
Abstract
In this study, RT-PCR assays were developed and applied to investigate the prevalence of grapevine virus E (GVE), grapevine virus F (GVF) and grapevine virus I (GVI) in Greek vineyards. Grapevine samples from different viticultural areas of Greece were tested and the presence of all three vitiviruses was revealed. These viruses were mainly detected in grafted indigenous grapevine cultivars, with GVF being the most prevalent (24.7%, 200/809) in contrast to GVI (2.2%, 12/554) and GVE (1.6%, 12/752). To further study their intraspecies genetic variability and the phylogenetic relationships of their populations, sequence similarity analyses of the replicase and the coat protein gene segments of each virus, as well as from a fragment of the movement protein gene of GVI, were performed. Results revealed genetic variability in all three virus populations and high molecular diversity between Greek isolates of GVE and GVF, in contrast to Greek GVI isolates, which showed high homogeneity. Overall, our study advanced the current knowledge on grapevine-infecting vitiviruses and highlighted that special attention should be given to the widely spread GVF and its impact on grapevine should be further investigated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Grapevine is an economically important and widespread crop in Greece. It is infected by numerous viruses and viroids, including viruses of the genus Vitivirus (Fuchs 2020). Since the establishment of the genus Vitivirus in 1997 (Martelli et al. 1997), 17 species have been recognized as members of the genus by the International Committee on Taxonomy of Viruses (ICTV, https://ictv.global/taxonomy), while seven more are expected to be officially recognized (Maree et al. 2020; Zhao et al. 2020; Massé et al. 2022; Read et al. 2022).
The genus Vitivirus of the family Betaflexiviridae (subfamily Trivirinae) includes plant viruses with + ssRNA genome (Adams et al. 2012). Vitiviruses have a 7,300 to 7,600 nucleotides long genome, which is encapsidated in non-enveloped flexuous filamented virions and organized in five open reading frames (ORFs), flanked by a 5’-end methylated cap and a 3’-end poly-A tail (Martelli et al. 2007; Hull 2014; Minafra et al. 2017). ORF1 encodes proteins responsible for viral replication; ORF2 encodes an 18–22 kDa protein with unknown function; ORF3 and ORF4 encode the movement protein (MP) and coat protein (CP), respectively, while ORF5 encodes a nucleic acid binding protein (NABP) (Minafra et al. 2017). All grapevine vitiviruses are transmitted by grafting whereas transmission by mealybugs (Hemiptera: Pseudococcidae) and soft scale insects (Hemiptera: Coccidae) have been demonstrated for grapevine virus A (GVA), grapevine virus B (GVB), grapevine virus E (GVE), grapevine virus G (GVG) and grapevine virus H (GVH) (Nakaune et al. 2008; Maliogka et al. 2015a; Jagunić et al. 2021, 2022). Three members of the genus Vitivirus, i.e., GVA, GVB and grapevine virus D (GVD), have been associated with the rugose wood (RW) disease complex, whereas the role of the newly described GVE, grapevine virus F (GVF) and grapevine virus I (GVI) in RW disease complex remains unknown (Al Rwahnih et al. 2012; Martelli 2014; Maliogka et al. 2015a).
High-Throughput Sequencing (HTS) is an important tool in detection of grapevine viruses, because of its sensitivity and wide detection range (Adams et al. 2009; Vončina and Almeida 2018). In recent years, HTS has contributed significantly to the identification of new grapevine viruses, including vitiviruses (Al Rwahnih et al. 2012; Blouin et al. 2018a, b; Candresse et al. 2018; Diaz-Lara et al. 2018; Alabi et al. 2019; Debat et al. 2019a). In Greece, HTS of a grapevine sample of cultivar ‘Dafnia’ (D2-1), from the Grapevine Institute in Lykovrisi Attica, revealed a high number of viruses including GVE, GVF and GVI, whose presence in Greek vineyards, until then, remained unknown (Panailidou et al. 2019; Lotos et al. 2020).
GVE and GVF were first detected in Japan in 2008 (Nakaune et al. 2008) and in California in 2012 (Al Rwahnih et al. 2012), respectively, and since then their presence has been reported in South Africa (Coetzee et al. 2010a, b; de Koker, 2012; Molenaar et al. 2015), China (Fan et al. 2013; Elbeaino et al. 2019), Tunisia (Selmi et al. 2017), Croatia (Vončina et al. 2017; Vončina and Almeida 2018), Hungary, Italy, Jordan and Malta (Elbeaino et al. 2019). GVE is also present in Poland (Komorowska et al. 2013), USA (Alabi et al. 2013; Vargas-Asencio et al. 2016; Hu et al. 2021; Yao et al. 2021), Korea (Jo et al. 2017), Argentina (Debat et al. 2019b), Palestine (Elbeaino et al. 2019), and Pakistan (Rasool et al. 2019) and GVF in Iran (Sabaghian et al. 2018), Afghanistan, Bulgaria, Lebanon (Elbeaino et al. 2019), Pakistan (Rasool et al. 2019) and Russia (Shvets et al. 2022). GVI was first reported in grapevine in New Zealand in 2018 (Blouin et al. 2018b) and shortly after in California (Diaz-Lara et al. 2019) and South Africa (Read et al. 2021).
