
Abstract
Phosphorus (P) exchange in clayey mud sediments was investigated with desorption–sorption isotherms at 22 coastal sites in the eutrophied brackish Gulf of Finland, the Baltic Sea. The aim was to gain information on P sorption potential and pattern of oxygenated surface sediments and to elucidate factors explaining them. Modified Freundlich equation was fitted to the isotherm data and used for calculation of the P exchange parameters EPC0 (equilibrium P concentration at zero net sorption), kEPC0, k15 and k100 (P buffering capacities at early, middle and high sorption stages, respectively). They were further used to assess the P sorption potential and pattern of the sediments. Sediment properties explaining the P sorption potential at different sorption stages were identified. The greatest sorption potentials were recorded in originally poorly oxic sediments in the inner archipelago with accumulated fine particulate material, high in adsorbents for P, transported from the watersheds. After oxygenation, they were high in amorphous Fe-oxyhydroxides, which explained their efficient P sorption (kEPC0 > 1.32 l g−1). Poorer P sorption (kEPC0 0.45–0.84 l g−1) was recorded in fine sediments abundant in Al-oxyhydroxides, presumably due to their higher original P occupation degree. The lowest sorption potentials were found in the outer archipelago sediments. Their lowest specific surface areas and highest original P contents referred to scarcity of adsorbents and high occupation degree of the existing ones. These results suggest that the coastal clayey mud sediments investigated possess high P sorption potential, which can be markedly diminished by eutrophication-induced hypoxia but recovered if oxygen conditions improve.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Excess of anthropogenic nutrients such as phosphorus (P) causes eutrophication in aquatic ecosystems worldwide, particularly in coastal marine areas [1]. The negative impacts of eutrophication include enhanced primary production and harmful algal blooms. The subsequent enhanced sedimentation of organic matter (OM) and its mineralization can lead to hypoxia and to decrease in biodiversity. Hypoxia further fuels the eutrophication by reducing iron (Fe) in Fe-oxyhydroxides, which results in release of P bound onto their surfaces [2, 3].
Coastal sediments receive P loads from watersheds via rivers mainly as suspended solids. Worldwide, ca. 95% of riverine P is associated with particles [4], the rest being in dissolved form. Similar shares are reported for the portion of particulate P (73–94%) in field runoff waters in the southern Finland [5]. Estuaries are considered as filtration areas for nutrients transported from land to coastal seas [6]. Sedimentation of riverine loads near the coastline increases due to hindered flow velocity. Furthermore, ionic strength being higher in marine than in river water enhances the flocculation of colloidal clayey particles rich in Fe- and Al-oxyhydroxides and OM [7,8,9].
In the sediments, the fate of dissolved and particulate P is highly affected by the conditions at the sea floor. Microbial mineralization of OM and reduction of Fe-oxyhydroxides under hypoxic conditions can transform particulate P into dissolved organic and inorganic forms [2, 10]. The dissolved inorganic P and some of the dissolved organic P forms are directly bioavailable [11, 12]. In undisturbed sediments, the soluble P in pore water diffuses towards lower concentrations in the overlying water [13]. Water movements (surface waves, bottom currents), ebullitions of gases (e.g. hydrogen sulphide or methane in reduced environments) and bioturbation can mix the sediment structure and cause resuspension, which further promote the P release [14, 15]. The properties of surface sediment, such as the presence of Fe- and Al-oxyhydroxides as well as Fe and Al associated with OM, influence the release of dissolved inorganic and organic P from the sediment [see, e.g. 16–18]. Under reduced conditions, however, the disruption of Fe(III)-oxyhydroxides lowers the P retention ability of the sediment surface, whereas the oxyhydroxides of Al are not affected [19].
Phosphorus retention onto and release from oxyhydroxides are dynamic ligand exchange reactions [16]. They continuously aim to maintain an equilibrium between P in the solid and the liquid phases. This means that P retention onto oxyhydroxides is ultimately regulated by the P saturation degree of sorption surfaces and the P concentration in the surrounding (ambient) water (i.e. pore water and water above the sea bottom). When the P concentration in the solution decreases (e.g. due to microbial uptake, diffusion- or mixing-induced dilution), some P adsorbed onto sediment surface will be released to water phase to maintain the equilibrium [9]. This means that some desorption of P from the sediments also occurs regardless of the prevailing oxygen (O2) conditions, unless P is very tightly bound, occluded inside the structure of Fe-oxyhydroxides [20].
Sorption of P in soils and aquatic sediments has been extensively studied using isotherms [21,22,23,24]. They are executed by equilibrating certain amounts of solids in solutions with varying P concentrations. Thereafter, the P concentration in the solution is measured and P desorbed from or sorbed by the sediment is calculated on the basis of the change in its concentration in the solution. The desorbed/sorbed P is presented in an isotherm graph as a function of measured equilibrium P concentration. Freundlich equation and its modifications are often used for empirical curve fitting for the P sorption [25, 26] or desorption–sorption data [27] as they describe multilayer sorption on heterogeneous surface, without taking any stand on the reaction mechanism [28].
The Baltic Sea, a large practically non-tidal [29] brackish water basin in the northern Europe, has suffered for decades from the impacts of eutrophication, Gulf of Finland (GoF) being one of its most heavily loaded basins [30]. Exchange of P is extensively investigated in calcium (Ca)-rich sediments in eutrophied coastal areas [31,32,33], but there are only few small-scale studies performed with non-calcareous sediments, such as those in the northern Baltic Sea [34, 35]. Sediments in the GoF vary substantially in terms of the extent and chemical composition of the sea floor P storage [36, 37] and, thus, presumably also in their P exchange patterns. During the last decade, various efforts have been made to diminish nutrient loading to the Baltic Sea [38]. Reduced loading will eventually lower the P concentrations in the water systems, which may further emphasize the role of sediments as a source of bioavailable P, also in oxic conditions.
Information on the P exchange properties of the coastal surface sediments is needed to understand their potential contribution to P sorption and their possible ability to act as sources of internal P loading. This study was undertaken to examine the P exchange properties of non-calcareous muddy clay sediments along the northern coast of the eutrophied GoF, the Baltic Sea. Our aim was to get information on the variation in P sorption potential and pattern of oxidized sediments in the inner and the outer archipelago to understand their contribution to P binding and exchange. We hypothesized that sediments collected from different sites in the archipelago differ in their P sorption potential and pattern and that these differences are related to varying properties of the sediments. This study provides new information on the wide variation in P sorption potential, and the P exchange parameters describing it, in the muddy clay coastal sediments in a eutrophied sea. The results highlight the complex impact of sediment environment on the P exchange properties of the sediments.
2 Materials and methods
2.1 Research area and sediment sampling
The GoF (60°N, 25°E) is a shallow brackish water bay with mean depth of 37 m, surface area of 29,600 km2 and catchment of 413,000 km2. Salinity of the bottom water ranges from ca. 6 to 10, and the average residence time of water in the bay is about 3 years [39]. In 2010, total phosphorus (TP) input to the GoF was about 6500 t [40] and the internal loading further supplies P to the water [41]. Bedrock in the northern archipelago of the GoF consists of Paleoproterozoic crystalline basement rocks. Due to complex and fragmented sea floor, many small semi-enclosed basins are characteristic in this area [42, 43].
