
Abstract
This study aims to describe the main cognitive and behavioral comorbidities of Dravet syndrome (DS) and Lennox–Gastaut syndrome (LGS), their impact on the health-related quality of life (QOL) of patients and their caregivers, and provide a summary of the neuropsychological tools available for the evaluation of these comorbidities. The cognitive and behavioral comorbidities in patients with DS and LGS have a profound effect on the QOL of affected individuals and their caregivers and, as patients grow, tend to surpass the impact of the seizures. DS is a genetic condition associated with loss-of-function mutations in the SCNA1 sodium channel gene; LGS is an etiologically heterogeneous condition that is often secondary to structural brain abnormalities. The first seizures associated with DS typically present in the first year of life, and developmental delay becomes progressively evident thereafter. LGS usually starts between the ages of 3 and 8 years, with cognitive impairment becoming clinically evident in most patients within 5 years from the onset. In both DS and LGS, cognitive impairment is generally moderate to severe and is often accompanied by behavioral problems such as hyperactivity and inattention. In addition to optimal seizure control, regular assessment and active management of cognitive and behavioral comorbidities are required to meet the complex needs of patients with DS or LGS.
Similar content being viewed by others

Introduction
Dravet syndrome (DS) and Lennox–Gastaut syndrome (LGS) are severe developmental and epileptic encephalopathies (DEEs) that begin in childhood and persist throughout adulthood [1,2,3,4]. In both conditions, epileptiform activity, seizures, and the underlying genetic, structural, or metabolic defect can result in neurodevelopmental delay or regression [5,6,7]. DS is a genetic condition that is primarily associated with loss-of-function mutations in the SCNA1 gene for voltage-gated sodium channels, which result in loss of action firing in gamma-aminobutyric acid (GABA)-ergic interneurons [8, 9]. In contrast, LGS has a relatively heterogeneous etiology [2, 10, 11] with an identifiable (genetic, structural, or metabolic) cause in 65–75% of patients [1, 2, 9,10,11]. LGS occurs most often as a result of a structural brain abnormality, such as tuberous sclerosis complex, cerebral malformation, or hypoxic-ischemic injury, and very rarely as a result of a progressive metabolic disorder [2, 9,10,11,12].
LGS accounts for an estimated 1–10% of childhood epilepsies, and the reported incidence of DS is between 1 in 15,700 and 1 in 40,000 live births, so these are rare diseases [2, 3, 13, 14]. However, the morbidity associated with LGS and DS leads to disproportionately large costs to the healthcare system and society in general [7, 13].
Treatment of DS and LGS focuses on alleviation of seizure burden, but some patients may have treatment-resistant seizures. In addition, DS and LGS are associated with multiple comorbidities, which present further management challenges [4, 10, 15,16,17]. Despite their different etiologies, DS and LGS share several comorbid features, including developmental delays in several domains: social–emotional, cognitive, motor, adaptive, and communicative [18,19,20,21,22]. Thorough and accurate early assessment and diagnosis, including careful assessment of clinical and electroencephalography (EEG) features, are important to ensure that patients receive the most appropriate treatment and support as soon as possible, to optimize long-term neurodevelopmental outcome [4, 19, 23].
In this review, we describe the most common cognitive and behavioral comorbidities of DS and LGS and their impact on the health-related quality of life (HRQOL) of patients and their caregivers. Management implications and neuropsychological tools for the evaluation of cognitive and behavioral aspects of LGS and DS are also discussed.
Cognitive and Behavioral Comorbidities of Dravet Syndrome and Lennox–Gastaut Syndrome
Cognitive Impairment
Moderate-to-severe cognitive impairment is common in DS and LGS and is a diagnostic feature of both disorders [2,3,4, 15]. Cognitive delay generally occurs earlier in life with DS than with LGS [2, 19], but cognitive impairment is not necessarily apparent at the onset of seizures in either syndrome [1, 21, 24].
Dravet Syndrome
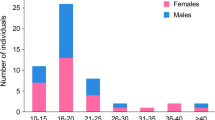
Intellectual disability (ID) is a comorbidity of DS, but it is still unclear if ID results from damage caused by seizures, the abnormal levels of Nav1.1 protein, the use of inappropriate medications, such as sodium channel inhibitors, or a combination of these factors [25]. DS typically presents in the first year of life in children with no pre-existing developmental problems [3, 21, 26, 27]. It was generally thought that DS patients show typical cognitive development until the second year of life, followed by a gradual decline which leads to moderate or severe ID by 6 years of age (Fig. 1) [26,27,28,29,30,31,32]. However, recent data indicate variability in cognitive development among DS children tested before 24 months of age, suggesting that cognitive delay may begin before 2 years of age in some patients [33]. A study by Nabbout et al. [31] in 67 children with DS showed that, after 6 years of age, IQ decreased with age, but patients continued acquiring new skills during the first decade. Therefore, the progression of developmental delay seen from 2 years of age [22, 26, 27, 31,32,33,34,35] could reflect an early arrest of cognitive development, followed by an increasing discrepancy between developmental age and chronological age [26], rather than consistent cognitive regression.

Cross-sectional analysis of cognitive development over time in 241 patients (aged 6 months to 42 years) with SCN1A mutation-positive Dravet syndrome, reproduced with permission [28]
In the literature, many authors have found that seizure frequency and severity, the presence of status epilepticus (SE), or EEG activity may influence the cognitive outcome in patients with DS [28, 29, 31, 32, 36, 37]. However, in other studies, there was no significant correlation between the severity of cognitive delay and seizure activity [31, 38]. Preclinical studies suggest that SCN1A mutation by itself may play a role in determining the final cognitive outcome [26, 31]. The pattern of cognitive deficits observed in patients with DS is consistent with cerebellar cognitive syndrome, as language comprehension tends to be better preserved than language production and visuospatial functions [22, 31, 34, 35, 37, 39, 40].
Lennox–Gastaut Syndrome
In contrast to DS, 20–60% of LGS patients have cognitive impairment before the onset of seizures, particularly when LGS is a consequence of an identifiable etiology [15, 19, 41]. Children with LGS of unknown origin may seem to develop normally before the appearance of the first seizures [15, 19]. In general, cognitive impairment becomes progressively evident over time [1, 2, 15], and the proportion of patients with serious ID may increase to 75–95% at 5 years after the onset of seizures [42]. The cognitive outcome of patients with LGS is variable [43], and there is no evidence that it correlates only with epilepsy severity. LGS is characterized by alterations in specific thalamocortical networks, which normally support a broad range of cognitive functions [44]. Disruption of normal interactions within and between brain networks that support key cognitive processes has been reported in an EEG-functional magnetic resonance imaging study of 15 adult patients with LGS [45]. It has been proposed that epileptic activity in LGS, which intersects with cognitive networks, may initiate sustained abnormal network behavior, and the persistence of these abnormal cognitive network interactions in the absence of detectable epileptic activity potentially contributes to impaired cognition [45]. The relatively preserved cognitive status observed in late-onset LGS may, in part, be because these patients did not experience epileptic activity during sensitive periods of neurodevelopment when cognitive networks are most vulnerable to disrupted maturation [36, 45,46,47].