In this study, RT-PCR assays were developed for the detection of GVE, GVF and GVI in self-rooted and grafted cultivars in Greek vineyards, as well as in a small number of rootstocks. In addition, their genetic variability and phylogenetic relationships were studied, in order to gain a deeper understanding of their populations’ structure and its implication in the viruses’ spread and epidemiology.
Materials and methods
Plant material and total RNA extraction
From 2009 to 2020, grapevine samples were collected from nine geographic regions of Greece (Table 1). Plant material of forty-three grafted cultivars and six rootstocks were sampled from the grapevine germplasm collection of the Aristotle University of Thessaloniki (AUTH), while seven grafted cultivars and six rootstocks came from the corresponding collection of the Agricultural University of Athens (AUA). Furthermore, plant material of 142 grafted Greek cultivars were collected from the national grapevine germplasm collection of the Viticulture Department of Athens, Institute of Olive Tree, Subtropical Crops and Viticulture (ΙΟSV) (ELGO-DEMETER) in Lykovrisi Attica. In total, 752 samples were tested for the presence of GVE, 809 samples for GVF and 554 samples for GVI (Table 1). It was not possible to test all samples for the three viruses as due to a technical problem, a number of them (plant tissue and/or RNA) was destroyed.
To test grapevine samples for the presence of the three viruses, total RNA was extracted from leaves, stems or phloem scrapings (depending on sampling date) applying a column-based method with a guanidine hydrochloride-based lysis buffer as described by Chatzinasiou et al. (2010) with the modifications proposed by Maliogka et al. (2015b) using premade spin-columns (Biocomma Limited). To validate the quality of extracted RNA, a fragment of the endogenous gene phosphoenolpiruvate carboxylase (PEP) of grapevine (354 base pairs - bp) was amplified in one-step RT-PCR by using the primers PepSfw/PepSrev designed by Oláh et al. (2017).
Total RNA extraction for HTS
Total RNA was extracted from leaf, petiole or phloem scraping tissues subjected to a slightly modified extraction protocol of Ruiz-García et al. (2019), as descripted in Panailidou et al. (2023).
RT-PCR detection assays
For the detection of GVE, a one-step RT-PCR was applied, using GVE up/GVE down primers (Panailidou et al. 2019), which amplify a 575 nucleotides (nt) fragment of the nearly complete CP encoding ORF4 (Table 2; Fig. 1), while a set of primers (GVF_F_4521/GVF_R_5190, Table 2; Fig. 1) (Panailidou et al. 2019) was used in a two-step RT-PCR to amplify a 670 nt part of ORF1 which encodes GVF’s replicase (no conserved domain) (Supplementary Text). For GVI, a set of primers (GVI_F_6122/GVI_R_6765) was designed (Table 2; Fig. 1) to amplify a 644 nt long partial sequence of ORF3, which encodes the MP and used in a two-step RT-PCR reaction (Supplementary Text).

Genome organization of (a) grapevine virus E (GVE), (b) grapevine virus F (GVF), and (c) grapevine virus I (GVI). Black arrows with hyphens indicate targeted genomic regions for virus detection and normal black arrows target genomic regions for sequencing (Table 2). Replicase domains abbreviations: Mtr: methyltranferase, Hel:helicase, AlkB: alkylation B, RdRp: RNA dependent RNA polymerase; MP: movement protein; CP: coat protein; NABP: nucleic-acid-binding protein
Genetic variability of GVE, GVF and GVI
Viral genotypes subjected to Sanger sequencing
To study the genetic variability of the three viruses, partial sequences were obtained from six samples infected with GVE, sixteen infected with GVF and six with GVI using Sanger sequencing (Suppl. Table 1). All sequences of GVE, four of GVF and five of GVI came from samples from the collection of the Viticulture Department of ΙΟSV in Lykovrisi Attica, while eleven GVF genotypes originated from Nemea, from samples collected in 2016 to 2017. In addition, a GVI infected sample from Kavala and a GVE infected sample from Thebes were also analyzed.