Sediments were collected from 22 sites using a Gemax gravity twin corer (Ø 90 mm, length 60 cm) by Geological Survey of Finland in the autumn (Sept–Nov) 2002 during the cruises of R/V Kaita. The sampling sites #1–13 are located in the inner and the sites #14–22 in the outer archipelago (Fig. 1). Underwater camera was used to identify the prevailing bottom conditions. One core was sectioned into 1-cm intervals, and the parallel one was vertically split for visual assessment of oxygenation status by measuring thickness of the light brown oxic surface layer. The sediments having 1.5–3-cm-thick light brown surface were considered originally oxic (Table 1). The remaining ones with a thinner light brown surface were considered poorly oxic. The samples were immediately frozen after sampling (− 20 °C) and kept frozen until freeze-dried (− 60 °C) within a few months after sampling. The freeze-dried samples were stored in plastic containers in dark at room temperature prior to the analyses. All the sediments were assumed to become oxidized during the sampling, sample handling and storing. Total contents of various elements were analyzed in December 2003. All the other analyses were carried out during the years 2011–2012 from the combined 2-cm surface layers.

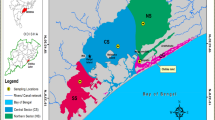
Location of the sampling sites in the inner (#1–13; circle) and the outer (#14–22; diamond and cross) archipelago in the northern Gulf of Finland, the Baltic Sea
Site-specific exposure index data were obtained from the Finnish Environment Institute and used as one parameter describing the sampling sites. The index (called hereafter the “exposure index”) was calculated from a simplified wave model, for detailed information see studies of Isaeus and Wijkmark and Isaeus [44, 45]. It describes the susceptibility of a certain sampling site to wind-induced water mixing, which at shallow sites predicts susceptibility of the sediments for resuspension. The exposure index values are based on parameters that describe mean wind speed of a certain area and the distance of open water over which the wind can develop waves [45]. Generally, the value is small in sheltered locations, i.e. in the places where an obstacle like an island hampers the wind blowing and thus the formation of waves. In the open locations where the wind can blow unrestricted for long distances, the value is high. The exposure index, water depth and bottom water salinity at the sampling sites are presented in Table 1.
2.2 Determination of physical and chemical properties of the sediments
Water content of the sediments (Table 1) was determined as weight loss during freeze-drying. Specific surface area (SSA) of the sediments was determined by nitrogen gas (N2) adsorption method (PANK2401, Finnish Pavement Technology Advisory Council) to describe the total surface area of the particles potentially available for sorption reactions. Total carbon (TC) and nitrogen (TN) in the sediments were determined with LECO CNH-600 analyzer, and carbon-to-nitrogen ratio (C:N) was calculated on the basis of their total contents. Since the sediments in the study area are typically poor in carbonate minerals [46], TC was used as an estimate for OM. Total contents of phosphorus (TP), iron (TFe), aluminium (TAl), manganese (TMn), calcium (TCa) and sulphur (TS) were analyzed using inductively coupled plasma atomic emission spectroscopy (ICP-AES) after digestion with hydrofluoric acid—perchloric acid.
2.3 Sequential extraction of Fe-, Al- and Mn-oxyhydroxides
A three-step sequential extraction was performed to determine the amount of oxyhydroxides of Fe, Al as well as Mn potentially participating in P sorption (Table 2). The first extractant, ascorbate solution buffered with ascorbic acid to pH 8.0 (Asc, Table 2), was targeted to dissolve amorphous easily reducible Fe-oxyhydroxides such as ferrihydrite [47]. The second extractant, sodium dithionite buffered with sodium bicarbonate to pH 7.0 (Dith; Table 2), was assumed to extract amorphous Fe-oxyhydroxides other than those dissolved by ascorbate [48]. Acid ammonium oxalate (Oxal, pH 3; Table 2) used in the third step was mainly targeted to extract amorphous Al-oxyhydroxides [49], as most of the amorphous Fe-oxyhydroxides were considered to have dissolved during the first and the second steps. However, oxalate is also shown to dissolve Fe and Al from organic complexes [50] as well as Fe from crystallized oxyhydroxides [47] and from FeS compounds [51]. Thus, the results of the extraction procedure are operationally defined and not expected to represent accurate phases.
For the extraction, duplicate 0.3 g (dry weight, dw) portions of sieved (Ø 2 mm) sediments were subsequently shaken in 30 ml of each extractant (2 h, 200 rpm, 22 °C). During the last extraction step with oxalate, the samples were protected from light. After each step, the mixtures were centrifuged for 15 min at 3000 rpm and filtered (0.40 µm, Whatman, Nuclepore Polycarbonate, PC, membrane filter). All the filtrates in the metal oxyhydroxide extractions were acidified (pH < 2) with concentrated nitric acid (65% HNO3, Suprapur, Merck®) for preservation prior to analyses using an inductively coupled plasma optical emission spectroscopy ICP-OES. The total amount of extracted Fe (Fe∑Ext), Al (Al∑Ext) and Mn (Mn∑Ext) was calculated as a sum of the molar concentration of each element in the extractants.
2.4 Phosphorus exchange isotherms
Two or three replicates of 0.3 g (dw) sieved (Ø 2 mm) sediment samples were weighed into 100 ml acid-washed polypropylene centrifuge tubes, rewetted to their ambient water content using Milli-Q water and allowed to moisten overnight (~ 17 h) on an orbital shaker (50 rpm, dark, + 5 °C). Rewetted sediments were equilibrated (200 rpm, dark, + 5 °C) for 48 h in 60 ml of artificial sea water (ASW) containing 0, 5, 10, 15, 20, 50, 75 or 100 µmol P l−1 as K2HPO4. The ASW was diluted from the stock solution (210 ppt) to correspond the site-specific bottom water salinity. The ASW stock consisted of sodium chloride (NaCl; 190 g l−1), magnesium sulphate (MgSO4; 61 g l−1) and sodium bicarbonate (NaHCO3; 1.2 g l−1) in Milli-Q water (modified from [52]). The P addition range was chosen to represent the near-bottom and pore water P concentrations in the GoF [36]. Two replicate blank samples of each P addition level were included in the experiments. Solid-to-solution ratio 1:200 (corresponding to particle concentration 5 g dw l−1) was used, because our previous studies revealed a risk of underestimation of desorbable P when using a sediment-to-solution ratio greater than that (Vaalama unpublished data).
After equilibration, the samples were centrifuged (3000 rpm, 15 min, 22 °C) and filtered (0.40 µm Nuclepore PC membrane filter, Whatman). The filtrates were analyzed for phosphate (PO4–P) using molybdenum blue method [53] and a spectrophotometer (Genesys 10 uv, Thermo Spectronic equipped with a 5-cm flow-injection cuvette; detection limit of 0.05 µmol P l−1; Thermo Fisher Scientific, Waltham, MA, USA). Control solutions containing 0.42, 4.2 and 10.5 µmol P l−1 were prepared from a commercial stock solution (Merck®, 1000 mg P l−1) and routinely included in the P analyses to assure the measurement quality. The pH of the centrifuged extracts obtained at P additions 0 and 100 µmol l−1 (P0 and P100, respectively) was measured prior to filtration. To investigate the potential impact of silicate (SiO4–Si) on P sorption, the corresponding filtrates were frozen and analyzed later spectrophotometrically for SiO4–Si (referred later as Si–P0 and Si–P100) carried out using a Lachat QuickChem 8000 autoanalyzer (Lachat Instruments, Hach Co., Loveland, CO, USA) and a method [54].