Behavioral Problems
In DS and LGS, cognitive impairment is frequently associated with social–emotional problems, but there is not necessarily a linear relationship between the severity of the cognitive disability and the frequency of behavioral difficulties [4, 20, 48,49,50]. Attention and hyperactivity problems, as well as autistic traits, are commonly observed in both DEEs [2,3,4, 15, 16, 51].
Dravet Syndrome
Behavioral difficulties in children with DS usually include attention disorders and hyperactivity, aggressive behavior, and social problems [16, 33]. These problems, which do not appear to be related to the severity or frequency of epilepsy, seem to increase in childhood, peak in adolescence, and then plateau or decrease in adulthood, while autistic traits tend to persist [28, 49, 52,53,54]. Parents and caregivers often report high levels of attention difficulties in patients affected by DS, which have been confirmed in several studies, but the prevalence of ADHD among children with DS is unknown [16, 50, 53].
DS patients may display autistic traits, such as stereotyped, repetitive and disinhibited behavior, obsessions, and poor peer relationships [38, 50,51,52, 54, 55], but the prevalence of autism spectrum disorder (ASD) in DS varies significantly across studies. One reason may be that communicative skills are relatively preserved, leading to an underestimation of ASD prevalence among patients with DS [38, 50,51,52]. When gold-standard ASD assessment tools were used, ASD prevalence ranged from 22 to 39% [16, 51]. Studies in mice show that impairment of sodium channel function and the resulting GABAergic signaling dysfunction likely contribute to behavioral and cognitive impairment in DS [8, 56, 57].
Lennox–Gastaut Syndrome
Hyperactivity/inattention, anxiety, agitation, depression, and aggression are commonly seen in patients with LGS [1, 2, 15, 42, 58, 59]. Autistic behavior has also been reported, but only in a few cases of LGS, and is less common in LGS than DS, despite the similar prevalence and severity of IDs in the two forms of encephalopathy [42]. It has been suggested that factors other than cognitive impairment are involved in the pathogenesis of autistic traits in LGS and DS [42], such as epilepsy itself, abnormal brain neural network characteristics, and the effect of medications [1].
Other Comorbidities of Dravet Syndrome and Lennox–Gastaut Syndrome
Patients with DS often develop disabling motor impairment, involving all motor domains (balance, coordination, visuomotor integration, power, and locomotion) [60]. As a result, walking becomes increasingly ataxic, and gait tends to worsen with age [3, 17, 20, 21, 28, 30, 50, 61,62,63], such that, from adolescence, many patients require the use of a wheelchair for longer distances [20]. A “crouch gait” (defined as the increased ankle, knee, and hip flexion during the whole gait cycle, plus rotations of the femur and/or tibia, and muscle retraction) is not specific to DS but often appears by 13 years of age [61, 63]. In fact, a study by Di Marco et al. [64] showed that the crouch gait pattern could appear as early as 4 years of age in patients with DS. By adolescence (age > 13 years), some patients have developed a flexed gait pattern with passive knee extension deficit and bony malalignment. In adulthood, extrapyramidal signs and Parkinsonian gait become evident [17, 18, 61,62,63].
Gitiaux et al. proposed that to understand gait disturbance, we should consider DS as a sodium channel interneuronopathy causing complex clinical presentations of varying nature [17]. So depending on the site or structure where the voltage-gated sodium channel type 1 (NaV1.1) is expressed, different movement disorders are induced. In particular, motor neuron dysfunction could partially explain the gait features observed at first as mild distal motor deficits, followed by proximal (crouch-like) deficits. In a genetic mouse model of DS, ataxia was caused by deficits in the cerebellar Purkinje neurons conveying output information on movement, coordination, and balance from the cerebellar cortex [65]. In contrast, mobility problems in LGS are often a direct result of seizures, particularly drop attacks, which are physically demanding and often result in injury [10, 48].
Sleep disturbances, which are common in DS and LGS, may contribute to cognitive, behavioral, and psychological issues [10, 48, 50, 55, 66,67,68]. Tonic seizures occurring during sleep, with the potential to disrupt the sleep cycle, are characteristic of LGS [48]. DS children and adults have more nighttime awakenings, reflective of poor quality of sleep [25]. SCNA1 mutations dysregulate neurological sleep networks [8, 66], as demonstrated in a mouse model, in which NaV1.1 channels encoded by SCN1A are expressed in the GABAergic neurons in the hypothalamus, thalamic reticular nucleus, and cortex [69]. In both DS [66] and LGS [70], nocturnal seizures, polypharmacy, developmental delay, and environmental factors (such as co-sleeping) may also contribute to a high frequency of sleep disturbances.
Epilepsy patients have a 24- to 28-fold higher rate of sudden death compared with the general population [71]. It has been proposed that channelopathy common to both epilepsy and cardiac disease may contribute to the increased risk of death via a lethal cardiac arrhythmia [72, 73]. Patients with DS have a genetic predisposition to cardiac autonomic dysfunction, heart rate abnormalities, and arrhythmias, reflecting SCN1A expression in the heart and contributing to a high risk of sudden unexpected death in epilepsy [74,75,76].
Impact of Comorbidities on Patients and Caregivers
Comorbidities of DS and LGS increase the complexity of the patient’s care needs and can disrupt daily activities, limit educational and social progress, and have a profound impact on the HRQOL of patients with DS or LGS and their families (Fig. 2) [20, 48,49,50, 54, 77,78,79,80]. Cognitive impairment and behavioral problems both independently predict reduced HRQOL, but behavioral problems can be particularly burdensome [20, 49, 53, 54, 81]. With their impact on mobility, motor impairments further detract from HRQOL [20, 78, 81].

Conceptual model of ways in which DS and LGS affect the HRQOL of patients, reproduced with permission [77]. DS, Dravet syndrome; HRQOL, health-related quality of life; LGS, Lennox–Gastaut syndrome
Whereas the nature of seizures can change and seizure burden can decrease, the burden of comorbidities in DS and LGS does not improve and increases with age [25, 29, 48, 50, 59]. Although seizures remain a prominent problem, cognitive, behavioral, and physical comorbidities can eclipse seizures as a higher management priority in adolescent and adult patients with LGS, reflecting the importance of education, independence, and the ability to work, interact with peers, and form relationships [15, 48, 59].