Viral isolate sequences retrieved from HTS
Sequences of GVE, GVF and GVI obtained by HTS from four grapevine samples (33.Ε3 − 2, 39.D3-2, 91.K10No2, 132.Κ1No8) arbitrarily selected in the context of grapevine virome studies (unpublished data), were also used herein (Suppl. Table 2). Sample 33.E3-2 was processed (rRNA depletion and library construction) and sequenced in LifeSequencing (S.L., Spain) in an Illumina NextSeq platform. The remaining three samples were sent to Macrogen (Korea) for rRNA depletion, library construction and sequencing on a NovaSeq 6000 platform (Illumina, Inc.). The resulting reads were analyzed in Geneious (Biomatters, Ltd.). De novo assembly was performed with SPAdes (Bankevich et al. 2012) and contigs larger than 240 nt were subjected to similarity search with BLASTn.
Amplification of genomic regions from the three viruses
For all three viruses, a part of the replicase (partial RdRp domain) and the complete CP gene were sequenced, whereas in the case of GVI a fragment of its MP gene (partial sequence missing approximately 200 nt from the 5’ end) was also included.
Primers were designed to amplify a 500 nt part of GVE’s replicase (GVE_F_4169/GVE_R_4669, Table 2) and an 842 nt segment of GVE’s genome containing the complete CP gene of the virus (GVE_F_6390/GVE_R_7231, Table 2; Fig. 1). In order to determine the partial nucleotide sequence of GVF replicase gene a new forward primer was designed, namely GVF_F_4795 (Table 2) and used in a PCR reaction together with GVF_R_5190 primer (Table 2; Fig. 1), which has been used in reverse transcription (RT) and PCR for GVF detection, while a set of primers were designed (GVF_F_6481/GVF_R_7345, Table 2; Fig. 1) for the amplification of the complete CP gene of GVF. Two sets of primers (GVI_Rep_F_2639/GVI_Rep_R_2998 and GVI_CP_F_6365/GVI_CP_R_7082) (Table 2; Fig. 1) were designed for the amplification of a 360 nt segment of the replicase and a 764 nt one of the CP gene of GVI, while primers GVI_F_6122/GVI_R_6765 (Table 2; Fig. 1) were used for sequencing a 644 nt fragment of the MP gene. Detailed information on how the reactions were conducted is given in the Supplementary text.
Sanger sequencing
In all cases, the sequences of each virus isolate were amplified in a final volume of 100 µl and the PCR products were purified using the NucleoTrap purification kit (Macherey-Nagel, Duren, Germany) according to the manufacturer’s instructions. Then, purified DNA was Sanger sequenced in both directions at Eurofins Genomics (Vienna, Austria).
Sequence analyses and construction of phylogenetic trees
The analysis of sequences was performed with the MEGA Χ software (Kumar et al. 2018), while the determination of similarities between the Greek isolates (sequenced by Sanger sequencing and HTS, Suppl. Tables 1 and 2) as well as among the Greek and foreign isolates for each virus, for which sequences are published in GenBank (https://www.ncbi.nlm.nih.gov/), was done in Geneious Prime software (Biomatters, Ltd.) after the alignment of available nucleotide sequences in each case by MAFFT. Every alignment was trimmed based on the size of the smallest available isolate (Greek or foreign) for each genomic region (251 nt and 471 nt for GVE replicase and CP alignments, respectively, 250 nt and 570 nt for GVF replicase and CP alignments, respectively, 204 nt, 506 nt and 478 nt for GVI replicase, MP and CP alignments respectively).
For the study of the phylogenetic relationships between isolates of GVE, GVF and GVI, seven phylogenetic trees were constructed, one for every virus targeted region, with Greek isolate sequences determined herein and foreign isolate sequences published in GenBank. More specifically, a 251 and 250 nt partial sequence of the replicase gene (domain of RdRp) and a 471 and 570 nt partial sequence of the CP gene were used to infer the replicase and CP phylogenetic trees, for GVE and GVF respectively. In the case of GVI, partial sequences of replicase (204 nt of AlKB domain), MP (sequence of 506 nt) and CP (part of 478 nt) were used to construct the corresponding phylogenetic trees of the virus. The alignment of nucleotide sequences was carried out in MEGA Χ software (Kumar et al. 2018), using the ClustalW algorithm, and the best nucleotide substitution model was found with the same program, using the option Find Best DNA/Protein Models (ML). Maximum Likelihood method was selected for constructing all phylogenetic trees, using the same software, while a Non-Parametric Bootstrap analyses of 1000 repetitions was performed for the evaluation of the reliability of the phylogenetic hypothesis.
