
Abstract
A rapid and sensitive liquid chromatography tandem mass spectrometry (LC–MS/MS) method for the simultaneous determination of Triamterene and Hydrochlorothiazide in human plasma was established. The analytes and the internal standards were separated on a Zorbax Eclipse Plus RRHD C18 column (2.1 mm × 50 mm, 1.7 μm) using isocratic elution with a mobile phase of 0.1% formic acid:methanol:acetonitrile 5:4:1 and 0.1% formic acid in water at a flow rate of 0.4 mL/min. The detection was performed on a triple quadrupole tandem mass spectrometer by multiple reaction monitoring mode to monitor the precursor-to-product ion transitions of m/z 254.0 → 237.1, 259.1 → 242.2 for Triamterene and Triamterene D5 in positive electrospray ionization mode, m/z 295.9 → 269.0, 300.9 → 271.0 for Hydrochlorothiazide and Hydrochlorothiazide 15N2-13C-D2 using a negative electrospray ionization mode. The method was validated over a concentration range of 0.5–200 ng/mL for Triamterene and 2.5–400 ng/mL for Hydrochlorothiazide. Total time for each chromatogram was 5.2 min (after 1.5 min switching to positive mode). The method was successfully applied to a bioequivalence study of Triamterene and Hydrochlorothiazide in healthy human volunteers.
Similar content being viewed by others

1 Introduction
The use of mixtures of diuretics is targeted at intensification of its efficiency by achieving synergic effects. In complex mixtures the combinations of thiazides and potassium-sparing diuretics are often used. The advantages of complex drugs include reduced dosages, while intensifying hypertension activity and reducing the hypokalemia symptoms [1].
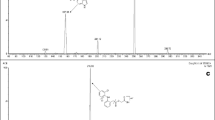
Triamterene (2, 4, 7-triamino-6-phenylpteridine) Fig. 1a is a potassium-sparing diuretic commonly used in combination with Hydrochlorothiazide (6-chloro-3, 4-dihydro-2H-1, 2, 4-benzothiadiazine-7-sulphonamide 1, 1-dioxide), a thiazide-type diuretic Fig. 1b in clinical management of edema and moderate hypertension.

a Chemical structure of Triamterene. b Chemical structure of Hydrochlorothiazide
Hydrochlorothiazide is incompletely but fairly rapidly absorbed from the gastrointestinal tract. Peak plasma concentrations occur between 60 and 120 min. It is excreted unchanged into urine, 50% being recovered within the first 12 h. The serum half-life is estimated to be in the range of 3–4 h.
Triamterene is rapidly absorbed from the gastrointestinal tract but to a variable extent (30–70% of the oral dose). The peak plasma concentration of Triamterene is reached 2–4 h after an oral dose and the half-life of the Triamterene in plasma ranges from 1.5 to 2 h. Approximately 50% of the Triamterene in plasma is protein bound. Triamterene undergoes metabolism in the liver. Triamterene and its metabolites are then excreted renally utilizing the processes of filtration and tubular secretion. About 20% of an oral dose appears unchanged in the urine, 70% as the sulphate ester of hydroxytriamterene, and 10% as free hydroxytriamterene and Triamterene glucuronide [2].
Various methods for the analysis of Triamterene and Hydrochlorothiazide have been published. These include HPLC–UV method [3, 4], HPTLC method [5], derivative Spectrophotometry method [1], liquid chromatography/tandem mass spectrometry method [6, 7]. Various available methods are intended to analyze single analyte and they did not have fast and sensitive enough method for simultaneous estimation. As these two analytes are having different ionization polarity in mass spectrometer, it is a challenge to develop simultaneous method. As per literature there is only one published procedure for simultaneous determination of both analytes in human plasma using LC–MS/MS method which has higher lower limit of quantification [2]. Therefore, the main objective of this study was to develop and validate a sensitive LC–MS/MS method for simultaneous quantification of Triamterene and Hydrochlorothiazide from human plasma, which could be applied to pilot bioequivalence study.