General Management Implications
Although seizures must be controlled, management of DS and LGS should focus not only on pharmacologic seizure control but should also address comorbidities [2, 4, 10, 20, 21, 48, 50, 79, 82]. Seizures may aggravate the cognitive and behavioral comorbidities of DS and LGS, and therefore, pharmacologic treatment should be tailored to each patient based on the type of seizure, age, and clinical history [83]. Moreover, currently used antiepileptic drugs (AEDs) may also contribute to cognitive impairment and behavioral disorders in these patients, and few data are available on the most appropriate antiepileptic therapy for patients with LGS or DS. Comorbidities and the risk of AED-related adverse events (AEs) can also influence the therapeutic choice [4, 10, 20, 41, 53, 79, 83, 84].
Valproic acid (VPA) is used in both syndromes; it is a broad-spectrum AED and is highly unlikely to aggravate seizures [10, 83]. In the pediatric LGS population, VPA has been associated with cognitive AEs (such as confused state or attention disturbances) or behavioral AEs (such as aggression or agitation); it is rarely associated with AEs such as abnormal behavior or hyperactivity [10]. However, VPA is known to be associated with two types of hepatotoxicity: dose dependent and reversible elevation of serum liver enzymes (type I), and rare but often fatal, idiosyncratic hepatotoxicity (type II) [85]. The risk of VPA-related hepatotoxicity is increased in children below 2 years of age and in patients receiving other anticonvulsants [86,87,88,89]. Also, VPA has been reported to cause some coagulopathies (von Willebrand’s disease, factor XIII deficiency, thrombocytopenia); however, their clinical relevance remains uncertain [85]. Clobazam (CLB) is a benzodiazepine with antiepileptic properties and is also used in both syndromes. It has been associated with cognitive and behavioral AEs, including slowed reaction time, confusion, irritability, and numbed emotions [10]. Stiripentol (STP) is an orally active unique AED, indicated for use with CLB and VPA as adjunctive therapy for refractory seizures in DS [90, 91]. AEs associated with STP treatment include somnolence and motor impairment such as ataxia or tremor. Felbamate (FLB), which is licensed in some European countries as adjunctive therapy in patients with LGS, showed adverse effects on cognition (abnormal thinking) in 6.5% of pediatric patients; behavioral AEs associated with FLB include agitation, aggression, or psychological disturbance [10]. Lamotrigine (LTG) was approved by the US Food and Drug Administration (FDA) in 1998 as an adjunctive treatment for patients with LGS aged > 2 years [83, 92]. It is considered a second-line AED after the failure of VPA. Common AEs associated with LTG include ataxia, rash, headache, anorexia, and drowsiness [83], or very rarely, confusion or hallucinations [10]. Behavioral AEs, such as aggression, irritability, and agitation, may also occur [10]. Rufinamide (RUF) was approved by the European Medicines Agency (in 2007) and by the US FDA (in 2008) as adjunctive seizure therapy in patients with LGS older than 4 years of age [93]. Anxiety is a common behavioral AE associated with RUF [10, 94, 95]. Topiramate (TPM) was approved by the US FDA in 2001 as adjunctive therapy for the treatment of seizures in patients with LGS aged ≥ 2 years [83]. Its tolerability profile in the pediatric population is characterized by AEs occurring early in the treatment course, and most are CNS related, including somnolence and psychomotor impairment [83]; depression has also been commonly observed [10].
In addition to optimal seizure control, long-term treatment goals should focus on maximizing developmental potential and HRQOL [4, 11, 19, 96].
Given the various comorbidities of DS and LGS, comprehensive multidisciplinary management involving different healthcare professionals is required to ensure that individual patients’ medical, educational, psychological, and social needs are met throughout their lives (Fig. 3) [4, 10, 11, 48, 50, 54, 77, 97]. Transitioning from pediatric to adult care can be particularly challenging as the provision of care becomes more dispersed and there are generally fewer resources available for adults compared with pediatric patients [10, 48, 50, 77, 96]. To ensure as smooth a transition as possible, patients and their families should receive appropriate educational and psychosocial support [19, 48].

Multidisciplinary division of care as patients with LGS transition from pediatric to adult services, reproduced with permission [48]. LGS, Lennox–Gastaut syndrome
Cannabidiol (CBD), a nonpsychoactive compound derived from Cannabis plants, is a recent addition to the treatment options for patients with DS and LGS [98]. In the USA, CBD (Epidiolex®, Greenwich Biosciences, Inc., Carlsbad, CA, USA) has been approved for the treatment of seizures associated with LGS or DS in patients aged ≥ 2 years [99]. In the EU, CBD (Epidyolex®, GW Pharmaceuticals, Cambridge, UK) has been approved for the adjunctive treatment (in combination with CLB) of seizures associated with LGS or DS in patients aged ≥ 2 years [100] and for seizures associated with tuberous sclerosis complex in patients aged ≥ 1 year [101]. There is some evidence that, in addition to reducing the frequency and severity of seizures, CBD improves mood and quality of life in patients with treatment-resistant epilepsy [87, 102]. In this population, long-term use of CBD does not appear to be associated with negative effects on cognitive functioning [103].
Neuropsychological Assessment in the Management of Cognitive and Behavioral Problems
Comprehensive care of patients with DS or LGS must involve a careful assessment of comorbidities, routine monitoring of development and behavior at clinic visits, a formal developmental or cognitive assessment before starting school or earlier if there are clinical concerns about development, regular school assessments, and reassessment when transitioning from pediatric to adult care [4, 48]. Table 1 summarizes the timepoints for conducting neuropsychological testing in pediatric patients with DS or LGS.
Active management of cognitive and behavioral problems is a high priority, but there are no neuropsychological assessment tools specifically for the evaluation of DS or LGS patients, and characterization of cognitive performance and behavior in these patients is challenging [15, 51]. General standardized assessment tools should be chosen according to the patient’s age and neurodevelopmental level, and the neuropsychological components that are typically compromised in children with DS or LGS (i.e., speech, attention, spatial, sensory-motor, and executive function). Many of the same tests are appropriate for use in both DS and LGS (Table 2).