Results
Prevalence of GVE, GVF and GVI in Greek vineyards
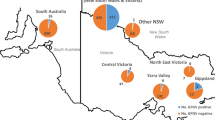
GVF was found in five of the nine sampling areas of Greece (Table 1), in samples originating from Peloponnese, Central and Western Macedonia, and in two grapevine collections (Viticulture Department of ΙΟSV (ELGO-DEMETER) and AUA), whereas GVI and GVE were found only in three and one regions of Greece, respectively and both were detected in the collection of Viticulture Department of ΙΟSV (ELGO-DEMETER) (Table 1; Fig. 2a). The majority of GVE and GVI positive samples were from the ELGO-DEMETER germplasm collection, with a few positive samples originating from vineyards located in Eastern Macedonia, Peloponnese, and Aegean Islands and in Central Greece, respectively (Fig. 2a).

(a) Map of Greece showing the areas where grapevine virus E (GVE), grapevine virus F (GVF) and grapevine virus I (GVI) were detected. In the map, the number of positives to the total of samples collected from commercial vineyards of every area is marked by plates in which the above information is presented for each virus by a different color (red for GVE, dark yellow for GVF and purple for GVI), while the same information is marked by bold plates in the case of samples collected from grapevine germplasm collections. (b) Presence of GVE, GVF and GVI in self-rooted, grafted grapevines and rootstocks. Bars with blue, orange, and grey colors represent the number of samples found positive to GVE, GVF and GVI, respectively
In particular, GVF was present in 24.7% of the tested samples (200 out of 809 tested vines), mostly in grafted Greek and foreign cultivars (30.2% − 194/643), especially in vineyards from Peloponnese (55.9% -118/211), and in Greek self-rooted grapevines (4.3% − 6/140) (Fig. 2b) (Table 1). GVI was detected in 2.2% (12/554) of samples originating from Greek self-rooted vines (1.2% − 1/87) and grafted indigenous and foreign cultivars (2.4% − 11/453) (Fig. 2b; Table 1). GVE was found in 1.6% (12/752) of the tested samples and only in grafted Greek grapevine cultivars (2% − 12/594) (Fig. 2b; Table 1). None of these viruses was detected in the tested non-grafted rootstocks (0/26 for GVE and GVF, 0/14 for GVI). In addition, mixed infections were observed, as one grapevine sample was infected by all three viruses, three samples by GVE and GVI, one by GVF and GVI and one by GVE and GVF, all of which were collected from the Viticulture Department of ΙΟSV.
HTS-derived near complete virus genomes
Regarding the virus sequences retrieved from the HTS analyses, sample 33.E3-2 yielded ~ 21 million pair-end reads ranging from 19 to 151 nt in length. De novo assembly produced 649 contigs, three of which (33.E3-2/5, 33.E3-2/9, 33.E3-2/10 GenBank accession No. OL690319, OL690320, OL690321, respectively) had higher BLASTn scores with the GVF sequences deposited in GenBank and represented three nearly complete genomes of GVF. Sample 39.D3-2 yielded ~ 56 million 101 nt pair-end reads which were assembled in 1243 contigs. BLASTn revealed the presence of three contigs, with each one covering almost the complete genome of GVE (39.D3-2/15, GenBank accession No. OL690333), GVF (39.D3-2/16, GenBank accession No. OL690318) and GVI (39.D3-2/17, GenBank accession No. OL770199). Moreover, similar to GVF, seven contigs were also found in 39.D3-2 which were assembled into a scaffold (39.D3-2/VS2, GenBank accession No. OL690331). The scaffold’s CP region was used for the phylogenetic and similarity analyses, as it derived from only one contig. Finally, samples 91.K10No2, and 132.Κ1No8 yielded ~ 54 and ~ 60 million 101 pair-end reads, respectively. In both samples multiple replicase contigs were present with only one contig covering the remaining four ORFs of the genome. Only replicase contigs larger than 3000 covering the RdRp region of the replicase were used for the phylogenetic analyses (Accession Numbers presented in Suppl. Table 2).
Sequence diversity and phylogenetic analyses of GVE, GVF and GVI populations
Grapevine virus E
In the case of GVE, sequence identity between Greek isolates for the part of the replicase gene (6 isolates, Suppl. Tables 1 and 2) was 72.9–100% in nt and 88–100% in amino acids (aa), while the identity among sequences from Greece and isolates retrieved by GenBank was 69.3–100% in nt and 84.3–100% in aa for the same genomic region (Suppl. Tables 3 and 4). In the CP gene all GVE sequences from Greece (8 isolates, Suppl. Tables 1 and 2) shared 77.3–100% nt (87.9–100% in aa) identity with each other and 70.5–100% nt (82.2–100% aa) with homologous sequences from other countries (Suppl. Tables 5 and 6). In both genes, one isolate sequenced from sample 109.B7-7_618 (Viticulture Department of ΙΟSV) was divergent from the other isolates from Greece, which were highly similar [95.6–100% nt and 100% aa identity in replicase (Suppl. Tables 3 and 4) and 98.3–100% nt and 99.4–100% aa identity in CP gene (Suppl. Tables 5 and 6)].