2 Materials and methods
2.1 Chemicals and reagents
Triamterene (purity 99.1%) and Hydrochlorothiazide (purity 99.8%) were received from the sponsor as an in-house standard. Internal standards (IS): Triamterene D5 (IS 1) (purity 99.0%) and Hydrochlorothiazide 15N2-13C-D2 (IS 2) (purity 98.6%) were obtained from TLC PharmaChem LTD (Ontario Canada). Two major metabolites of Triamterene—4-hydroxy triamterene (purity 95%) and 4-hydroxy triamterene sulfate sodium salt (purity 97%) were obtained from Santa Cruz Biotechnology, Inc. (California, US). Certified ACS grade formic acid and HPLC grade water were purchased from Carl Roth (Germany). HPLC grade acetonitrile, ethyl acetate, methanol and isopropanol were purchased from Fisher Scientific. Human Plasma (K3EDTA as an anticoagulant) was obtained from Research Center of Radiation and Burns, Armenia.
2.2 Instrumentation and chromatographic conditions
Liquid chromatography was performed on an Agilent Infinity 1290 ultra high performance liquid chromatography (UHPLC) (Agilent Technologies, Santa Clara, CA US) with a Zorbax Eclipse Plus RRHD C18 column (50 × 2.1 mm, 1.7 μm particle size) (Agilent Technologies, Santa Clara, CA USA). An isocratic program was employed for chromatographic separation with 65% solvent A (0.1% formic acid in water) and 35% solvent B (0.1% formic acid:methanol:acetonitrile 5:4:1). The flow rate was 0.4 mL/min and the injection volume was 3 μL. The column and sample temperature were maintained at 40 °C and 4 °C, respectively. An Sciex API 4000 triple quadrupole mass spectrometer equipped with an electro-spray ionization (ESI) source (Toronto, Canada) was used for mass spectrometric detection. The quantitative analysis of Triamterene was performed in positive mode and Hydrochlorothiazide in negative mode using multiple reaction monitoring (MRM) method. Details are presented in the Table 1. The data were acquired and analyzed by Analyst 1.6.1 Chromatographic Data System.
2.3 Preparation of calibration curve standards and
The stock solutions for Triamterene (0.400 mg/mL), Hydrochlorothiazide (0.400 mg/mL), IS 1 (0.200 mg/mL) and IS 2 (0.500 mg/mL) were prepared in methanol. Combined working solutions for calibration and controls were prepared from the stock solutions by dilution with water. The combined IS working solution (320 ng/mL IS 1; 600 ng/mL IS 2) was prepared from the IS stock solutions by dilution with water. Calibration curve standards were freshly spiked for every batch. Quality control (QC) samples were prepared in bulk by spiking appropriate amounts of working solutions into blank human plasma with 1:20 ratio. Final concentrations of the calibration curve standards in human plasma were 0.5, 1, 5, 10, 25, 50, 100 and 200 ng/mL for Triamterene and 2.5, 5, 10, 20, 50, 100, 200 and 400 ng/mL for Hydrochlorothiazide respectively. Five levels of QC samples were used: lower limit of quantitation quality control (LLOQ QC), low quality control (LQC), two medium quality control (MQC), high quality control (HQC) and out of calibration curve quality control (OCC QC). The concentrations of QC samples (LLOQ QC, LQC, MQC1, MQC2, HQC and OCC QC) in plasma were 0.5, 1.5, 20, 90, 180 and 400 ng/mL for Triamterene and 2.5, 7.5, 90, 180, 300 and 800 ng/mL for Hydrochlorothiazide respectively. All stock solutions and working solutions were stored at 4 °C, QC samples were stored at − 20 °C. Independent stock solutions were used to prepare calibration curve standards and QC samples.
2.4 Extraction solvent preparation (saturation of ethyl acetate with water)
Ethyl acetate and water were mixed in the separation funnel with 8:1 ratio. The mixture was shaken vigorously approximately for 1 min at room temperature so that the water breaks into tiny droplets in the ethyl acetate. After resting approximately 5 min on the rack, mixing step was repeated two more times. Before the separation of water phase the mixture was left on the rack for about 30 min [8].
2.5 Sample preparation
Before analysis, the plasma sample was thawed to room temperature. In a 15 mL centrifuge tube, 50 μL of the IS working solution was added to 200 μL of collected plasma sample. For calibration curve standard preparation 10 μL of calibration curve standard working solution was spiked with blank plasma. 10 μL of water was added to QC and clinical samples and the tubes were vortex mixed briefly. Analytes were extracted with 4 mL water saturated ethyl acetate by vortex mixing for about 5 min. After centrifugation at 4500 g for 10 min, organic layer was transferred into a clean glass tube and evaporated to dryness under the nitrogen stream at 55 °C. Residues were reconstituted in 200 μL of 0.7% formic acid:methanol (9:1) mixture. 3 μL was injected into the LC–MS/MS system for analysis.