The Wechsler Intelligence Scale for Children (WISC) and the Wechsler Preschool and Primary Scale of Intelligence (WPPSI) are useful for an initial general assessment of cognitive abilities in children with DS or LGS [40, 84]. The Leiter-R test is a nonverbal assessment of intellectual functioning, comprising visualization and reasoning, and attention and memory components [104], and is also recommended as an initial test at diagnosis/onset. The Griffiths or Bayley Scales can be used for very young DS children up to 3 years of age [40, 84]. From 3 years, the Visual–Motor Integration Test can be used to assess visual function, which is particularly compromised in DS and precedes detectable cognitive decline [22, 35, 40]. Other tests that can be used to assess specific skills in patients with DS or LGS include the Corsi Test for visuospatial memory, Bell’s Cancellation Test for attention, the Boston Naming Test for language, the Tower of London procedure for executive function [40] in patients with minimal cognitive impairment, and the MacArthur-Bates Communicative Development Inventories for assessment of language [105].
Behavior assessment may be performed using tools such as the Child Behavior Checklist (CBCL) for children aged 1–5 years (CBCL/1–5) or older children and adolescents (CBCL/6–18) [40]. The CBCL questionnaire is completed by parents/caregivers, thereby acknowledging the importance of the role of the caregiver in evaluating the patient. Another generalized behavior assessment tool that is suitable for use in patients with DS or LGS [38, 51] is the Vineland Adaptive Behavioral Scales (Vineland-3), which focus on age-dependent adaptive behavior, including communication, daily living, and socialization skills [106].
Conclusions
DS and LGS are multifaceted diseases with a range of clinical comorbidities that are not purely the result of poorly controlled seizures. Moderate-to-severe cognitive impairment and general behavioral problems are characteristic comorbidities in both DS and LGS. The profound impact of cognitive and behavioral comorbidities on the lives of patients with DS or LGS and their caregivers and families highlights the importance of active multidisciplinary management, careful treatment choice, and appropriate educational and psychosocial support systems. In particular, psychoeducation (e.g., cognitive behavioral therapy or mood management) and community support may help patients and their family/caregivers in coping and adapting to the distress of pervasive and severe behavioral comorbidities of DS and LGS. Therefore, in addition to the medical management of seizures, comprehensive assessment and careful management of comorbidities should be a core aspect of care (Table 3).
Data Availability
Not applicable.
Change history
19 October 2021
A Correction to this paper has been published: https://doi.org/10.1007/s42399-021-01077-x
References
Asadi-Pooya AA. Lennox-Gastaut syndrome: a comprehensive review. Neurol Sci. 2018;39(3):403–14. https://doi.org/10.1007/s10072-017-3188-y.
Camfield PR. Definition and natural history of Lennox-Gastaut syndrome. Epilepsia. 2011;52(Suppl 5):3–9. https://doi.org/10.1111/j.1528-1167.2011.03177.x.
Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52(Suppl 2):3–9. https://doi.org/10.1111/j.1528-1167.2011.02994.x.
Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the diagnosis and management of Dravet Syndrome: recommendations from a North American consensus panel. Pediatr Neurol. 2017;68:18–34 e3. https://doi.org/10.1016/j.pediatrneurol.2017.01.025.
Covanis A. Clinical management of epileptic encephalopathies of childhood and infancy. Expert Rev Neurother. 2014;14(6):687–701. https://doi.org/10.1586/14737175.2014.919854.
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58(4):512–21. https://doi.org/10.1111/epi.13709.
Stafstrom CE, Kossoff EM. Epileptic encephalopathy in infants and children. Epilepsy Curr. 2016;16(4):273–9. https://doi.org/10.5698/1535-7511-16.4.273.
Catterall WA. Dravet syndrome: a sodium channel interneuronopathy. Curr Opin Physiol. 2018;2:42–50. https://doi.org/10.1016/j.cophys.2017.12.007.
Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68(6):1327–32. https://doi.org/10.1086/320609.
Cross JH, Auvin S, Falip M, Striano P, Arzimanoglou A. Expert opinion on the management of Lennox-Gastaut syndrome: treatment algorithms and practical considerations. Front Neurol. 2017;8:505. https://doi.org/10.3389/fneur.2017.00505.
Mastrangelo M. Lennox-Gastaut syndrome: a state of the art review. Neuropediatrics. 2017;48(3):143–51. https://doi.org/10.1055/s-0037-1601324.
Guerrini R, Striano P, Catarino C, Sisodiya SM. Neuroimaging and neuropathology of Dravet syndrome. Epilepsia. 2011;52(Suppl 2):30–4. https://doi.org/10.1111/j.1528-1167.2011.02998.x.
Trevathan E, Murphy CC, Yeargin-Allsopp M. Prevalence and descriptive epidemiology of Lennox-Gastaut syndrome among Atlanta children. Epilepsia. 1997;38(12):1283–8. https://doi.org/10.1111/j.1528-1157.1997.tb00065.x.
Wu YW, Sullivan J, McDaniel SS, Meisler MH, Walsh EM, Li SX, et al. Incidence of Dravet syndrome in a US population. Pediatrics. 2015;136(5):e1310–5. https://doi.org/10.1542/peds.2015-1807.
Arzimanoglou A, French J, Blume WT, Cross JH, Ernst JP, Feucht M, et al. Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol. 2009;8(1):82–93. https://doi.org/10.1016/S1474-4422(08)70292-8.
Jansson JS, Hallbook T, Reilly C. Intellectual functioning and behavior in Dravet syndrome: a systematic review. Epilepsy Behav. 2020;108:107079. https://doi.org/10.1016/j.yebeh.2020.107079.
Wyers L, Van de Walle P, Hoornweg A, Tepes Bobescu I, Verheyen K, Ceulemans B, et al. Gait deviations in patients with Dravet syndrome: a systematic review. Eur J Paediatr Neurol. 2019;23(3):357–67. https://doi.org/10.1016/j.ejpn.2019.03.003.
Aljaafari D, Fasano A, Nascimento FA, Lang AE, Andrade DM. Adult motor phenotype differentiates Dravet syndrome from Lennox-Gastaut syndrome and links SCN1A to early onset parkinsonian features. Epilepsia. 2017;58(3):e44–e8. https://doi.org/10.1111/epi.13692.
Arzimanoglou A, Resnick T. All children who experience epileptic falls do not necessarily have Lennox-Gastaut syndrome... but many do. Epileptic Disord. 2011;13(Suppl 1):S3–13. https://doi.org/10.1684/epd.2011.0422.
de Lange IM, Gunning B, Sonsma ACM, van Gemert L, van Kempen M, Verbeek NE, et al. Outcomes and comorbidities of SCN1A-related seizure disorders. Epilepsy Behav. 2019;90:252–9. https://doi.org/10.1016/j.yebeh.2018.09.041.
Gataullina S, Dulac O. From genotype to phenotype in Dravet disease. Seizure. 2017;44:58–64. https://doi.org/10.1016/j.seizure.2016.10.014.
