficiency of the TiO2–DCP catalyst on the dye mixtures after six cycles under UV light irradiation after 180 min. TiO2–DCP catalyst demonstrates a slight reduction in the degradation efficiency on the dye mixtures with an increase in the number of cycles, and at the end of the sixth cycle, 80 and 90% removal was obtained for MO and MB in the dye mixtures. The recycled TiO2–DCP (after six reaction cycles) was analyzed via FTIR spectroscopy to elucidate the effect of recycling hampered the structure of the catalyst. The FTIR spectrum of the recycled TiO2–DCP (Fig. 9b) was very similar to that of the fresh one, highlighting that TiO2–DCP was stable after a repetitive cycle.

a Reusability of TiO2–DCP photocatalyst on the degradation of dye mixtures solution under UV light irradiation for 180 min and b FTIR spectra of recycled and fresh catalyst
3.3 Possible degradation mechanism of TiO2–DCP
The photocatalytic mechanism of the TiO2–DCP catalyst on the dye mixtures degradation can also be deduced from the band structure is presented in Fig. 10. The valence band edge of TiO2–DCP was calculated in line with the concepts of electronegativity [57]. Therefore the band potentials of a semiconductor can be calculated by the following empirical equation below [58]:

The proposed photocatalytic mechanism of TiO2–DCP on the degradation of the dye mixtures
In the equation above, EVB is valence band (VB) edge potential, ECB is a conduction band (CB) edge potential, Eg is the bandgap energy of the semiconductor, X is the electronegativity of the semiconductor, which is the geometric mean of the electronegativity of the constituent atoms, and Ee is the energy of free electrons on the hydrogen scale (about 4.5 eV). The X value of TiO2 is ca. 5.79 eV and the bandgap energy calculated from Taucs plots for TiO2–DCP is 2.94 eV, while the EVB and ECB of TiO2–DCP are estimated to be 2.71 eV and − 0.23 eV respectively. Based on the estimated values of EVB and ECB for TiO2–DCP, the reaction mechanism is proposed and also described by Eqs. (5) to (10). The dye mixtures solution is first adsorbed onto the surface of the TiO2–DCP catalyst. Then upon illumination with the UV light, TiO2 absorbs sufficient light photon with energy (hʋ) equal to its bandgap, which invariably results in the creation of electron–hole pairs. The excited electrons move from the valence band (VB) into the conduction band (CB), while the holes are left behind in the VB as shown in Fig. 10. The generation of superoxide radical (O ∙−2 ) is not allowed because the potential of the electrons at the CB of TiO2 is not negative enough than the redox potential of O2/O ∙−2 (− 0.33 eV). The excited electrons are also immediately transferred to DCP, here DCP in this system acts as an electron transporter, due to its high electron mobility of zeolites framework [59]. The transfer of the photogenerated electrons to the DCP framework occurs in femtoseconds, where a continuous transfer of electrons from the metal–semiconductor to the support material keeps the electrons away from TiO2 to avoid recombination with the hole. This excited electrons on HCP molecular surface are been trapped by O2 molecules in the aqueous solution to produce O ∙−2 radicals which attacks the dye mixtures. Furthermore, considering the potential of holes at the VB of TiO2 (2.79 eV) is more positive than the redox potential of OH−/∙OH (2.38 eV), the holes can easily oxidize H2O/OH− into hydroxyl (∙OH) radicals, which is another key radical species in oxidizing the dye mixtures (Fig. 10). Invariably, both O ∙−2 and OH∙ radicals generated from TiO2–DCP degrade the dye mixtures into CO2, H2O, and other products.
Overall, the abundance of active sites, reduced bandgap of TiO2 upon introduction of DCP as the catalyst support, also its ability (DCP) to transport and separate photoexcited charge carriers from TiO2 are all crucial physicochemical and electrochemical properties of TiO2–DCP with synergistic influence on enhanced degradation of dye mixtures in this present study.
4 Conclusions
Sol–gel route was employed for the synthesis of TiO2–DCP photocatalyst, its synergistic influence on catalytic applications based on physicochemical and electrochemical properties was evaluated. For this purpose, the degradation of dye mixtures (MO and MB) under UV light irradiation was investigated. Results from this study establish the synergistic influence of partial aggregation, increased crystallite size, extended absorption edge and higher separation efficiency of photogenerated charge carriers for the formation of reactive oxidative species, evidenced the enhanced catalytic activity of TiO2–DCP in comparison to TiO2 and HCP on the degradation of dye mixtures (83%—MO and 94%—MB). The kinetics study of dye mixtures photodegradation follows the pseudo-first-order reaction model. The TiO2–DCP photocatalyst had good reusability and stability for practical applications after six reaction cycles (as evidenced by FTIR spectra). The reaction mechanism for the TiO2–DCP photocatalyst with excellent photocatalytic activity was proposed via band structure analysis and elucidated in this study. The excellent activity of TiO2–DCP nanocomposite reveals a promising photocatalyst for various pollutants removal from wastewater.