2.6 Method validation
Method validation was performed to evaluate the sensitivity, selectivity, linearity, precision, accuracy, carry over, matrix effect, dilution integrity and various stability tests according to United States FDA guidelines on bioanalytical method validation [9].
2.6.1 Selectivity (blank check, interference check, metabolite interference)
Selectivity (blank check) refers to the ability of an analytical method to differentiate and quantify the analyte (Triamterene, Hydrochlorothiazide) in the presence of other components in the sample. Six independent blank lots were screened for possible interference at the retention time of the analyte and the IS. The interference in the blank was calculated using the mean peak area of the analyte and the IS at their respective retention times in the lower limit of quantification (LLOQ) sample.
An extracted solution of the analyte at approximately the highest standard concentration was prepared without the IS in duplicate and injected to observe any possible interference at the retention time of the IS.
An extracted solution of the IS at approximately the concentration equivalent to the IS concentration was prepared without the analyte in duplicate and injected to observe any possible interference at the retention time of the analyte. To check the interference, extracted LLOQ + IS samples were prepared in duplicate and injected.
Interference from Triamterene two main metabolites (4-Hydroxy-triamterene, 4-Hydroxy-triamterene sulfate) were evaluated by spiking Blank and LQC samples with the metabolite to obtain a final concentration of 1.0 μg/mL of each metabolite. Metabolite interference samples in three replicates per each metabolite were extracted and quantified to observe any possible interference at the retention time of the analyte.
2.6.2 Calibration curve and sensitivity
Calibration curves were constructed at 0.5–200 ng/mL for Triamterene and 2.5–400 ng/mL for Hydrochlorothiazide, and analyzed in nine runs. The calibration curves were fitted using a weighted (1/x2) least-squares linear regression method. The acceptance criterion for each back-calculated standard concentration was ± 15.0% deviation from the nominal value, except for the LLOQ, for which a deviation of ± 20.0% was permitted.
2.6.3 Precision and accuracy
Precision of an assay is defined as the closeness of the results when replicate assays of a test sample are carried out. The precision is expressed as %CV. The accuracy of a method is defined as the proximity of the determined values to actual concentrations of the samples. The accuracy is expressed as %Accuracy. Inter-run and intra-run precision and accuracy results of six replicate QC samples at LLOQ QC, LQC, MQC1, MQC2 and HQC concentration levels for four separate batches were performed. The criteria for acceptable accuracy for both Intra (within) and Inter (overall) run was the following: the mean value must be within ± 15.0% of the nominal value at each level, except for LLOQ QC where it must not deviate by more than ± 20.0%.
The criteria for acceptable precision for both Intra (within) and Inter (overall) batches was the following: the %CV must be within 15.0% at each level, except for LLOQ QC where it must not deviate by more than 20.0%.
2.6.4 Matrix effect
Matrix effects were investigated using six lots of blank matrix from individual donors. For each analyte and IS, the matrix factor (MF) was calculated for each lot of matrix, by calculating the ratio of the peak area in the presence of matrix (measured by analyzing blank matrix spiked with analyte after extraction), to the peak area in absence of matrix (pure solution of the analyte). The IS normalized MF was also calculated by dividing the MF of the analyte by the MF of the IS. This determination was done in three replicates at LQC and HQC concentrations.
2.6.5 Recovery
The recovery of analyte from human plasma was evaluated by comparing the peak area ratios of six replicates of extracted QC samples, versus the peak area ratios of six replicates of analyte and IS containing aqueous solutions representing 100% recovery. The evaluation was assessed at low, medium and high QC concentrations.
2.6.6 Carry over
The carry over effect was monitored prior to the injection of every analytical run. Additionally, the carry over effect was investigated by injection of Blank sample and reconstitution solution after OCC QC sample. OCC QC is 2 times of the upper limit of quantification (ULOQ).
2.6.7 Ruggedness
The ruggedness test was performed during precision and accuracy testing by different chemists on different columns.
2.6.8 Hemolysis effect
The impact of hemolysis was assessed by employing an additional lot of analyte free hemolyzed matrix for the preparation of each of the low and high QC level samples. Hemolyzed matrix was mixed with non-hemolyzed matrix in ratio of 1:49 (resembling 2% hemolysis) and 1:19 (resembling 5% hemolysis) and six replicates of LQC and HQC level samples were prepared and assayed.