Chieffo D, Battaglia D, Lettori D, Del Re M, Brogna C, Dravet C, et al. Neuropsychological development in children with Dravet syndrome. Epilepsy Res. 2011;95(1-2):86–93. https://doi.org/10.1016/j.eplepsyres.2011.03.005.
Bourgeois BF, Douglass LM, Sankar R. Lennox-Gastaut syndrome: a consensus approach to differential diagnosis. Epilepsia. 2014;55(Suppl 4):4–9. https://doi.org/10.1111/epi.12567.
Berg AT, Levy SR, Testa FM. Evolution and course of early life developmental encephalopathic epilepsies: focus on Lennox-Gastaut syndrome. Epilepsia. 2018;59(11):2096–105. https://doi.org/10.1111/epi.14569.
Selvarajah A, Zulfiqar-Ali Q, Marques P, Rong M, Andrade DM. A systematic review of adults with Dravet syndrome. Seizure. 2021;87:39–45. https://doi.org/10.1016/j.seizure.2021.02.025.
Ragona F, Brazzo D, De Giorgi I, Morbi M, Freri E, Teutonico F, et al. Dravet syndrome: early clinical manifestations and cognitive outcome in 37 Italian patients. Brain and Development. 2010;32(1):71–7. https://doi.org/10.1016/j.braindev.2009.09.014.
Wolff M, Casse-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia. 2006;47(Suppl 2):45–8. https://doi.org/10.1111/j.1528-1167.2006.00688.x.
Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain. 2012;135(Pt 8):2329–36. https://doi.org/10.1093/brain/aws151.
Darra F, Battaglia D, Dravet C, Patrini M, Offredi F, Chieffo D, et al. Dravet syndrome: early electroclinical findings and long-term outcome in adolescents and adults. Epilepsia. 2019;60(Suppl 3):S49–58. https://doi.org/10.1111/epi.16297.
Jansen FE, Sadleir LG, Harkin LA, Vadlamudi L, McMahon JM, Mulley JC, et al. Severe myoclonic epilepsy of infancy (Dravet syndrome): recognition and diagnosis in adults. Neurology. 2006;67(12):2224–6. https://doi.org/10.1212/01.wnl.0000249312.73155.7d.
Nabbout R, Chemaly N, Chipaux M, Barcia G, Bouis C, Dubouch C, et al. Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J Rare Dis. 2013;8:176. https://doi.org/10.1186/1750-1172-8-176.
Ragona F, Granata T, Dalla Bernardina B, Offredi F, Darra F, Battaglia D, et al. Cognitive development in Dravet syndrome: a retrospective, multicenter study of 26 patients. Epilepsia. 2011;52(2):386–92. https://doi.org/10.1111/j.1528-1167.2010.02925.x.
Verheyen K, Wyers L, Del Felice A, Schoonjans AS, Ceulemans B, Van de Walle P, et al. Independent walking and cognitive development in preschool children with Dravet syndrome. Dev Med Child Neurol. 2021;63(4):472–9. https://doi.org/10.1111/dmcn.14738.
Chieffo D, Ricci D, Baranello G, Martinelli D, Veredice C, Lettori D, et al. Early development in Dravet syndrome: visual function impairment precedes cognitive decline. Epilepsy Res. 2011;93(1):73–9. https://doi.org/10.1016/j.eplepsyres.2010.10.015.
Ricci D, Chieffo D, Battaglia D, Brogna C, Contaldo I, De Clemente V, et al. A prospective longitudinal study on visuo-cognitive development in Dravet syndrome: is there a “dorsal stream vulnerability”? Epilepsy Res. 2015;109:57–64. https://doi.org/10.1016/j.eplepsyres.2014.10.009.
Dupont S, Banica-Wolters R, An-Gourfinkel I, Lambrecq V, Navarro V, Adam C, et al. Understanding Lennox-Gastaut syndrome: insights from focal epilepsy patients with Lennox-Gastaut features. J Neurol. 2017;264(7):1388–96. https://doi.org/10.1007/s00415-017-8535-7.
Olivieri G, Battaglia D, Chieffo D, Rubbino R, Ranalli D, Contaldo I, et al. Cognitive-behavioral profiles in teenagers with Dravet syndrome. Brain and Development. 2016;38(6):554–62. https://doi.org/10.1016/j.braindev.2015.12.014.
Villeneuve N, Laguitton V, Viellard M, Lepine A, Chabrol B, Dravet C, et al. Cognitive and adaptive evaluation of 21 consecutive patients with Dravet syndrome. Epilepsy Behav. 2014;31:143–8. https://doi.org/10.1016/j.yebeh.2013.11.021.
Chieffo D, Battaglia D, Lucibello S, Gambardella ML, Moriconi F, Ferrantini G, et al. Disorders of early language development in Dravet syndrome. Epilepsy Behav. 2016;54:30–3. https://doi.org/10.1016/j.yebeh.2015.10.027.
Battaglia D, Chieffo D, Siracusano R, Waure C, Brogna C, Ranalli D, et al. Cognitive decline in Dravet syndrome: is there a cerebellar role? Epilepsy Res. 2013;106(1-2):211–21. https://doi.org/10.1016/j.eplepsyres.2013.03.012.
Resnick T, Sheth RD. Early diagnosis and treatment of Lennox-Gastaut syndrome. J Child Neurol. 2017;32(11):947–55. https://doi.org/10.1177/0883073817714394.
He N, Li BM, Li ZX, Wang J, Liu XR, Meng H, et al. Few individuals with Lennox-Gastaut syndrome have autism spectrum disorder: a comparison with Dravet syndrome. J Neurodev Disord. 2018;10(1):10. https://doi.org/10.1186/s11689-018-9229-x.
Archer JS, Warren AE, Jackson GD, Abbott DF. Conceptualizing lennox-gastaut syndrome as a secondary network epilepsy. Front Neurol. 2014;5:225. https://doi.org/10.3389/fneur.2014.00225.
Warren AEL, Abbott DF, Jackson GD, Archer JS. Thalamocortical functional connectivity in Lennox-Gastaut syndrome is abnormally enhanced in executive-control and default-mode networks. Epilepsia. 2017;58(12):2085–97. https://doi.org/10.1111/epi.13932.
Warren AE, Abbott DF, Vaughan DN, Jackson GD, Archer JS. Abnormal cognitive network interactions in Lennox-Gastaut syndrome: a potential mechanism of epileptic encephalopathy. Epilepsia. 2016;57(5):812–22. https://doi.org/10.1111/epi.13342.
Chourasia N, Maheshwari A, Kalamangalam G. Cognitive and functional status in late-onset Lennox-Gastaut syndrome: variation on a classic phenotype. Epilepsy Behav. 2020;102:106660. https://doi.org/10.1016/j.yebeh.2019.106660.