References
Tonlé IK, Ngameni E, Tcheumi HL, Tchiéda V, Carteret C, Walcarius AJ (2008) Sorption of methylene blue on an organoclay bearing thiol groups and application to electrochemical sensing of the dye. Talanta 74:489–497
Chen C, Lu C (2007) Photocatalytic degradation of basic violet 4: degradation efficiency, product distribution, and mechanisms. J Phys Chem C 111:13922–13932
Mu Y, Rabaey K, Rozendal RA, Yuan Z, Keller J (2009) Decolorization of azo dyes in bioelectrochemical systems. Environ Sci Technol 43:5137–5143
Joseph S, Mathew B (2015) Microwave-assisted green synthesis of silver nanoparticles and the study on catalytic activity in the degradation of dyes. J Molec Liq 204:184–191
Houas A, Bakir I, Ksibi M, Elaloui E (1999) Étude de l’élimination de bleu de méthylène dans l’eau par le charbon actif commercial CECA40. J Chim Phys Phys Chim Biol 96:479–486
Hammami S, Bellakhal N, Oturan N, Oturan MA, Dachraoui M (2008) Degradation of acid orange 7 by electrochemically generated ∙OH radicals in acidic aqueous medium using a boron-doped diamond or platinum anode: a mechanistic study. Chemosphere 73:678–684
Barrera-Dıˇıaz C, Linares-Hernaˇındez I, Roa-Morales G, Bilyeu B, BalderasHernaˇındez P (2009) Removal of biorefractory compounds in industrial wastewater by chemical and electrochemical pretreatments. Ind Eng Chem Res 48:1253–1258
More AT, Vira A, Fogel S (1989) Biodegradation of trans-l,2-dichloroethylene by methaneutilizing bacteria in an aquifer simulator. Environ Sci Technol 23:403–406
Slokar YM, Le Marechal AM (1998) Methods of decoloration of textile wastewaters. Dyes Pigm 37(4):335–356
Wu J-S, Liu C-H, Chu KH, Suen S-Y (2008) Removal of cationic dye methyl violet 2B from water by cation exchange membranes. J Memb Sci 309:239–245
Damodar RA, You S-J, Ou S-H (2010) Coupling of membrane separation with photocatalytic slurry reactor for advanced dye wastewater treatment. Sep Purif Technol 76:64–71
Arslan I, Balcioglu IA, Tuhkanen T, Bahnemann D (2000) H2O2/UV-C and Fe2+/H2O2/UV-C versus TiO2/UV-A treatment for reactive dyewastewater. J Environ Eng 126:903–911
Stock N, Peller J, Vinodgopal K, Kamat PV (2000) Combinative sonolysis and photocatalysis for textile dye degradation. Environ Sci Technol 34:1747–1750
Jiang Z, Wang M, Cheng H, Li J, Husnu A, Lv H, Yao Y, Shao L, Huang Y, Dong MJ (2015) Facile preparation of TiO2 nanoclusters on graphene templates for photodegradation of organic compounds. J Mater Sci Technol 31:840–844
Lergini O, Oliveros E, Barun AM (1993) Photochemical processes for water treatment. Chem Rev 95(93):671–710
Uygur A (1997) An overview of oxidative and photooxidative decolorisation treatments of textile waste-waters. J Soc Dyers Colour 113:211–217
Guillard C, Lachheb H, Honas A, Ksibi M, Herrmann JM (2003) Influence of chemical structure of dyes, of pH and of inorganic salts on their photocatalytic degradation by TiO2 comparison of the efficiency of powder and supported TiO2. J Photochem Photobiol A Chem 158:27–36
Robert D (2007) Photosensitization of TiO2 by MxOy and MxSy nanoparticles for heterogeneous photocatalysis applications. Catal Today 122:20–26
Aleksić M, Kušić H, Koprivanac N, Leszczynska D, Božić AL (2010) Heterogeneous Fenton type processes for the degradation of organic dye pollutant in water—the application of zeolite assisted AOPs. Desalination 257(1–3):22–29
Park Y, Lee SH, Kang SO, Choi WY (2010) Organic dye-sensitized TiO2 for the redox conversion of water pollutants under visible light. Chem Commun 46:2477–2479
Liu ZY, Zhang XT, Nisimoto S, Murakami T, Fujishima A (2008) Efficient photocatalytic degradation of gaseous acetaldehyde by highly ordered TiO2 nanotube arrays. Environ Sci Technol 42:8547–8551
Guesh K, Márquez-Álvarez C, Chebude Y, Díaz I (2016) Enhanced photocatalytic activity of supported TiO2 by selective surface modification of zeolite Y. Appl Surf Sci 378:473–478