The phylogenetic trees constructed based on the partial nucleotide sequences of the replicase and the CP gene of GVE have the same topology, grouping the virus isolates in five distinct groups (I-V) (Fig. 3). Most of the GVE sequences from Greece were classified into group I, while genotype 109.B7-7_618 was classified in group V apart from the other Greek isolates.

Maximum likelihood phylogenetic trees based on partial nucleotide sequences of the replicase (a) and the coat protein gene (b) of grapevine virus E (GVE). Sequences of Greek GVE isolates (black circle for sequences obtained by Sanger sequencing, black square for sequences obtained by HTS, and back triangle for sequences retrieved from GenBank) and other GVE sequences from different countries (referred with GenBank accession and their origin) were used. The percentage of 1000 repetitions of the bootstrap analysis, which supports grouping at each node, is indicated. The scale bar represents the number of nucleotide substitutions per position, while the length of the branches is proportional to the genetic distances that were calculated. An isolate of GVI (GenBank accession number MF927925) was used as an outgroup in both phylogenetic trees
Grapevine virus F
Sequence identity among the replicase gene of GVF isolates from Greece [34 isolates (16 from Sanger sequencing and the rest from the HTS data where multiple isolates were found co-infecting the same grapevine), Suppl. Tables 1 and 2] was 82.8–100% at the nt level (96.4–100% aa) while identity between isolates from Greece and elsewhere was 79.4–97.3% in nt (87.2– 100% aa) (Suppl. Tables 7 and 8). The GVF CP sequences from Greece (15 isolates, Suppl. Tables 1 and 2) shared 84–99.8% nt (90.5–100% aa) identity between them and 83.9–98.2% nt (87.4–99.5% aa) with the foreign sequences retrieved from GenBank (Suppl. Tables 9 and 10).
In the replicase based tree of GVF, 5 phylogenetic groups (I-V) were clearly formed with the virus sequences from Greece classified mainly in group I, apart from 33.E3-2/5 which is a member of the group III (Fig. 4a). In addition, GVF sequences retrieved from the same sample, like 33.E3-2/5, 33.E3-2/9 and 33.E3-2/10 (group I and II), were not classified in the same phylogenetic group, whereas others, such as Mpak5/F and Mpak5/R, clustered together (group I). In contrast to the formation of phylogroups based on replicase, two main phylogenetic groups (A and B) (Fig. 4b) were set up by CP sequences of GVF. Group A included only one Bulgarian isolate (LT960646.1) and group B all the Greek isolates and the majority of published GVF isolates. Further separation of GVF isolates of group B into subgroups is not possible as there is no clear topology.

Maximum likelihood phylogenetic trees based on partial nucleotide sequences of the replicase (a) and coat protein gene (b) of grapevine virus F (GVF). Sequences of Greek GVF isolates (black cycle for isolates obtained by Sanger sequencing, black square for isolates obtained by HTS, and black triangle for isolates retrieved by GenBank) and other GVF sequences from different countries (referred with GenBank accession and their origin) were used. The percentage of 1000 repetitions of the bootstrap analysis, which supports grouping at each node, is indicated. The scale bar represents the number of nucleotide substitutions per position, while the length of the branches is proportional to the genetic distances that were calculated. An isolate of GVA (GenBank accession numbers DQ787959) was used as an outgroup in each phylogenetic tree
Grapevine virus I
Sequences of partial GVI replicase gene from Greece (6 isolate, Suppl Tables 1 and 2) shared 97.5–100% nt (100% aa) identities between them and 77.9–99% in nt (91.2–100% aa) with the corresponding virus sequences retrieved from GenBank (Suppl. Tables 11 and 12). The GVI MP gene sequences from Greece (7 isolates, Suppl. Tables 1 and 2) were highly identical to each other (99–100% nt and 98.8–100% aa). However, they shared 85.7–98.4% nt and 92.2–100% aa identities with the homologous region of the GVI isolates from New Zealand and South Africa (Suppl. Tables 14 and 13). In pairwise comparisons, the GVI CP sequences from Greece (6 isolates, Suppl. Tables 1 and 2) shared 99.2–100% nt and 100% aa identities with each other and 84.7–99% nt and 95.6–100% aa identities with sequences retrieved from GenBank (Suppl. Tables 15 and 16).