2.5 Phosphorus exchange parameters
The amount of P sorbed onto or desorbed from the sediments was calculated as a change of P concentration in the solution and plotted as a function of the P concentration in the equilibrium solution (Fig. 2). Modified Freundlich equation Q = Q0 + aIb was fitted to the data, and the isotherm graphs were drawn using SigmaPlot 10.0 (Systat Software Inc., Chicago, IL, USA). In the equation, Q stands for the quantity of P sorbed onto or desorbed from the sediment surface and I for the P concentration in the equilibrium solution. The parameter a is a Freundlich constant and b describes the measure of nonlinearity involved [55], while Q0 is a theoretical estimate for the total amount of desorbable P in the sediments [56, 57].

Graphical illustration of P exchange isotherm and the parameters EPC0 (µmol l−1), kEPC0, k15 and k100 (l g−1) calculated from the Freundlich equation. Intensity (I on the x-axis) represents the equilibrium P concentration (µmol l−1), quantity (Q on the y-axis) the amount of P desorbed from or sorbed onto the sediments (µmol g−1) and Q0 the amount of potentially desorbable P in sediments
The P exchange parameters EPC0, kEPC0, k15 and k100 were calculated from the Freundlich equation to study the differences among the isotherms (Fig. 2). Equilibrium P concentration EPC0 (i.e. I at Q = 0; µmol l−1) stands for the crossover P concentration at which no P release from or sorption to sediment occurs [22]. The kEPC0 (l g−1) is the slope of the tangent fitted to the isotherm equation at EPC0, calculated as Kd in [31]. It describes the P equilibrium buffering capacity [22] at early sorption stage. The k15 and k100 (l g−1) are calculated slopes of the tangents at two observation points Q1/I1 and Q2/I2 that stand for the P additions 15 and 100 µmol l−1, respectively (Fig. 2). The k15 describes the sediment P buffering capacity at the middle sorption stage (i.e. at the point where, in most of the studied sediments, the sorption isotherms turned from a linear to a bending sorption pattern). The k100 describes the P buffering capacity at the high sorption stage at the end of the isotherm (i.e. when the sorption weakens as a result of increased P saturation degree of the sediment surface). The amount of P sorbed by the sediments at the middle and at the high sorption stages was calculated as percentages from the P addition levels 15 and 100 µmol l−1 (S%P15 and S%P100, respectively). Finally, the amount of P released from the sediments in equilibration with P-free ASW (desorbable P; µmol l−1) was used as an estimate for the easily exchangeable, loosely bound P in the sediments.
However, in the sediment #21 the Freundlich equation did not converge properly, and the equation parameters Q0, a and b resulted in calculated EPC0 that deviated markedly from the P exchange data. Therefore, EPC0 for this sediment was determined from the linear part of the isotherm using linear fitting (Q = kI + b), while the other P exchange parameters were calculated from the Freundlich equation.
2.6 Statistical analysis
Spearman correlation was used to investigate the statistically significant relations within the variables in the dataset. Differences between distinguished sediment groups were studied using either the nonparametric Mann–Whitney U or Kruskal–Wallis test (non-normal and homoscedastic data) and Kolmogorov–Smirnov test (non-normal and heteroscedastic data). Hierarchical cluster analysis was used to classify the sediments according to their P sorption pattern using the P exchange parameters (EPC0, kEPC0, k15 and k100) and the sorption percentages (S%15 and S%100) as variables. Non-normal variables were log-transformed prior to analyses. Discriminant analysis was used to discover the possible multivariate effect of different factor combinations on the single P exchange parameters describing the early (EPC0, kEPC0) and the middle (k15) sorption stages. For the analysis, each parameter was divided into groups: “high”, “moderate” and “low” according to their values. Outliers of the predictor variables were identified using box plots and replaced by the mean value of the variable in question. The qualification of the analysis was ensured by equal log determinants and similar covariance matrices (Box’s M test, p > 0.05). The statistical tests were performed using R 3.5.2 (2018-12-20, package “Hmisc” for the correlation analysis) and SPSS (IBM SPSS Statistics 24) programs, with significance level p = 0.05.
3 Results
3.1 Physical properties and content of total elements in the sediments
As for the sampling sites, five oxic and eight poorly oxic sites were located in the inner and five oxic and four poorly oxic ones in the outer archipelago. The pH measured from P0 isotherm extracts was significantly higher in the originally oxic (6.9–7.3) than in the originally poorly oxic sediments (5.6–7.2) (D = 0.667, p ≤ 0.04) and higher in P0 than in P100 solutions (results not shown). At the inner archipelago, the underwater camera recorded in situ gas ebullitions (i.e. bubbling) that resuspended the sediment surface at the sites #6, 9, 10 and 12. The sediment at the site #8 contained plenty of gas holes, while laminated sediments were recorded at the site #3 in the inner and at the sites #14 and 17 in the outer archipelago. The SSA of the sediments varied from 2.4 to 26.3 m2 g−1, being significantly higher in the inner than outer archipelago (W = 107.5, p < 0.001).
Content of TC ranged from 811 to 6432 µmol g−1 and that of TN from 132 to 828 µmol g−1 (Fig. 3a, b). They correlated positively with each other and TS (Online Resource 1; Table 1). Poorly oxic sediments were somewhat higher in TC and TS (average ± sd, 4440 ± 1290 and 181.5 ± 77.5 µmol g−1, respectively) than the originally oxic ones (3720 ± 1420 and 122.7 ± 62.8 µmol g−1, respectively), but the difference was not significant. However, C:N ratios were significantly higher in the originally poorly oxic than in the oxic sediments and also higher in the inner than in the outer archipelago (D = 0.6, p = 0.023 and D = 0.735, p = 0.003, respectively). Total Fe (280–1236 µmol g−1) and Al (1714–2648 µmol g−1) correlated positively with SSA and negatively with exposure index (Online Resource 1; Table 1). In the inner archipelago, the sediments were significantly higher in TFe (W = 110, p < 0.001) and TAl (W = 93, p = 0.021) than in the outer archipelago. Content of TMn was lower than that of TFe or TAl (Fig. 3d). Total P ranged between 21 and 72 µmol g−1, excluding the exceptionally high content (181 µmol g−1) in the sediment #1 (Fig. 3d). No significant difference in TP was found between either the inner and the outer archipelago sediments or the originally oxic and the poorly oxic sediments.