Dong H, Zeng G, Tang L, Fan C, Zhang C, He X, He Y (2015) An overview on limitations of TiO2-based particles for photocatalytic degradation of organic pollutants and the corresponding countermeasures. Water Res 79:128–146
Sanni S, Viljoen E, Ofomaja A (2019) Accelerated electron transport and improved photocatalytic activity of Ag/AgBr under visible light irradiation based on conductive carbon derived biomass. Catal Lett 149:3027–3040
Takeda N, Torimoto T, Sampath S, Kuwabata S, Yoneyama H (1995) Effect of inert supports for titanium dioxide loading on enhancement of photodecomposition rate of gaseous propionaldehyde. J Phys Chem B 99:9986–9991
Takeda N, Iwata N, Torimoto T, Yoneyama H (1998) Influence of carbon black as an adsorbent used in TiO2 photocatalyst films on photodegradation behaviors of propyzamide. J Catal 177:240–246
Fukahori S, Ichiura H, Kitaoka T, Tanaka H (2003) Photocatalytic decomposition of bisphenol A in water using composite TiO2–zeolite sheets prepared by a papermaking technique. Environ Sci Technol 37:1048–1051
Vohra MS, Tanaka K (2003) Photocatalytic degradation of aqueous pollutants using silica-modified TiO2. Water Res 37:3992–3996
Xu Y, Langford CH (1997) Photoactivity of Titanium dioxide Supported on MCM-41, Zeolite X, and Zeolite Y. J Phys Chem B 101:3115–3121
Tryba B, Morawski AW, Inagaki M (2003) Application of TiO2-mounted activated carbon to the removal of phenol from water. Appl Catal B Environ 41:427–433
Yazawa T, Machida F, Kubo N, Jin T (2009) Photocatalytic activity of transparent porous glass supported TiO2. Ceram Int 35:3321–3325
Shankar MA, Anandan S, Venkatachalam N, Arabindoo B, Murugesan V (2006) Fine route for an efficient removal of 2,4-dichlorophenoxyacetic acid (2,4-D) by zeolite-supported TiO2. Chemosphere 63(6):1014–1021
Shimizu K, Kaneko T, Fujishima T, Kodama T, Yoshida H, Kitayama Y (2002) Selective oxidation of liquid hydrocarbons over photoirradiated TiO2 pillared clays. Appl Catal A Gen 225:185–191
Chen HM, Matsumoto A, Nishimiy N, Tsutsumi K (1999) Preparation and characterization of TiO2 incorporated Y-zeolite. Coll Surf A Physicochem Eng Aspects 157(157):295
Liu X, Iu K-K, Thomas JK (1993) Photophysical properties of pyrene in zeolites. Observation of pyrene anion radicals in zeolites x and y. Chem Phys Lett 204:163–167
Alzeer MI, MacKenzie KJ, Keyzers RA (2017) Facile synthesis of new hierarchical aluminosilicate inorganic polymer solid acids and their catalytic performance in alkylation reactions. Microp Mesop Mat 241:316–325
Sene RA, Sharifnia S, Moradi G (2018) On the impact evaluation of various chemical treatments of support on the photocatalytic properties and hydrogen evolution of sonochemically synthesized TiO2/Clinoptilolite. Int J Hydrog Ener 43:695–707
You SJ, Park ED (2014) Effects of dealumination and desilication of H-ZSM-5 on xylose dehydration. Microp Mesop Mat 186:121–129
Verma P, Samanta SK (2018) Facile synthesis of TiO2–PC composites for enhanced photocatalytic abatement of multiple pollutant dye mixtures: a comprehensive study on the kinetics, mechanism, and effects of environmental factors. Res Chem Intermed 44:1963–1988
Tariq MK, Riaz A, Khan R, Wajid A, Haq H-U, Javed S, Akram MA, Islam M (2019) Comparative study of Ag, Sn or Zn doped TiO2 thin films for photocatalytic degradation of methylene blue and methyl orange. Mater Res Exp 6:106435
Saheed SO, Modise SJ, Sipamla AM. TiO2 supported clinoptilotile: characterization and optimization of operational parameters for methyl orange removal. In: Adv Mater Res Trans Tech Publ 2249–2252
Hsien YH, Chang CF, Chen YH, Cheng S (2001) Photodegradation of aromatic pollutants in water over TiO2 supported on molecular sieves. Appl Catal B Environ 31:241–249