The phylogenetic trees constructed based on the partial replicase, MP and CP genes of Greek and foreign GVI sequences revealed the formation of two distinct phylogenetic groups (I and II) in all cases (Fig. 4). The grouping of isolates from New Zealand and USA in a different clade (group I) from Greek isolates was already mentioned by Lotos et al. (2020) for replicase and CP gene. Isolates from South Africa grouped together with the new reported Greek isolates in replicase and CP based phylogenetic trees, while the phylogenetic tree of GVI’s MP follows the clustering of the other two.
In all cases attempts were made to sequence additional isolates of each virus from other regions of the country, which however were unsuccessful.
Discussion
The most prevalent virus in this study was GVF which was found in 24.5% of the samples tested exhibiting a wide geographical distribution (Table 1; Fig. 2), in contrast to GVI and GVE, which were identified in a very small number of samples (1.7% and 2.4%, respectively), mainly in the collection of the Viticulture Department of ΙΟSV in Lykovrisi Attica. It is also noteworthy that GVF had a very high prevalence (55.9% 118 positive samples in 211 total) in the viticulture region of Nemea in Peloponnese. These positives were found in ten different vineyards in grapevines of different age, variety and rootstock indicating that the virus is endemic in this region and has potentially found a competent vector. Surveys performed in other countries showed variable prevalence of the three vitiviruses, which can be attributed to a variety of factors. Low incidence of GVE was also reported in Poland (Komorowska et al. 2013), New York and California (Vargas-Asencio et al. 2016), China (Fang et al. 2020) and Pakistan (Rasool et al. 2019), as well as for GVI in California (Diaz-Lara et al. 2019), whereas GVF incidence was high (41%) in Iranian vineyards (Sabaghian et al. 2018). Elbeaino et al. (2019), reported the presence of GVF and GVE at 11.5% and 14.7%, respectively, in vines from different Mediterranean countries, whereas Shvets et al. (2022), found GVF in 2% of the tested grapevine samples from Russia. In addition, these three viruses were detected in grafted Greek grapevine cultivars, while GVF and GVI were found, also, in a limited number of grafted foreign cultivars and in a smaller number of Greek autochthonous grapevines, most of which were collected between 2009 and 2012. Consequently, we can deduce that these three viruses were present in the Greek vineyards for several years before their first identification in 2017 by HTS (Panailidou et al. 2019; Lotos et al. 2020) but with their initial entry time being currently unknown. GVF and GVI were detected in self-rooted vines collected from Cyclades islands in 2009, indicating the existence of the two viruses in Greece for at least the past 12 years. However, GVE’s presence in Greek vineyards could be much shorter, given that it was found only in grafted vines which were collected in 2016 and 2018. The virus has extremely limited spread with the majority of positive samples (11/12) originating from the germplasm collection of the Viticulture Department of ΙΟSV, where plant material with diverse geographic origin exists, with only one positive sample identified in a commercial vineyard.
Coexistence of more than one virus sequences in the same grapevine sample was noticed both in Sanger sequencing and HTS analyses of the Greek GVE and GVF isolates. More specifically, up to three different sequences of GVE and up to five GVF sequences, were identified in the same sample by HTS. This trait was not observed in the case of GVI. Coexistence of more than one genotypes of GVA, GVB and GVL in the same vine has been previously reported (Goszczynski and Jooste 2003; Shi et al. 2004; Panailidou et al. 2023), which indicates that this is a common phenomenon among vitiviruses, as well as their coexistence with ampeloviruses [grapevine leafroll-associated virus 1 (GLRaV-1) and grapevine leafroll-associated virus 3 (GLRaV-3)] (Rowhani et al. 2018; Diaz-Lara et al. 2019) since they use the same vectors. However, the effect of coexistence of divergent viral variants on the symptomatology of infected grapevines remains to be explored.
The Greek isolates of GVE and GVF exhibited intraspecies genetic variability in every genomic region studied, whereas GVI isolates were almost identical throughout their genomes (Table 3). The majority of Greek isolates grouped together forming phylogenetic groups with small genetic distances, except for a few divergent isolates which were placed in different and more distant phylogenetic groups, like isolates 109.B7-7_618 of GVE and 33.E3-2/5 of GVF (Figs. 3, 4 and 5). Our results are in accordance with previous studies which also reported similar levels of genetic variability of GVE (Nakaune et al. 2008; Vargas-Asencio et al. 2016; Elbeaino et al. 2019; Fang et al. 2020; Maree et al. 2020), GVF (Elbeaino et al. 2019) and GVI isolates (Diaz‑Lara et al. 2019; Lotos et al. 2020). In addition, their findings support the formation of the phylogenetic groups presented in our study for the replicase and CP regions of GVE (Nakaune et al. 2008; Coetzee et al. 2010b; Vargas-Asencio et al. 2016; Maree et al. 2020) and GVI (Lotos et al. 2020), but not for GVF where different topologies were formed compared to previous studies. More specifically, the isolates grouped herein in phylogenetic group A were previously classified in two distinct groups (Elbeaino et al. 2019; Shvets et al. 2022).