Goldsmith IL, Zupanc ML, Buchhalter JR. Long-term seizure outcome in 74 patients with Lennox-Gastaut syndrome: effects of incorporating MRI head imaging in defining the cryptogenic subgroup. Epilepsia. 2000;41(4):395–9. https://doi.org/10.1111/j.1528-1157.2000.tb00179.x.
Kerr M, Kluger G, Philip S. Evolution and management of Lennox-Gastaut syndrome through adolescence and into adulthood: are seizures always the primary issue? Epileptic Disord. 2011;13(Suppl 1):S15–26. https://doi.org/10.1684/epd.2011.0409.
Sinoo C, de Lange IM, Westers P, Gunning WB, Jongmans MJ, Brilstra EH. Behavior problems and health-related quality of life in Dravet syndrome. Epilepsy Behav. 2019;90:217–27. https://doi.org/10.1016/j.yebeh.2018.11.029.
Villas N, Meskis MA, Goodliffe S. Dravet syndrome: characteristics, comorbidities, and caregiver concerns. Epilepsy Behav. 2017;74:81–6. https://doi.org/10.1016/j.yebeh.2017.06.031.
Ouss L, Leunen D, Laschet J, Chemaly N, Barcia G, Losito EM, et al. Autism spectrum disorder and cognitive profile in children with Dravet syndrome: delineation of a specific phenotype. Epilepsia Open. 2019;4(1):40–53. https://doi.org/10.1002/epi4.12281.
Berkvens JJ, Veugen I, Veendrick-Meekes MJ, Snoeijen-Schouwenaars FM, Schelhaas HJ, Willemsen MH, et al. Autism and behavior in adult patients with Dravet syndrome (DS). Epilepsy Behav. 2015;47:11–6. https://doi.org/10.1016/j.yebeh.2015.04.057.
Lagae L, Brambilla I, Mingorance A, Gibson E, Battersby A. Quality of life and comorbidities associated with Dravet syndrome severity: a multinational cohort survey. Dev Med Child Neurol. 2018;60(1):63–72. https://doi.org/10.1111/dmcn.13591.
Brunklaus A, Dorris L, Zuberi SM. Comorbidities and predictors of health-related quality of life in Dravet syndrome. Epilepsia. 2011;52(8):1476–82. https://doi.org/10.1111/j.1528-1167.2011.03129.x.
Knupp KG, Scarbro S, Wilkening G, Juarez-Colunga E, Kempe A, Dempsey A. Parental perception of comorbidities in children with Dravet syndrome. Pediatr Neurol. 2017;76:60–5. https://doi.org/10.1016/j.pediatrneurol.2017.06.008.
Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, et al. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489(7416):385–90. https://doi.org/10.1038/nature11356.
Tatsukawa T, Ogiwara I, Mazaki E, Shimohata A, Yamakawa K. Impairments in social novelty recognition and spatial memory in mice with conditional deletion of Scn1a in parvalbumin-expressing cells. Neurobiol Dis. 2018;112:24–34. https://doi.org/10.1016/j.nbd.2018.01.009.
Ferlazzo E, Nikanorova M, Italiano D, Bureau M, Dravet C, Calarese T, et al. Lennox-Gastaut syndrome in adulthood: clinical and EEG features. Epilepsy Res. 2010;89(2-3):271–7. https://doi.org/10.1016/j.eplepsyres.2010.01.012.
Vignoli A, Oggioni G, De Maria G, Peron A, Savini MN, Zambrelli E, et al. Lennox-Gastaut syndrome in adulthood: long-term clinical follow-up of 38 patients and analysis of their recorded seizures. Epilepsy Behav. 2017;77:73–8. https://doi.org/10.1016/j.yebeh.2017.09.006.
Verheyen K, Verbecque E, Ceulemans B, Schoonjans AS, Van De Walle P, Hallemans A. Motor development in children with Dravet syndrome. Dev Med Child Neurol. 2019;61(8):950–6. https://doi.org/10.1111/dmcn.14147.
Fasano A, Borlot F, Lang AE, Andrade DM. Antecollis and levodopa-responsive parkinsonism are late features of Dravet syndrome. Neurology. 2014;82(24):2250–1. https://doi.org/10.1212/WNL.0000000000000521.
Gitiaux C, Chemaly N, Quijano-Roy S, Barnerias C, Desguerre I, Hully M, et al. Motor neuropathy contributes to crouching in patients with Dravet syndrome. Neurology. 2016;87(3):277–81. https://doi.org/10.1212/WNL.0000000000002859.
Rodda JM, Scheffer IE, McMahon JM, Berkovic SF, Graham HK. Progressive gait deterioration in adolescents with Dravet syndrome. Arch Neurol. 2012;69(7):873–8. https://doi.org/10.1001/archneurol.2011.3275.
Di Marco R, Hallemans A, Bellon G, Ragona F, Piazza E, Granata T, et al. Gait abnormalities in people with Dravet syndrome: a cross-sectional multi-center study. Eur J Paediatr Neurol. 2019;23(6):808–18. https://doi.org/10.1016/j.ejpn.2019.09.010.
Kalume F, Yu FH, Westenbroek RE, Scheuer T, Catterall WA. Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci. 2007;27(41):11065–74. https://doi.org/10.1523/JNEUROSCI.2162-07.2007.
Licheni SH, McMahon JM, Schneider AL, Davey MJ, Scheffer IE. Sleep problems in Dravet syndrome: a modifiable comorbidity. Dev Med Child Neurol. 2018;60(2):192–8. https://doi.org/10.1111/dmcn.13601.
Schoonjans AS, De Keersmaecker S, Van Bouwel M, Ceulemans B. More daytime sleepiness and worse quality of sleep in patients with Dravet Syndrome compared to other epilepsy patients. Eur J Paediatr Neurol. 2019;23(1):61–9. https://doi.org/10.1016/j.ejpn.2018.09.012.
Van Nuland A, Ivanenko A, Meskis MA, Villas N, Knupp KG, Berg AT. Sleep in Dravet syndrome: a parent-driven survey. Seizure. 2021;85:102–10. https://doi.org/10.1016/j.seizure.2020.12.021.
Kalume F, Oakley JC, Westenbroek RE, Gile J, de la Iglesia HO, Scheuer T, et al. Sleep impairment and reduced interneuron excitability in a mouse model of Dravet syndrome. Neurobiol Dis. 2015;77:141–54. https://doi.org/10.1016/j.nbd.2015.02.016.
Amir N, Shalev RS, Steinberg A. Sleep patterns in the Lennox-Gastaut syndrome. Neurology. 1986;36(9):1224–6. https://doi.org/10.1212/wnl.36.9.1224.