Total content of C and Ca (a), N and S (b), Al and Fe (c) as well as P and Mn (d) in the sediments (µmol g−1). The line between the sediments #13 and 14 separates the sediments locating in the inner (#1–13) and in the outer (#14–22) archipelago. Note the different scales on the y-axes
3.2 Oxyhydroxides of Fe, Al and Mn
The investigated sediments were higher in FeƩExt (148.9 ± 124.2 µmol g−1) than in AlƩExt (84.2 ± 34.8 µmol g−1) or MnƩExt (5.18 ± 7.92 µmol g−1). In most of them, FeDith (65.0 ± 76.7 µmol g−1) comprised the greatest fraction of FeƩExt, while the contents of FeAsc and FeOxal were rather similar (45.4 ± 34.5 and 38.6 ± 19.0 µmol g−1, respectively). Of the Al-fractions, AlOxal was the largest one (68.4 ± 27.5 µmol g−1), followed by AlAsc and AlDith (10.6 ± 6.6 and 5.2 ± 4.0 µmol g−1, respectively). In the inner archipelago, the sediments were significantly higher in FeDith and FeOxal (D ≥ 0.812, p ≤ 0.001) and in all extracted Al-fractions (D ≥ 0.590, p ≤ 0.028) than those in the outer archipelago. Furthermore, the originally poorly oxic sediments were significantly higher in AlAsc than the oxic ones (D = 0.55, p = 0.049). Each Al- and Fe-fraction correlated positively with SSA (Online Resource 1; Table 1), and AlAsc correlated with C:N ratio (ρ = 0.728, p < 0.001). No differences in Mn were recorded either between the inner and the outer archipelago sediments or the originally oxic and the poorly oxic ones.
3.3 Phosphorus exchange parameters
Parameters EPC0 and kEPC0 varied widely among the investigated sediments (0.18–38.2 µmol l−1 and 0.03–7.42 l g−1, respectively). The parameters k15 (0.07–2.36 l g−1) and k100 (0.01–0.18 l g−1) varied relatively less. All the P exchange parameters correlated significantly with exposure index (Online Resource 1; Table 2). Furthermore, the parameters EPC0, kEPC0 and k15 correlated strongly with desorbable P. High kEPC0, k15 and k100 values were related to the abundance of AlAsc, AlDith, FeDith and FeAsc+Dith as well as to large SSA of the sediment material. The correlations of these Al- and Fe-fractions and P buffering capacities became stronger with increasing P sorption: the kEPC0 had the lowest correlations (Spearman’s rho) with AlAsc, TFe, FeDith, FeAsc+Dith and AlDith, whereas k100 had the greatest ones (Online Resource 1; Table 2). In addition, FeOxal, FeƩExt, AlƩExt and C:N ratio correlated significantly with the buffering capacity at k15 and especially at k100. The k100 was the only parameter that correlated significantly with FeAsc and AlOxal.
According to the discriminant analysis, the sediment groups of low, moderate and high EPC0 values were most clearly separated by using either the predictor variable combination Ʃ(desorbable P, AlƩExt, exposure index, Si–P0,) or that of Ʃ(desorbable P, C:N ratio, exposure index, water depth) (Fig. 4a, b). For summaries of the discriminant functions, see Online Resource 2 (Tables 1–3). The predictor variables are presented in decreasing order of their absolute values of the standardized function coefficients (Fig. 5, Online Resource 2: Table 3). A negative sign in the standardized function coefficient refers to a negative correlation of predictor variables with the discriminating function. The sediment groups of high, moderate and low kEPC0 values were most clearly separated with the variable combination Ʃ(desorbable P, AlOxal, SiP0 and exposure index) and with that of Ʃ(desorbable P, AlƩExt and TP) (Fig. 4c, d). The first significant discriminant function obtained in analyses with the used variable combinations explained ≥ 94% and ≥ 86% of the variances among the EPC0 and the kEPC0 groups, respectively, calculated from the squared canonical correlations (Online Resource 2; Table 1). Furthermore, the sediment groups of low, moderate and high k15 values were separated by variable combinations Ʃ(AlƩExt, desorbable P and TP) and Ʃ(AlOxal, desorbable P, FeAsc+Dith and exposure index) (Fig. 4e, f). The first discriminating functions obtained in both variable combinations explained ≥ 83% the variance among the k15 groups.

Variable combinations discriminating the groups of low, moderate and high EPC0 (a. and b.), kEPC0 (c. and d.) and k15 (e. and f). Standardized canonical function coefficients that describe the relative importance of each predictor are presented in brackets (EI = exposure index)

Phosphorus exchange isotherms of the sediments and their separation into clusters A and B and subgroups a–d by the hierarchical cluster analysis. The originally oxic sediments are highlighted with circles. Note the different scales on the x-axis
3.4 Phosphorus exchange patterns of the sediments
The hierarchical cluster analysis divided the sediments into the main clusters A and B according to their sorption patterns (Fig. 5). The cluster A was further divided into subgroups a and b and the cluster B into subgroups c and d. The main clusters differed significantly (D ≥ 0.727, p ≤ 0.006) in all their P exchange parameters (Table 3). In the cluster A, the sediments had lower EPC0 and higher kEPC0, k15 and k100 values than those in the cluster B and, thus, produced isotherms of steep sorption pattern. In the cluster B, in turn, the opposite parameter values resulted in isotherms with gently sloping sorption pattern. The sediments in the cluster A sorbed 47–66% of the maximum amount of the added P (100 µmol P l−1), while the corresponding portions in the cluster B were 32–38% and 9–24% for the subgroups c and d, respectively (Fig. 6).

Phosphorus sorption percentages (S%100), desorbable P (µmol l−1), SSA (m2 g−1) as well as extracted metals FeAsc, FeDith, FeOxal, AlAsc. AlDith and AlOxal (µmol g−1) of the sediments in the main cluster A, with the steep sorption pattern, and in the subgroups c and d, with the gently sloping sorption patterns
In the cluster A, eight out of the 11 sediments had been collected from the inner and the rest three ones from the outer archipelago. Four sediments were originally oxic and the rest seven ones poorly oxic (Fig. 5). As for the sediments in the cluster B, those in the subgroup c were all collected from the inner archipelago. They were originally poorly oxic, except #13. The sediments in the subgroup d were, in turn, collected from the outer archipelago and were originally oxic (except #14). The sediments in the cluster A were significantly lower in desorbable P than those in the subgroups c and d (D = 0.818, p = 0.001). However, the sediments in the cluster A and the subgroup c were quite similar in terms of their SSAs and contents of FeDith, FeOxal, AlAsc, AlDith and AlOxal, while in the subgroup d sediments these parameters and C:N ratios were significantly lower (D ≥ 0.800, p ≤ 0.048) (Fig. 6). Furthermore, in the sediments of subgroup d also FeAsc were lower than in the other groups, but the difference was significant only between the subgroups c and d (D = 0.833, p = 0.026).
4 Discussion
4.1 Sedimentation environment
The material transported and deposited to the sea floor as well as the prevailing conditions at the sedimentation environment dictate the P sorption properties of the surface sediments. In the present study at the topographically fractured northern coast of the GoF, the sedimentation environments differed according to their location in the inner or the outer archipelago. According to the classification presented by Wijkmark and Isaeus [45], all the inner archipelago sites of this study are located in areas classified as “sheltered”, “very sheltered” or “extremely sheltered”. In these types of locations, the free exchange of water and suspended material is restricted between the inner and the outer archipelago. In the sheltered locations, calm sedimentation conditions favour the sedimentation of light slowly depositing fine particulate OM as well as that of clay particles high in Fe- and Al-oxyhydroxides providing sorption surface for P. In fact, in the investigated sediments, the large SSA was strongly related to the amount of Fe- and Al-oxyhydroxides in the sediments being significantly higher in the inner than in the outer archipelago.