2.6.9 Hyperlipidemia effect
The impact of the high level of lipids was assessed by employing an additional lot of analyte free lipemic matrix for the preparation of each of the low and high QC samples. Six replicates at each of the low and high QC sample concentration levels were prepared and assayed for this matrix lot. Plasma with triglyceride concentration of more than 300 mg/dL is considered a lipemic matrix.
2.6.10 Dilution integrity
Dilution integrity was validated by diluting six replicates of OCC QC with blank biological matrix using dilution factor 5.
2.6.11 Stability
The stability of the analytes in human plasma, whole blood, processed sample and analytical solutions was assessed. Freeze–thaw (3 cycles), short-term room temperature (12 h), long-term (25 days at − 20 °C) stability of analytes in human plasma were assessed by analyzing six replicates (stored in six different tubes) of QC samples at the low, high and OCC (two times of ULOQ) concentrations. The stability of analyte in human whole blood was assessed by comparing six replicates of stability whole blood samples, maintained in an ice water bath for 2 h, versus six replicates of comparison samples at both low and high QC concentrations taken from aliquots at the beginning of experiment.
Processed sample stability at room temperature (6 h) and autosampler stability of Triamterene and Hydrochlorothiazide were assessed by six replicates of QC samples at low and high concentrations stored in autosampler (4 °C nominal for 77 h) along with a freshly extracted calibration curve and QC samples.
Stock and working solution stability of analytes and ISs at room temperature (17 h) and in refrigerator (35 days for analytes stocks and working solutions, 51 days for ISs stocks and ISs working solutions) was performed by comparing the peak area ratios of stability and comparison samples.
2.6.12 Maximum batch size
The maximum batch size was tested by including in one analytical batch more samples than the expected maximal batch size during subject sample analysis of pharmacokinetic study. Total amount of injections was 130 samples.
3 Results and discussion
3.1 Method development and optimization
The method development was initiated to develop a simple, rugged, rapid and selective LC–ESI–MS method for quantification of Triamterene and Hydrochlorothiazide in human plasma. Two analytes have different ionization modes therefore, it was essential to have a good peak resolution for switching between polarity modes since switching time in API 4000 is about 700 ms. To achieve complete separation and desired resolution under isocratic conditions, several columns (C18, C8, Biphenyl, PFP) under different chromatographic conditions with different mobile phases were tested.
Protein precipitation with acetonitrile, methanol and acidified (with 0.1–1% formic acid) acetonitrile/methanol were tested. To attain required sensitivity liquid–liquid extraction method was chosen. Different extraction solvents (ethyl acetate, diethyl ether, dichloromethane and hexane) with different ratios were tested. The best results were observed with ethyl acetate, however ethyl acetate is soluble in water to some extent, which resulted in dirtier extracts requiring filtering step. To overcome this problem ethyl acetate was saturated with water prior to extraction.
The highest extraction yields for both analytes with the best reproducibility results were achieved with water saturated ethyl acetate.
The other challenging step was the elimination of carry-over of Triamterene. The lowest carry-over was achieved with isopropanol:water 50:50 needle wash solution with low injection volume (3 μL) and by using 0.7% formic acid:methanol (9:1) mixture for the reconstitution of the residue after evaporation.
3.2 Method validation
3.2.1 Selectivity (blank check, interference check, metabolite interference)
Six lots of blank plasma were screened and interference at retention time of analytes and ISs was evaluated. No significant interference was observed at the retention time of Triamterene and Hydrochlorothiazide and their ISs in all the batches screened. Figures 2, 3, 4 and 5 illustrate the representative chromatograms for Blank and LLOQ samples.

Representative chromatogram of blank sample for Triamterene

Representative chromatogram of LLOQ sample for Triamterene

Representative chromatogram of blank sample for Hydrochlorothiazide

Representative chromatogram of LLOQ sample for Hydrochlorothiazide
There was no interference at the retention time of the analyte due to the IS and at retention time of the IS due to the analyte.
There was no significant interference from Triamterene two main metabolites (4-Hydroxy-triamterene, 4-Hydroxy-triamterene sulfate) at the retention time of analyte and IS.