Tomson T, Surges R, Delamont R, Haywood S, Hesdorffer DC. WHO to target in sudden unexpected death in epilepsy prevention and how? Risk factors, biomarkers, and intervention study designs. Epilepsia. 2016;57(Suppl 1):4–16. https://doi.org/10.1111/epi.13234.
Partemi S, Vidal MC, Striano P, Campuzano O, Allegue C, Pezzella M, et al. Genetic and forensic implications in epilepsy and cardiac arrhythmias: a case series. Int J Legal Med. 2015;129(3):495–504. https://doi.org/10.1007/s00414-014-1063-4.
Coll M, Striano P, Ferrer-Costa C, Campuzano O, Mates J, Del Olmo B, et al. Targeted next-generation sequencing provides novel clues for associated epilepsy and cardiac conduction disorder/SUDEP. PLoS One. 2017;12(12):e0189618. https://doi.org/10.1371/journal.pone.0189618.
Lyu SY, Nam SO, Lee YJ, Kim G, Kim YA, Kong J, et al. Longitudinal change of cardiac electrical and autonomic function and potential risk factors in children with Dravet syndrome. Epilepsy Res. 2019;152:11–7. https://doi.org/10.1016/j.eplepsyres.2019.02.018.
Delogu AB, Spinelli A, Battaglia D, Dravet C, De Nisco A, Saracino A, et al. Electrical and autonomic cardiac function in patients with Dravet syndrome. Epilepsia. 2011;52(Suppl 2):55–8. https://doi.org/10.1111/j.1528-1167.2011.03003.x.
Frasier CR, Zhang H, Offord J, Dang LT, Auerbach DS, Shi H, et al. Channelopathy as a SUDEP biomarker in Dravet syndrome patient-derived cardiac myocytes. Stem Cell Rep. 2018;11(3):626–34. https://doi.org/10.1016/j.stemcr.2018.07.012.
Gallop K, Wild D, Verdian L, Kerr M, Jacoby A, Baker G, et al. Lennox-Gastaut syndrome (LGS): development of conceptual models of health-related quality of life (HRQL) for caregivers and children. Seizure. 2010;19(1):23–30. https://doi.org/10.1016/j.seizure.2009.10.007.
Nabbout R, Auvin S, Chiron C, Irwin J, Mistry A, Bonner N, et al. Development and content validation of a preliminary core set of patient- and caregiver-relevant outcomes for inclusion in a potential composite endpoint for Dravet syndrome. Epilepsy Behav. 2018;78:232–42. https://doi.org/10.1016/j.yebeh.2017.08.029.
Nabbout R, Auvin S, Chiron C, Thiele E, Cross H, Scheffer IE, et al. Perception of impact of Dravet syndrome on children and caregivers in multiple countries: looking beyond seizures. Dev Med Child Neurol. 2019;61(10):1229–36. https://doi.org/10.1111/dmcn.14186.
Nolan KJ, Camfield CS, Camfield PR. Coping with Dravet syndrome: parental experiences with a catastrophic epilepsy. Dev Med Child Neurol. 2006;48(9):761–5. https://doi.org/10.1017/S0012162206001629.
Campbell JD, Whittington MD, Kim CH, VanderVeen GR, Knupp KG, Gammaitoni A. Assessing the impact of caring for a child with Dravet syndrome: results of a caregiver survey. Epilepsy Behav. 2018;80:152–6. https://doi.org/10.1016/j.yebeh.2018.01.003.
Samanta D. Management of Lennox-Gastaut syndrome beyond childhood: a comprehensive review. Epilepsy Behav. 2021;114(Pt A):107612. https://doi.org/10.1016/j.yebeh.2020.107612.
Verrotti A, Striano P, Iapadre G, Zagaroli L, Bonanni P, Coppola G, et al. The pharmacological management of Lennox-Gastaut syndrome and critical literature review. Seizure. 2018;63:17–25. https://doi.org/10.1016/j.seizure.2018.10.016.
Battaglia D, Ricci D, Chieffo D, Guzzetta F. Outlining a core neuropsychological phenotype for Dravet syndrome. Epilepsy Res. 2016;120:91–7. https://doi.org/10.1016/j.eplepsyres.2015.11.020.
Gerstner T, Bell N, Konig S. Oral valproic acid for epilepsy--long-term experience in therapy and side effects. Expert Opin Pharmacother. 2008;9(2):285–92. https://doi.org/10.1517/14656566.9.2.285.
Bryant AE 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996;46(2):465–9. https://doi.org/10.1212/wnl.46.2.465.
Gaston TE, Ampah SB, Martina Bebin E, Grayson LP, Cutter GR, Hernando K, et al. Long-term safety and efficacy of highly purified cannabidiol for treatment refractory epilepsy. Epilepsy Behav. 2021;117:107862. https://doi.org/10.1016/j.yebeh.2021.107862.
Lattanzi S, Zaccara G, Russo E, La Neve A, Lodi MAM, Striano P. Practical use of pharmaceutically purified oral cannabidiol in Dravet syndrome and Lennox-Gastaut syndrome. Expert Rev Neurother. 2021;21(1):99–110. https://doi.org/10.1080/14737175.2021.1834383.
Thiele EA, Marsh ED, French JA, Mazurkiewicz-Beldzinska M, Benbadis SR, Joshi C, et al. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391(10125):1085–96. https://doi.org/10.1016/S0140-6736(18)30136-3.
Frampton JE. Stiripentol: A review in Dravet syndrome. Drugs. 2019;79(16):1785–96. https://doi.org/10.1007/s40265-019-01204-y.
Wheless JW, Fulton SP, Mudigoudar BD. Dravet syndrome: a review of current management. Pediatr Neurol. 2020;107:28–40. https://doi.org/10.1016/j.pediatrneurol.2020.01.005.
Weisler RH, Calabrese JR, Bowden CL, Ascher JA, DeVeaugh-Geiss J, Evoniuk G. Discovery and development of lamotrigine for bipolar disorder: a story of serendipity, clinical observations, risk taking, and persistence. J Affect Disord. 2008;108(1-2):1–9. https://doi.org/10.1016/j.jad.2007.09.012.
Coppola G, Besag F, Cusmai R, Dulac O, Kluger G, Moavero R, et al. Current role of rufinamide in the treatment of childhood epilepsy: literature review and treatment guidelines. Eur J Paediatr Neurol. 2014;18(6):685–90. https://doi.org/10.1016/j.ejpn.2014.05.008.