On the other hand, in sheltered coastal environments, subjected to high external nutrient loading, mineralization of OM can cause depletion of O2 at the sea bottom. This, together with temperature stratification further hindering mixing of water, explains the occurrence of poorly oxic sediments in the inner archipelago. At these sites, TC and C:N ratios in the sediments were higher than in the originally oxic ones, which indicates conditions favourable for accumulation of (terrestrial) OM as well as possible hypoxia/anoxia-induced N2 losses from the sediments [see 58]. The originally oxic inner archipelago sediments #2 and 4 located at clearly shallower water depths (< 7 m) and, thus, they were more prone to the turnover of the water column than the other sediments in the inner archipelago. This explains their good O2 status. On the contrary, the high exposure indexes on the outer archipelago sediments indicated their susceptibility to waves and wind-induced water mixing. However, most of the sediments on this area are located at depths > 30 m, i.e. deeper than the estimated water mixing depth (25–30 m) on the open Baltic during storm events [59]. This can explain the poor oxygen conditions found at the deep sites #14, 17, 18 and 22. Of all the outer archipelago sediments, the originally oxic ones (#15, 16, 19, 20 and 21) had a lower SSA, C:N ratio and TFe content than the poorly oxic ones. This indicates that these areas did not efficiently accumulate fine particulate inorganic and organic material. These findings suggest that the sediment material differed between the inner and the outer archipelago, being more fine-textured in the former and coarser in the latter one.
4.2 Phosphorus exchange at the early sorption stage (EPC0 and k EPC0)
In the studied sediments, the variation in the EPC0 0.2–38.2 µmol l−1 corresponds to that reported in earlier studies on Ca-rich environments. For instance, in the surface sediments of Florida Bay lagoon the range in EPC0 is reported to be 0.4–27.8 µmol l−1 [31], in those of mesotrophic and hypereutrophic lake sediments 0.71–6.97 µmol l−1 [60] and in plateau lake sediments in China 1.26–2.26 µmol l−1 [24]. However, in the present study the range of kEPC0 values (0.03–7.4 l g−1) was clearly higher than, for example, that recorded on the Florida Bay (0.033–0.716 l g−1) [31]. This difference is likely due to dissimilar sediment properties, as our sediments were on average more than tenfold higher in FeAsc and FeDith than the calcareous Florida Bay sediments (dithionite and ascorbic acid extractable Fe 0.81–4.21 µmol g−1) [61].
In the present study, the original P content (on the oxyhydroxide surfaces) in the sediments was the strongest single factor explaining the P sorption at the early sorption stage, recorded as the strong correlation between desorbable P and EPC0 and kEPC0. This finding agrees with the results obtained in a study of Zhang and Huang with calcareous (CaCO3 > 81 weight-%) Florida Bay sediments [31]. Their study revealed significant correlations between MgCl2-extractable (i.e. loosely bound) P and EPC0 and Kd values. In general, a low amount of desorbable P indicates either low content of P in the sediments or that the P is strongly bound onto the particle surfaces or occluded in the particle structures. Thus, it can result from the small amount of adsorbents or from the fact that they are rather empty of P. These factors result in low EPC0. For instance, Villnäs et al. [35] reported high EPC0 (10.93 µmol l−1) for the sandy sediment in the northern GoF that was likely attributable to coarse sediment material poor in the P sorption sites. All the above-referred EPC0 and Kd values are from studies executed with freeze-dried sediments, which renders them relatively well comparable to those obtained in our study. However, the comparison of the P exchange parameters reported in various studies is always guiding, as experimental conditions used often differ.
Altogether, the low EPC0 and the high kEPC0 indicate the presence of substantial amount of available unoccupied sorption sites. In the present study, all sediments in the groups of low EPC0 (< 1 µmol l−1) and high kEPC0 (2.60–7.42 l g−1) were characterized by steep sorption patterns and the highest sorption potentials (the cluster A sediments). They were lower in desorbable P than others and had significantly lower ratios of desorbable P/SSA (Chi-squared = 16.35, df = 2, p < 0.001) than the sediments with the gently sloping sorption pattern. This outcome indicates that their P occupation degree was the lowest one. They also possessed large SSAs, which, together with the low P occupation degree, explains their low EPC0 and high kEPC0.
The strong positive correlation between FeDith and kEPC0 reflected the abundance of amorphous Fe-oxyhydroxides particularly in the originally poorly oxic sediments. Most of the sediments of steep sorption pattern were originally poorly oxic, wherefore Fe in the sediments had been reduced to soluble Fe(II) form unable to bind P. The sediments had been oxidized during sampling, handling and drying. These changes allowed the oxygenation of Fe(II) to amorphous Fe(III)-oxyhydroxides, which explains the abundance of FeDith. This was related to high buffering capacities as well as to low occupation degrees of the sediments. The hypoxia-induced lower P occupation degree agrees also with results reported by Villnäs et al. [35] that hypoxia lowered EPC0 in the surface sediment. Furthermore, resuspension of sediment caused by gas ebullitions (recorded in situ at the sites #6, 9, 10) can promote the P release from the particle surfaces. This reaction pattern is attributable to a lowered ionic strength and P concentration of the surrounding solution as a result of increasing water volume that favours the release of P from the oxyhydroxides, see Yli-Halla and Hartikainen [62]. The AlAsc- and AlDith-fractions correlated also strongly positively with the kEPC0, but they were less abundant than the FeDith in the sediments. Furthermore, their origin is uncertain, wherefore it is not known whether they acted as real adsorbents for P or were they, for instance, liberated inside Fe-containing oxyhydroxides along with the dissolution of Fe in the analysis [37, 63].
The highest desorbable P contents and P occupation degrees were recorded in the sediments with gently sloping sorption pattern. Thus, the sorption of added P started at a higher P concentration of the solution, i.e. resulting in a high EPC0 (≥ 1.97 µmol l−1) and low kEPC0 (≤ 0.84 l g−1). Of the sediments of gently sloping sorption pattern, those in the subgroup c were mostly hypoxic and had more adsorbents than the mostly oxic sediments in the subgroup d. The subgroup c sediments had high SSAs and they were abundant in oxyhydroxides, particularly in those of Al (AlOxal). This explains their P occupation degree being higher than in the hypoxic sediments with the steep sorption pattern. It seems that the redox-resistant Al-oxyhydroxides were able to keep P bound to them despite the poor oxygen status at the time of sampling. The sediments of the subgroup d, in turn, possessed a limited amount of adsorbents due to their coarser material with lower contents of oxyhydroxides. Furthermore, these sediments had the highest desorbable P/SSA ratios and P contents. It seems that the oxic conditions that prevailed at the time of sampling in most of these sediments had allowed P accumulation which further lowered their P sorption ability.