3.2.2 Calibraton curve and sensitivity
The calibration curve was linear over the concentration range of 0.5–200 ng/mL for Triamterene and 2.5–400 ng/mL for Hydrochlorothiazide. The average %Accuracy for calibration curve standards was between 95.0 and 104.9% (Triamterene) and 97.3–102.0% (Hydrochlorothiazide). The highest %CVs of the calibrators were 4.3% (Triamterene) and 5.7% (Hydrochlorothiazide). The lower limit of quantification (LLOQ) was established as 0.5 ng/mL for Triamterene, and 2.5 ng/mL for Hydrochlorothiazide. In the present study, the precision and accuracy of LLOQ were acceptable, with %CV values less than 2.0% and %Accuracy values within 100.7–102.0% for both analytes.
3.2.3 Precision and accuracy
Four different precision and accuracy batches were run on different days. The %CV and %Accuracy of samples per each group and QC level in each batch (intra) and among four batches (inter) were within acceptable limits. Results are presented in Table 2.
For intra-day precision and accuracy testing %Accuracy of LLOQ QC was within 102.5–110.1% and 95.5–115.7% ranges, and the highest %CV was 4.7% and 11.9% for Triamterene and Hydrochlorothiazide correspondingly. For the rest of QCs %Accuracy was within 97.2–110.3% and 99.4–110.8% ranges, the highest %CV was 2.8% and 4.1% for Triamterene and Hydrochlorothiazide correspondingly.
The %CV and %Accuracy of QCs at each level among four batches (inter-assay) were within acceptable limits for both Triamterene and Hydrochlorothiazide. The %Accuracy of LLOQ QC was 106.0% and 104.5%, %CV was 4.2% and 11.8% for Triamterene and Hydrochlorothiazide correspondingly. For the rest of QCs %Accuracy was within 97.2–111.8% and 102.6–107.2% ranges and the highest %CV was 2.6% and 4.5% for Triamterene and Hydrochlorothiazide correspondingly.
3.2.4 Matrix effect
The %CV of IS-normalized MF for each LQC and HQC samples prepared from six matrices was 5.0% (LQC) and 1.0% (HQC) for Triamterene and 2.5% (LQC) and 1.8% (HQC) for Hydrochlorothiazide. These data indicated that ion suppression or enhancement from the human plasma was insignificant.
3.2.5 Recovery
The mean recoveries for Triamterene ranged from 99.4 to 104.0% and 100.9 to 105.2% for Hydrochlorothiazide across the concentration range. In addition, the mean recovery of the IS 1 was 100.5% and 99.0% for IS 2.
3.2.6 Carry over
The maximal carry over was 4.6% for Triamterene and 0.4% for Hydrochlorothiazide after OCC QC sample. For the IS carry over test it was 0.0% for both ISs.
3.2.7 Ruggedness
The Ruggedness test was performed during precision and accuracy testing by three different chemists on three different columns.
3.2.8 Hemolysis effect
The mean values of 2% and 5% Hemolytic LQC and HQC samples were in ranges of 94.8–109.3% (Triamterene) and 103.0–107.5 (Hydrochlorothiazide) for %Accuracy and NMT 3.2% (Triamterene) and 4.7% (Hydrochlorothiazide) for Precision. Hence, slightly (NMT 5%) hemolyzed samples can be assayed for Triamterene and Hydrochlorothiazide determination.
3.2.9 Hyperlipidemia effect
The mean values of the lipemic LQC and HQC samples were 100.2% and 90.0% (Triamterene) and 98.4% and 99.3% (Hydrochlorothiazide) for %Accuracy and NMT 3.9% (Triamterene) and 3.4% (Hydrochlorothiazide) for Precision. Hence, lipemic samples can be assayed for Triamterene and Hydrochlorothiazide determination.
3.2.10 Dilution integrity
The mean value of diluted samples at each level was within ± 15.0% (108.8% for Triamterene and 110.8% for Hydrochlorothiazide) of the nominal value and the %CV at each level was within 15.0% (1.1% for Triamterene and 2.4% for Hydrochlorothiazide). Thus, when the calculated concentration of the samples are above upper limit of quantification samples can be diluted up to 5 times, if necessary, to fit within the calibration curve.