Balagura G, Riva A, Marchese F, Verrotti A, Striano P. Adjunctive rufinamide in children with Lennox-Gastaut syndrome: a literature review. Neuropsychiatr Dis Treat. 2020;16:369–79. https://doi.org/10.2147/NDT.S185774.
Operto FF, Verrotti A, Marrelli A, Ciuffini R, Coppola G, Pastorino GMG, et al. Cognitive, adaptive, and behavioral effects of adjunctive rufinamide in Lennox-Gastaut syndrome: a prospective observational clinical study. Epilepsy Behav. 2020;112:107445. https://doi.org/10.1016/j.yebeh.2020.107445.
Connolly MB. Dravet syndrome: diagnosis and long-term course. Can J Neurol Sci. 2016;43(Suppl 3):S3–8. https://doi.org/10.1017/cjn.2016.243.
Ziobro J, Eschbach K, Sullivan JE, Knupp KG. Current treatment strategies and future treatment options for Dravet syndrome. Curr Treat Options Neurol. 2018;20(12):52. https://doi.org/10.1007/s11940-018-0537-y.
Rosenberg EC, Tsien RW, Whalley BJ, Devinsky O. Cannabinoids and epilepsy. Neurotherapeutics. 2015;12(4):747–68. https://doi.org/10.1007/s13311-015-0375-5.
Greenwich Biosciences, Inc. EPIDIOLEX®: Highlights of prescribing information. Greenwich Biosciences, Inc., Carlsbad, California, USA 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210365s002lbl.pdf. Accessed 22 July 2020 2020.
European Medicines Agency (EMA). Epidyolex: EPAR - product information. European Medicines Agency (EMA). 2019. https://www.ema.europa.eu/en/documents/product-information/epidyolex-epar-product-information_en.pdf. Accessed 22 July 2020.
Thiele EA, Bebin EM, Bhathal H, Jansen FE, Kotulska K, Lawson JA, et al. Add-on cannabidiol treatment for drug-resistant seizures in tuberous sclerosis complex: a placebo-controlled randomized clinical trial. JAMA Neurol. 2021;78(3):285–92. https://doi.org/10.1001/jamaneurol.2020.4607.
Gaston TE, Szaflarski M, Hansen B, Bebin EM, Szaflarski JP, Program UC. Quality of life in adults enrolled in an open-label study of cannabidiol (CBD) for treatment-resistant epilepsy. Epilepsy Behav. 2019;95:10–7. https://doi.org/10.1016/j.yebeh.2019.03.035.
Martin RC, Gaston TE, Thompson M, Ampah SB, Cutter G, Bebin EM, et al. Cognitive functioning following long-term cannabidiol use in adults with treatment-resistant epilepsy. Epilepsy Behav. 2019;97:105–10. https://doi.org/10.1016/j.yebeh.2019.04.044.
Roid G, Pomplun M, Martin J. Nonverbal intellectual and cognitive assessment with the Leiter international performance scale-revised (Leiter-R). In: Naglieri JA, Goldstein S, editors. Practitioner’s guide to assessing intelligence and achievement. Hoboken: John Wiley and Sons; 2009.
Fenson L, Marchman VA, Thal DJ, Dale PS, Reznick J, Bates E. MacArthur-Bates communicative development inventories. 2nd ed. Paul H. Brookes: Baltimore; 2007.
Sparrow SS, Cicchetti DV, Saunier CA. Vineland Adaptive Behavior Scales. 3rd ed. (Vineland-3). Pearson: San Antonio; 2016.
Italian Medicines Agency. Valproic acid: summary of product characteristics. 2017. https://farmaci.agenziafarmaco.gov.it/aifa/servlet/PdfDownloadServlet?pdfFileName=footer_001392_036334_RCP.pdf&retry=0&sys=m0b1l3. Accessed 27 July 2020.
Italian Medicines Agency. Epaclob: summary of product characteristics. 2019. https://farmaci.agenziafarmaco.gov.it/aifa/servlet/PdfDownloadServlet?pdfFileName=footer_002352_045041_RCP.pdf&retry=0&sys=m0b1l3. Accessed 27 July 2020.
European Medicines Agency (EMA). Diacomit: EPAR - all authorised presentations. European Medicines Agency (EMA). 2009. https://www.ema.europa.eu/en/documents/product-information/diacomit-epar-product-information_en.pdf. Accessed 22 July 2020 2020.
Italian Medicines Agency. Taloxa: summary of product characteristics. 2020. https://farmaci.agenziafarmaco.gov.it/aifa/servlet/PdfDownloadServlet?pdfFileName=footer_001117_030822_RCP.pdf&retry=0&sys=m0b1l3. Accessed 27 July 2020.
European Medicines Agency (EMA). Lamictal: EPAR - all authorised presentations. European Medicines Agency (EMA). 2008. https://www.ema.europa.eu/en/documents/referral/lamictal-article-30-referral-annex-i-ii-iii_en.pdf. Accessed 27 July 2020 2020.
European Medicines Agency (EMA). Inovelon: EPAR - all authorised presentations. European Medicines Agency (EMA). 2009. https://www.ema.europa.eu/en/documents/product-information/inovelon-epar-product-information_en.pdf. Accessed 27 July 2020 2020.
European Medicines Agency (EMA). Topamax: EPAR - all authorised presentations. European Medicines Agency (EMA). 2009. https://www.ema.europa.eu/en/documents/referral/topamax-article-30-referral-annex-i-ii-iii_en.pdf. Accessed 27 July 2020 2020.
Acknowledgements
This work was developed within the framework of the DINOGMI Department of Excellence of MIUR 2018–2022 (legge 232 del 2016). We thank Jo Dalton for her medical writing assistance provided on behalf of Springer Healthcare Communications, Tracy Harrison of Springer Healthcare Communications for editing assistance prior to submission, and Catherine Rees of Springer Healthcare Communications for post-submission revisions; all writing and editing assistance was funded by GW Pharmaceuticals.
Code Availability
Not applicable.
Funding
The publication was made possible by an unconditioned educational grant from GW Pharmaceuticals.
Author information
Authors and Affiliations
Contributions
Francesca Marchese and Pasquale Striano drafted the manuscript, and the other authors modified the drafts; all authors approved the final version.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
P.S. has received speaker fees and participated at advisory boards for Biomarin, Zogenyx, and GW Pharmaceuticals and has received research funding from ENECTA BV, GW Pharmaceuticals, Kolfarma srl., and Eisai. The other authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Medicine
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marchese, F., Cappelletti, S., Filippini, M. et al. Comorbidities in Dravet Syndrome and Lennox–Gastaut Syndrome. SN Compr. Clin. Med. 3, 2167–2179 (2021). https://doi.org/10.1007/s42399-021-00989-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42399-021-00989-y