The examination of the effect of various variable combinations on the P sorption at the early sorption stage revealed that the amount of Al-oxyhydroxides was the second strongest variable, after desorbable P, explaining the sediment grouping according to their EPC0 and kEPC0 values. The stronger explaining power of Al- than that of Fe-oxyhydroxides in the discriminant analysis was probably attributable to the smaller variation in the Al- than Fe-fractions within the sediment groups. Furthermore, concentration of silicate (SiO4–Si) desorbed from the sediments into P-free ASW (Si–P0) and C:N ratio in the sediments affected the sediment grouping according to their EPC0 and kEPC0 values. This result implies that the presence of reactive silicate lowers the P buffering capacity at the early sorption stages, particularly if present concomitantly with abundant desorbable P, as these anions tend to occupy the same sorption sites [64]. The contribution of C:N ratio in separating the EPC0 and kEPC0 groups, in turn, may reflect the original O2 conditions, as the ratio was higher in the originally poorly oxic than in the oxic sediments, as discussed above.
4.3 Phosphorus exchange at the middle and the high sorption stages (k 15 and k 100)
The stronger positive correlation between SSA and k100 than that between SSA and k15 suggests that large SSA was necessary for the highest P sorption potentials. The strong positive correlation between the P buffering capacities and the SSAs agrees with the findings of earlier studies. For instance, Meng et al. [65] showed that P sorption isotherms were the steepest in the fine silt fraction and decreased in the order: medium silt, coarse silt and sand. In the investigated sediments, the high SSAs of the sediments with steep sorption pattern were reflected as significant positive correlations between FeDith, AlAsc, AlDith and FeOxal and the buffering capacities at the middle (k15) and the high sorption stages (k100) as well as that between AlOxal and k100. These results indicate that a variety of sorption components, represented as Fe and Al extracted from oxyhydroxides of differing redox sensitivity, participated in efficient P sorption. Chemical extraction does not distinguish between certain compounds but rather between mixed oxyhydroxides of variable stability. For instance, the FeOxal-fraction may be overestimated, as oxalate is reported to extract Fe also from FeS compounds [51]. In our samples, the originally poorly oxic sediments with steep P sorption patterns were high in TS and it is possible that part of the TS originated in FeS compounds.
All the sediments with the highest k15 values (1.08–2.36 l g−1) showed steep sorption patterns. Of the sediments with gently sloping sorption pattern, most of those, again, in the subgroup c possessed moderate k15 values (0.29–0.94 l g−1), whereas all the subgroup d sediments had low k15 values (≤ 0.24 l g−1). The AlƩExt and AlOxal explained most strongly the separation of the sediments into groups of high, moderate and low k15 values. This result suggests that Al-oxyhydroxides had an important role along with the Fe-oxyhydroxides at all the sorption stages.
4.4 Potential impacts of freeze-drying on the P exchange in sediments
As the study was performed with oxygenated freeze-dried sediments, the results are not fully equivalent to the P exchange properties of fresh oxic samples. Previous studies report freeze-drying to change the partitioning of Fe and other trace metals [66], to promote the formation of Fe oxyhydroxides and to increase their crystallization [67]. Furthermore, P exchange experiments with air-dried sediments have been found to produce more gently sloping isotherm graphs than those with fresh sediments [68, 69]. Previously we investigated P exchange in fresh (protected from atmospheric O2) and freeze-dried oxic and hypoxic sediments collected from the same sea area (the experiment and its results are presented in Vaalama et al., under revision). Our results revealed that the EPC0 values were 0.1–0.5 µmol l−1 higher in the freeze-dried samples than in the fresh ones. Furthermore, the kEPC0 values of the freeze-dried samples were approximately a tenth of the kEPC0 values of the fresh samples. This outcome is presumably attributable to drying-induced structural changes in P sorption components. Our findings indicate that the P sorption potentials of the freeze-dried sediments are somewhat lower than they would be in fresh sediment samples. However, the purposes of the present study were to investigate the potential differences in sorption properties among various coastal sediments (pre-treated similarly) and to unravel how their physico-chemical characteristics explain their differing P sorption behaviour. Thus, freeze-drying, which is a common pre-treatment method used for preservation purposes in sediments analyses, see, e.g. [31, 32, 35], seems an appropriate pre-treatment method in this kind of investigations. However, according to our knowledge, the effect of long-term storing of freeze-dried sediments on their P exchange properties has not been investigated.
4.5 Importance of the results concerning eutrophied coastal seas
The results suggest that temporarily hypoxic sheltered bays accumulating fine particulate material and being subjected to P inputs from the watersheds are at elevated risk of internal P loading. During hypoxia, the ability of surface sediments to act as a buffering layer for P releasing from the deeper layers will be lowered. Furthermore, the results reveal that a high occupation degree of P on the existing adsorbents lowers the P buffering ability of oxic sediments, which suggest that good O2 conditions do not guarantee any complete P retention onto the sediments. They also agree with the study of Puttonen et al. [70], where the shallow inner archipelago sediments in the northern Baltic Sea (in the Archipelago sea) showed to be an important and probably underestimated sources for aquatic P. However, in case the poor O2 conditions improve, the sediments in the inner archipelago seem to possess potential to efficiently bind P. In the future, the P exchange parameters EPC0 and kEPC0 of oxygenated coastal surface sediments could be used, together with the information on the O2 conditions near the sea floor, when estimating their potential risk of internal P loading.
5 Conclusions
The sediments with the highest P sorption potentials showed steep sorption patterns, characterized by low EPC0 (< 1 µmol l−1) and high kEPC0 (≥ 1.32 l g−1) and k15 (≥ 0.60 l g−1) values. They had high SSAs and a variety of sorption components (amorphous Fe- and Al-oxyhydroxides, FeOxal, AlAsc and AlDith) that were attributable to fine-textured sediment material being characteristics of sheltered environments in the inner archipelago near the coastline. Such environments are subjected to riverine supply of terrestrial sorption components from the watersheds. The greatest P sorption potentials were recorded in the originally poorly oxic sediments with low P occupation degree of the adsorbents. This can be attributable to oxidation-induced formation of new Fe-oxyhydroxides, with high affinity for P, in the sediment sampling, handling and storing. The Al-oxyhydroxides seemed to act as important sorption sites for P in the reduced sediments collected from the poorly oxygenated areas in the inner archipelago. The sediments with low P sorption potentials showed gently sloping sorption patterns, characterized by high EPC0 (≥ 2.0 µmol l−1) as well as low kEPC0 (< 0.80 g−1) and k15 values (< 0.40 l g−1). The low sorption potentials resulted from the rather high P occupation degree of the adsorbents. Furthermore, the lowest sorption potentials were recorded in the coarse-textured sediments in the outer archipelago, which refers to scarcity of adsorbents for P. Our results highlight the complex contribution of physico-chemical properties of the sedimentation environment on the P exchange in the sediments. They also suggest that when hypoxic sediment surface is allowed to oxidize, its P sorption ability can improve, if adsorbents for P are available. Thus, the clayey mud sediments in hypoxic eutrophic coastal areas may recover their ability to bind P if the oxygen conditions improve.