3.2.11 Stability
The stability of the analytes was investigated under a variety of storage and process conditions. All established stability for the analytes were summarized in Tables 3 and 4. The data showed that the stability of QC samples at room temperature for 12 h, 77 h in the auto-sampler (4 °C), three freeze–thaw cycles, long-term storage in freezer (25 days at − 20 °C), 2 h in the human whole blood and 6 h in the processed sample was acceptable. The results showed that both analytes and their ISs stock and working solutions were stable at room temperature for 17 h. Analytes stock and working solutions were stable in refrigerator for 35 days and the ISs stock and working solutions for 51 days.
3.2.12 Maximum batch size
%CV of all QC samples in the maximum batch size study was less than 15.0% (highest %CV was 2.9% for Triamterene, and 4.5% for Hydrochlorothiazide) and the %Accuracy was within ± 15.0% (102.3–106.6% for Triamterene and 103.6–109.3% for Hydrochlorothiazide). This indicates that up to 130 samples can be included within the same batch.
3.2.13 Application to pharmacokinetic study
In the present study, a specific, sensitive and rapid method based on LC–MS/MS was developed and fully validated to quantify Triamterene and Hydrochlorothiazide in human plasma samples. This method was successfully applied to determine the pharmacokinetic profile of Triamterene and Hydrochlorothiazide in 12 healthy adult subjects after a single oral dose (75/50 mg) administration under fasting conditions. The study was approved by the Ethics Committee of Ministry of Health of Republic of Armenia, and all subjects signed the Informed Consent Form before the study. The main pharmacokinetic parameters’ results are presented in the Table 5. The mean plasma concentration–time curve is shown in Figs. 6 and 7.

Mean plasma Triamterene concentrations versus time plot for the test (1) and reference (2) drug

Mean plasma Hydrochlorothiazide concentrations versus time plot for the test (1) and reference (2) drug
4 Conclusion
Since Triamterene and Hydrochlorothiazide is commonly used combination as a potassium-sparing and thiazide-type diuretic, it is desired to have method for simultaneous determination of both analytes for pharmacokinetic studies. In the current article we discussed all issues which we faced during method development. As a result:
-
LC–MS/MS method for simultaneous quantification of Triamterene and Hydrochlorothiazide in human plasma has been successfully developed and validated,
-
An LLOQ of 0.5 ng/mL for Triamterene and 2.5 ng/mL for Hydrochlorothiazide was achieved,
-
The validated method was successfully applied to pilot bioequivalence study.
References
Stolarczyk M et al (2008) Simultaneous determination of triamterene and hydrochlorothiazide in tablets using derivative spectrophotometry. Acta Pol Pharm 65(3):283–287
Sable D, Bagul ATM, Goswami S (2016) Simultaneous determination of Triamterene and Hydrochlorothiazide in human plasma by liquid chromatography-mass spectrometry. Am J Pharm Technol Res 6(5):153–170
Hamidi M, Shahbazi MA, Azimi K (2011) Bioequivalence evaluation of a triamterene–hydrochlorothiazide generic product: a new bioequivalence index for fixed-dose combinations. Regul Toxicol Pharmacol 59(1):149–156
Li H et al (2011) Simultaneous analysis of hydrochlorothiazide, triamterene and reserpine in rat plasma by high performance liquid chromatography and tandem solid-phase extraction. J Sep Sci 34(5):542–547
Rote AR, Sonavane PR (2012) Development and validation of bioanalytical method for determination of Telmisartan and Hydrochlorothiazide using HPTLC in human plasma. Am J Anal Chem 3(11):774–778
Liu F et al (2007) Determination of hydrochlorothiazide in human plasma by liquid chromatography/tandem mass spectrometry. J Pharm Biomed Anal 44(5):1187–1191
Ramakrishna NV et al (2005) Sensitive liquid chromatography-tandem mass spectrometry method for quantification of hydrochlorothiazide in human plasma. Biomed Chromatogr 19(10):751–760
Yeung YG, Stanley ER (2010) Rapid detergent removal from peptide samples with ethyl acetate for mass spectrometry analysis. Curr Protoc Protein Sci Chapter 16: p. Unit 16 12
U.S. Food and Drug Administration (2018) Bioanalytical method validation; guidance for industry. https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf. Accessed 26 Oct 2018
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Margaryan, T., Mikayelyan, A., Zakaryan, H. et al. Simultaneous determination of Triamterene and Hydrochlorothiazide in human plasma by liquid chromatography tandem mass spectrometry and its application to a bioequivalence study. SN Appl. Sci. 1, 653 (2019). https://doi.org/10.1007/s42452-019-0672-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-019-0672-4