Pasandideh EK, Kakavandi B, Nasseri S, Mahvi AH, Nabizadeh R, Esrafili A, Kalantary RR (2016) Silica-coated magnetite nanoparticles core-shell spheres (Fe3O4@SiO2) for natural organic matter removal. J Environ Health Sci Eng 14:21
Qian T, Yin X, Li J, Xu H, Deng Y, Wang X (2017) Nano-TiO2 decorated radial-like mesoporous silica: preparation, characterization, and adsorption-photodegradation behavior. J Mater Sci Technol 33:1314–1322
Mehrabadi Z, Faghihian H (2018) Elimination of highly consumed herbicide; 2, 4-dichlorophenoxyacetic acid from aqueous solution by TiO2 impregnated clinoptilolite, study of degradation pathway. Spectrochim Acta Part A Molec Biomolec Spectro 204:248–259
Rasalingam S, Peng R, Koodali RT (2014) Removal of hazardous pollutants from wastewaters: applications of TiO2–SiO2 mixed oxide materials. J Nanomater 617405
Moshoeu DE, Sanni SO, Oseghe EO, Msagati TA, Mamba BB, Ofomaja AE (2020) Morphological influence of TiO2 nanostructures on charge transfer and tetracycline degradation under LED light. Chem Select 5:1037–1040
Nezamzadeh-Ejhieh AM, Moeinirad S (2011) Heterogeneous photocatalytic degradation of furfural using NiS-clinoptilolite zeolite. Desalination 273:248–257
Garcia- Basabe Y, Rodriguez-Iznaga I, Menorval LCD, Llewellyn P, Maurin G, Lewis DW, Binions R, Autie M, Ruiz-Salvador AR (2010) Step-wise dealumination of natural clinoptilolite: structural and physicochemical characterization. Micropor Mesopor Mater 135(135):187
Castaldi P, Santona L, Cozza C, Giuliano V, Abbruzzese C, Nastro V, Melis P (2005) Thermal and spectroscopic studies of zeolites exchanged with metal cations. J Molec Struct 734:99–105
Pham XN, Pham DT, Ngo HS, Nguyen MB, Doan HV (2020) Characterization and application of C–TiO2 doped cellulose acetate nanocomposite film for removal of Reactive Red-195. Chem Engineer Commun 1–14
Khan MM, Lee J, Cho MH (2014) Au@ TiO2 nanocomposites for the catalytic degradation of methyl orange and methylene blue: an electron relay effect. J Ind Eng Chem 20:1584–1590
Nezamzadeh-Ejhieh A, Karimi-Shamsabadi M (2014) Comparison of photocatalytic efficiency of supported CuO onto micro and nano particles of zeolite X in photodecolorization of Methylene blue and Methyl orange aqueous mixture. Appl Catal A Gen 477:83–92
Saravanan R, Sacari E, Gracia F, Khan MM, Mosquera E, Gupta VK (2016) Conducting PANI stimulated ZnO system for visible light photocatalytic degradation of coloured dyes. J Molec Liq 221:1029–1033
Wang S, Teng F, Zhao Y (2015) Effect of the molecular structure and surface charge of a bismuth catalyst on the adsorption and photocatalytic degradation of dye mixtures. RSC Adv 5:76588–76598
Saravanan R, Thirumal E, Gupta V, Narayanan V, Stephen A (2013) The photocatalytic activity of ZnO prepared by simple thermal decomposition method at various temperatures. J Molec Liq 177:394–401
Nethercot AH Jr (1974) Prediction of Fermi energies and photoelectric thresholds based on electronegativity concepts. Phys Rev Lett 33:1088
Cui W, Li X, Gao C, Dong F, Chen X (2017) Ternary Ag/AgCl–(BiO)2CO3 composites as high-performance visible-light plasmonic photocatalysts. Catal Today 284:67–76
Anandan S, Yoon M (2003) Photocatalytic activities of the nano-sized TiO2-supported Y-zeolites. J Photochem Photobiol C Photochem Rev 4:5–18
Acknowledgements
The authors gratefully acknowledge the financial support of the National Research Foundation of South Africa (Grant No. 99330 and 111330), the Sasol University Collaboration Program and the Vaal University of Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sanni, S.O., Modise, S.J., Viljoen, E.L. et al. Enhanced degradation of dye mixtures: physicochemical and electrochemical properties of titania dispersed on clinoptilolite, synergistic influence. SN Appl. Sci. 2, 1668 (2020). https://doi.org/10.1007/s42452-020-03398-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-03398-3