References
Rabalais NN, Turner RE, Díaz RJ, Justic D (2009) Global change and eutrophication of coastal waters. ICES J Mar Sci 66:1528–1537
Mortimer CH (1941) The exchange of dissolved substances between mud and water in lakes, I. J Ecol 29:280–329
Conley DJ, Björck S, Bonsdorff E, Carstensen J, Destouni G, Gustafsson BG, Hietanen S, Kortekaas M, Kuosa H, Meier HEM, Müller-Karulis B, Nordberg K, Norkko A, Nürnberg G, Pitkänen H, Rabalais NN, Rosenberg R, Savchuk OP, Slomp CP, Voss M, Wulff F, Zillén L (2009) Hypoxia-related processes in the Baltic Sea. Environ Sci Technol 43:3412–3420
Meybeck M (1982) Carbon, nitrogen, and phosphorus transport by world rivers. Am J Sci 282:401–450
Uusitalo R, Turtola E (2003) Determination of redox-sensitive phosphorus in field runoff without sediment preconcentration. J Environ Qual 32:70–77
Asmala E, Carstensen J, Conley DJ, Slomp CP, Stadmark J, Voss M (2017) Efficiency of the coastal filter: nitrogen and phosphorus removal in the Baltic Sea. Limnol Oceanogr 62:S222–S238
Sholkovitz ER (1976) Flocculation of dissolved organic and inorganic matter during the mixing of river water and seawater. Geochim Cosmochim Acta 40:831–845
Lukkari K, Leivuori M, Hartikainen H (2008) Vertical distribution and chemical character of sediment phosphorus in two shallow estuaries in the Baltic Sea. Biogeochemistry 90:171–191
Froelich PN (1988) Kinetic control of dissolved phosphate in natural rivers and estuaries: a primer on the phosphate buffer mechanism. Limnol Oceanogr 33:649–668
Krom MD, Berner RA (1980) The diagenesis of phosphorus in a nearshore marine sediment. Geochim. et Cosmochim. Acta 45:207–216
Bjorkman K, Karl DM (1994) Bioavilability of inorganic and organic phosphorus compounds to natural assemblages of microorganisms in Hawaiian coastal waters. Mar Ecol Prog Ser 111:265–274
Lønborg C, Davidson K, Álvarez-Salgado XA, Miller AEJ (2009) Bioavailability and bacterial degradation rates of dissolved organic matter in a temperate coastal area during an annual cycle. Mar Chem 113:219–226
Berner RA (1979) Kinetics of nutrient regeneration in anoxic marine sediments. Phys Chem Earth 11:279–292
Boström B, Andersen JM, Fleischer S, Jansson M (1988) Exchange of phosphorus across the sediment-water interface. Hydrobiologia 170:229–244
Koski-Vähälä J, Hartikainen H, Kairesalo T (2000) Resuspension in regulating sedimentation dynamics in Lake Vesijarvi. Arch Hydrobiol 148:357–381
Parfitt RL, Atkinson RJ, Smart R (1975) The mechanism of phosphate fixation by iron oxides. Soil Sci Soc Am Proc 39:837–841
Gerke JHR (1992) Adsorption of orthophosphate to humic- Fe- complexes and to amorphous Fe- oxide. J Plant Nutr Soil Sci 155:233–236
Ruttenberg KC, Sulak DJ (2011) Sorption and desorption of dissolved organic phosphorus onto iron (oxyhydr)oxides in seawater. Geochim Cosmochim Acta 75:4095–4112
Kopácek J, Ulrich K-, Hejzlar J, Borovec J, Stuchlík E (2001) Natural inactivation of phosphorus by aluminum in atmospherically acidified water bodies. Water Res 35:3783–3790
Gálvez N, Barrón V, Torrent J (1999) Preparation and properties of hematite with structural phosphorus. Clays Clay Min 47:375–385
Nair PS, Logan TJ, Sharpley AN, Sommers LE, Tabatabai MA, Yuan TL (1984) Interlaboratory comparison of a standardized phosphorus adsorption procedure. J Environ Qual 13:591–595
Hartikainen H (1991) Potential mobility of accumulated phosphorus in soil as estimated by the indices of Q/I plots and by extractant. Soil Sci 152:204–209
Slomp CP, Malschaert JFP, Van Raaphorst W (1998) The role of adsorption in sediment-water exchange of phosphate in North Sea continental margin sediments. Limnol Oceanogr 43:832–846
Huang W, Lu Y, Li J, Zheng Z, Zhang J, Jiang X (2015) Effect of ionic strength on phosphorus sorption in different sediments from a eutrophic plateau lake. RSC Adv 5:79607–79615
Mead JA (1981) A comparison of the langmuir, freundlich and temkin equations to describe phosphate adsorption properties of soils. Aust J Soil Res 19:333–342
Giesler R, Andersson T, Lövgren L, Persson P (2005) Phosphate sorption in aluminum- and iron-rich humus soils. Soil Sci Soc Am J 69:77–86
Peltovuori T, Soinne H (2005) Phosphorus solubility and sorption in frozen, air-dried and field-moist soil. Eur J Soil Sci 56:821–826
Foo KY, Hameed BH (2010) Insights into the modeling of adsorption isotherm systems. Chem Eng J 156:2–10
Alenius P, Myrberg K, Nekrasov A (1998) The physical oceanography of the Gulf of Finland: a review. Boreal Environ Res 3:97–125
Łysiak-Pastuszak E, Carstens M, Leppänen J, Leujak W, Nausch G, Murray C, Andersen JH (2014) Eutrophication status of the Baltic Sea 2007–2011—a concise thematic assessment. HELCOM Balt Sea Environ Proc 143:1–41
Zhang J-J, Huang X-L (2007) Relative importance of solid-phase phosphorus and iron on the sorption behavior of sediments. Environ Sci Technol 41:2789–2795
Wang Q, Li Y (2010) Phosphorus adsorption and desorption behaviour on sediments of different origins. J Soils Sediments 10:1159–1173
Li M, Whelan MJ, Wang GQ, White SM (2013) Phosphorus sorption and buffering mechanisms in suspended sediments from the Yangtze Estuary and Hangzhou Bay, China. Biogeosciences 10:3341–3348
Carman R, Wulff F (1989) Adsorption capacity of phosphorus in Baltic Sea sediments. Estuar Coast Shelf Sci 29:447–456
Villnäs A, Norkko J, Lukkari K, Hewitt J, Norkko A (2012) Consequences of increasing hypoxic disturbance on benthic communities and ecosystem functioning. PLoS ONE 7:e44920
Lukkari K, Leivuori M, Kotilainen A (2009) The chemical character and behaviour of phosphorus in poorly oxygenated sediments from open sea to organic-rich inner bay in the Baltic Sea. Biogeochemistry 96:25–48
Lukkari K, Leivuori M, Vallius H, Kotilainen A (2009) The chemical character and burial of phosphorus in shallow coastal sediments in the northeastern Baltic Sea. Biogeochemistry 94:141–162
Svendsen LM, Staaf H, Pyhälä M, Kotilainen P, Bartnicki J, Knuuttila S, Durkin M (2012) Fifth Baltic sea pollution load compilation (PLC-5) an executive summary. HELCOM Balt Sea Environ Proc 128A:1–32
Kullenberg G (1981) Physical oceanography. In: Voipio A (ed) The Baltic Sea. Elsevier, Amsterdam, pp 135–181
HELCOM (2019) Developments in nutrient inputs http://www.helcom.fi/baltic-sea-trends/eutrophication/inputs-of-nutrients/developments-in-nutrient-inputs/. Accessed 4 April 2019