Maximum likelihood phylogenetic trees based on partial nucleotide sequences of the replicase (a), the movement protein (b) and the coat protein gene (c) of grapevine virus I (GVI). Sequences of Greek GVI isolates (black cycle for isolates obtained for Sanger sequencing and black square for isolates obtained by HTS) and other GVI sequences from different countries (referred with GenBank accession and their origin) were used. The percentage of 1000 repetitions of the bootstrap analysis, which supports grouping at each node, is indicated. The scale bar represents the number of nucleotide substitutions per position, while the length of the branches is proportional to the genetic distances that were calculated. Isolate WAHH2 of GVE (GenBank accession number JX402759) was used as an outgroup in each phylogenetic tree
The spread, prevalence, molecular and phylogenetic characteristics of GVE and GVI in Greece resemble those of the newly identified GVL (Panailidou et al. 2023) indicating that these phylogenetically close species possibly share common epidemiological characteristics. Moreover, it is evident that even though the entry of new grapevine vitiviruses in Greece is a common event, the impact most of them have on the crop is minimal, possibly due to limitations in their transmission and/or adaptation to the cultivars used.
Particular attention should be paid to the case of GVO isolates from South Africa (GenBank No. MZ682356 and MZ682357). Read et al. (2022) presented GVO as a potential novel member of the genus Vitivirus, having the typical genome organization of the other members of the GVE-clade and being more closely related to GVE isolates. Interestingly, based on our findings the taxonomic position of the GVO isolates should be reconsidered as they might represent divergent isolates of GVE, given that they formed a new phylogroup (IV) in both phylogenetic trees of GVE (Fig. 3).
In conclusion, GVF seems to be the virus among the three vitiviruses studied herein that exhibits a high epidemiological potential as it was found to be widespread in commercial vineyards in different geographic regions of Greece. However, more information on the biological characteristics of all three vitiviruses is required to better understand their epidemiology and achieve their effective management in the field.
Data Availability
The nucleotide sequences reported here have been deposited in the GenBank database under accession numbers: OL690318 to OL690333, MT324251 to MT324261, MT332358 to MT332395, MT332385 to MT332390.
References
Adams IP, Glover RH, Monger WA et al (2009) Next generation sequencing and metagenomics: a universal diagnostic tool in plant virology. Mol Plant Pathol 10:537–545. https://doi.org/10.1111/j.1364-3703.2009.00545.x
Adams M, Candresse T, Hammond J et al (2012) Family Betaflexiviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (Eds), Virus taxonomy: ninth report of the international committee on taxonomy of viruses, pp. 920–941
Al Rwahnih M, Sudarshana MR, Uyemoto JK, Rowhani A (2012) Complete genome sequence of a novel vitivirus isolate from grapevine. J Virol 86:9545. https://doi.org/10.1128/JVI.01444-12
Alabi OJ, Poojari S, Sarver K, Martin RR, Naidu RA (2013) Complete genome sequence analysis of an american isolate of Grapevine virus E. Virus Genes 46:563–566. https://doi.org/10.1007/s11262-012-0872-0
Alabi OJ, McBride S, Appel DN, Al Rwahnih M, Pontasch FM (2019) Grapevine virus M, a novel vitivirus discovered in the american hybrid bunch grape cultivar Blanc du Bois in Texas. Arch Virol 164:1739–1741. https://doi.org/10.1007/s00705-019-04252-7
Bankevich A, Nurk S, Antipov D et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Blouin AG, Keenan S, Napier KR, Barrero RA, MacDiarmid RM (2018a) Identification of a novel vitivirus from grapevines in New Zealand. Arch Virol 163:281–284. https://doi.org/10.1007/s00705-017-3581-0
Blouin AG, Chooi KM, Warren B, Napier KR, Barrero RA, MacDiarmid RM (2018b) Grapevine virus I, a putative new vitivirus detected in co-infection with Grapevine virus G in New Zealand. Arch Virol 163:1371–1374. https://doi.org/10.1007/s00705-018-3738-5