Conley DJ, Stockenberg A, Carman R, Johnstone RW, Rahm L, Wulff F (1997) Sediment-water nutrient fluxes in the Gulf of Finland, Baltic Sea. Estuar Coast Shelf Sci 45:591–598
Kaskela AM, Kotilainen AT, Al-Hamdani Z, Leth JO, Reker J (2012) Seabed geomorphic features in a glaciated shelf of the Baltic Sea. Estuar Coast Shelf Sci 100:150–161
Vallius H (2015) Sediment and carbon accumulation rates off the southern coast of Finland. Baltica 28:81–88
Isaeus M (2004) Factors structuring Fucus communities at open and complex coastlines in the Baltic Sea. Dissertation, Stockholm University
Wijkmark N, Isaeus M (2010) Wave exposure calculations for the Baltic Sea. AquaBiota Water Res Rep 2:1–37
Winterhalter B, Flodén T, Ignatius H, Axberg S, Niemistö L (1981) Geology of the Baltic Sea. In: Voipio A (ed) the Baltic Sea. Elsevier, Amsterdam, pp 1–121
Kostka JE, Luther GW III (1994) Partitioning and speciation of solid phase iron in saltmarsh sediments. Geochim Cosmochim Acta 58:1701–1710
Anschutz P, Zhong S, Sundby B, Mucci A, Gobeil C (1998) Burial efficiency of phosphorus and the geochemistry of iron in continental margin sediments. Limnol Oceanogr 43:53–64
Schwertmann U (1964) Differenzierung der Eisenoxide des Bodens durch Extraktion mit Ammoniumoxalat-Lösung. J Plant Nutr Soil Sci 105:194–202
Farmer VC, Russell JD, Smith BFL (1983) Extraction of inorganic forms of translocated Al, Fe and Si from a podzol Bs horizon. Eur J Soil Sci 34:571–576
Jan J, Borovec J, Kopácek J, Hejzlar J (2013) What do results of common sequential fractionation and single-step extractions tell us about P binding with Fe and Al compounds in non-calcareous sediments? Water Res 47:547–557
Koroleff F (1976) Determination of nutrients. In: Grasshoff K (ed) Methods of seawater analyses, Verlag Chemie Gmbh, Weinheim, pp 117–181
Koroleff F (1983) Determination of phosphorus. In: Grasshoff K, Ehrhardt M, Kremling K (eds) Methods of seawater analysis, 2nd edn. Verlag Chemie Gmbh, Weinheim, pp 125–142
Hansen HP, Koroleff F (1999) Determination of nutrients. In: Grasshoff K, Kremling K, Ehrhardt M (eds) Methods of seawater analyses, 2nd edn. Verlag Chemie Gmbh, Weinheim, pp 159–228
Stumm W, Morgan JJ (1996) The Solid-solution interface. In: Schnoor JL, Zehnder A (eds) Aquatic chemistry, 3rd edn. Wiley, New York, pp 516–613
Barrow NJ, Bolland MDA, Allen DG (1998) Effect of previous additions of superphosphate on sorption of phosphate. Aust J Soil Res 36:359–372
White RE, Beckett PHT (1964) Studies on the phosphate potentials of soils—Part I—the measurement of phosphate potential. Plant Soil 20:1–16
Hietanen S, Lukkari K (2007) Effects of short-term anoxia on benthic denitrification, nutrient fluxes and phosphorus forms in coastal Baltic sediment. Aquat Microb Ecol 49:293–302
Björkqvist J-V, Lukas I, Alari V, van Vledder GP, Hulst S, Pettersson H, Behrens A, Männik A (2018) Comparing a 41-year model hindcast with decades of wave measurements from the Baltic Sea. Ocean Eng 152:57–71
Dong L, Yang Z, Liu X (2011) Phosphorus fractions, sorption characteristics, and its release in the sediments of Baiyangdian Lake, China. Environ Monit Assess 179:335–345
Zhang J-J, Fischer CJ, Ortner PB (2004) Potential availability of sedimentary phosphorus to sediment resuspension in Florida Bay. Global Biogeochem Cycles 18:1–14
Yli-Halla M, Hartikainen H (1996) Release of soil phosphorus during runoff as affected by ionic strength and temperature. Agric Food Sci Finl 5:193–202
Adra A, Morin G, Ona-Nguema G, Menguy N, Maillot F, Casiot C, Bruneel O, Lebrun S, Juillot F, Brest J (2013) Arsenic scavenging by aluminum-substituted ferrihydrites in a circumneutral pH river impacted by acid mine drainage. Environ Sci Technol 47:12784–12792
Hartikainen H, Pitkänen M, Kairesalo T, Tuominen L (1996) Co-occurrence and potential chemical competition of phosphorus and silicon in lake sediment. Water Res 30:2472–2478
Meng J, Yao Q, Yu Z (2014) Particulate phosphorus speciation and phosphate adsorption characteristics associated with sediment grain size. Ecol Eng 70:140–145
Rapln F, Tessler A, Campbell PGC, Carignan R (1986) Potential artifacts in the determination of metal partitioning in sediments by a sequential extraction procedure. Environ Sci Technol 20:836–840
Raiswell R, Vu HP, Brinza L, Benning LG (2010) The determination of labile Fe in ferrihydrite by ascorbic acid extraction: methodology, dissolution kinetics and loss of solubility with age and de-watering. Chem Geol 278:70–79
Baldwin DS (1996) Effects of exposure to air and subsequent drying on the phosphate sorption characteristics of sediments from a eutrophic reservoir. Limnol Oceanogr 41:1725–1732
Dieter D, Herzog C, Hupfer M (2015) Effects of drying on phosphorus uptake in re-flooded lake sediments. Environ Sci Pollut Res 22:17065–17081
Puttonen I, Mattila J, Jonsson P, Karlsson OM, Kohonen T, Kotilainen A, Lukkari K, Malmaeus JM, Rydin E (2014) Distribution and estimated release of sediment phosphorus in the northern Baltic Sea archipelagos. Estuar Coast Shelf Sci 145:9–21
Acknowledgements
Open access funding provided by Finnish Environment Institute (SYKE). We thank Maj and Tor Nessling foundation for the financial support of this project. Maa- ja vesitekniikan tuki ry, The Finnish Foundation for Nature Conservation, Otto A. Malm Foundation and The Finnish Concordia Fund are acknowledged for funding the writing of this article. We also thank Geological Survey of Finland for sediment sampling, Hannu Rita and Hanna Sinkko for statistical advices and Elina Miettunen for the help with processing the ArcGIS data.
Funding
This study was funded by Maj and Tor Nessling foundation (Grant number 2011401), Maa- ja vesitekniikan tuki ry (Grant number 27931), The Finnish Foundation for Nature Conservation (Grant number not available), Otto A. Malm Foundation (Grant number not available) and The Finnish Concordia Fund (Grant number not available).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Vaalama, A., Hartikainen, H., Vallius, H. et al. Phosphorus exchange in eutrophied coastal brackish water sediments—sorption pattern, potential and factors affecting them. SN Appl. Sci. 1, 1372 (2019). https://doi.org/10.1007/s42452-019-1374-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-019-1374-7