Candresse T, Theil S, Faure C, Marais A (2018) Determination of the complete genomic sequence of Grapevine virus H, a novel vitivirus infecting grapevine. Arch Virol 163:277–280. https://doi.org/10.1007/s00705-017-3587-7
Chatzinasiou E, Dovas CI, Papanastassopoulou M et al (2010) Assessment of bluetongue viraemia in sheep by real-time PCR and correlation with viral infectivity. J Virol Methods 169:305–315. https://doi.org/10.1016/j.jviromet.2010.07.033
Coetzee B, Freeborough MJ, Maree HJ, Celton JM, Jasper D, Rees G, Burger JT (2010a) Deep sequencing analysis of viruses infecting grapevines: Virome of a vineyard. Virology 400:157–163. https://doi.org/10.1016/j.virol.2010.01.023
Coetzee B, Maree HJ, Stephan D, Freeborough MJ, Burger JT (2010b) The first complete nucleotide sequence of a Grapevine virus E variant. Arch Virol 155:1357–1360. https://doi.org/10.1007/s00705-010-0685-1
Debat H, Zavallo D, Brisbane RS et al (2019a) Grapevine virus L: a novel vitivirus in grapevine. Eur J Plant Pathol 155:319–328. https://doi.org/10.1007/s10658-019-01727-w
Debat H, Zavallo D, Luna F, Moyano S, Asurmendi S, Gomez-Talquenca S (2019b) First report of Grapevine virus E infecting grapevine in Argentina. J Plant Path 101:1221–1222. https://doi.org/10.1007/s42161-019-00282-x
Diaz-Lara A, Golino D, Al Rwahnih M (2018) Genomic characterization of Grapevine virus J, a novel virus identified in grapevine. Arch Virol 163:1965–1967. https://doi.org/10.1007/s00705-018-3793-y
Diaz-Lara A, Brisbane RS, Aram K, Golino D, Al Rwahnih M (2019) Detection of new vitiviruses infecting grapevine in California. Arch Virol 164:2573–2580. https://doi.org/10.1007/s00705-019-04355-1
Elbeaino T, Chammema H, Alsahelia Z, Slimena AB, Digiaro M (2019) Development of RT-PCR assays for the detection and the resultant phylogenetic analysis of four grapevine vitiviruses based on the coat protein sequences. J Virol Methods 273:1–5. https://doi.org/10.1016/j.jviromet.2019.113712
Fan XD, Dong YF, Zhang ZP, Ren F, Hu GJ, Zhu HJ (2013) First report of Grapevine virus E from grapevines in China. J Plant Pathol 95:659–668
Fang REN, Zhang ZP, Fan XD, Hu GJ, Zhang MY, Dong YF (2020) A sensitive SYBR Green RT-qPCR method for grapevine virus E and its application for virus detection in different grapevine sample types. J Integr Agric 19:1834–1841. https://doi.org/10.1016/S2095-3119(19)62784-X
Fuchs M (2020) Grapevine viruses: a multitude of diverse species with simple but overall poorly adopted management solutions in the vineyard. J Plant Pathol 102:643–653. https://doi.org/10.1007/s42161-020-00579-2
Goszczynski DE, Jooste AEC (2003) Identification of divergent variants of Grapevine virus A. Eur J Plant Pathol 109:397–403. https://doi.org/10.1023/A:1023555018700
Hu R, Dias NP, Soltani N et al (2021) Cultivated and wild grapevines in Tennessee possess overlapping but distinct virus populations. Plant Dis 105:2785–2791. https://doi.org/10.1094/PDIS-11-20-2483-SC
Hull R (2014) Plant Virology, 5th edn. Elsevier Academic Press, Norwich, UK
Jagunić M, Lazarević B, Nikolić K, Stupić D, Preiner D, Vončina D (2021) Detection, transmission, and characterization of grapevine virus H in Croatia. Pathogens 10:1578. https://doi.org/10.3390/pathogens10121578
Jagunić M, De Stradis A, Preiner D, La Notte P, Al Rwahnih M, Almeida RP, Vončina D (2022) Biology and ultrastructural characterization of Grapevine Badnavirus 1 and Grapevine Virus G. Viruses 14:2695. https://doi.org/10.3390/v14122695
Jo Y, Song MK, Choi H et al (2017) Genome sequence of Grapevine virus K, a novel vitivirus infecting grapevine. Genome Announc 5:e00994–e00917. https://doi.org/10.1128/genomeA.00994-17
Komorowska B, Berniak H, Golis T (2013) Detection of grapevine viruses in Poland. J Plant Pathol 162:326–331. https://doi.org/10.1111/jph.12186
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Lotos L, Ruiz-García AB, Panailidou P, Olmos A, Katis NI, Maliogka VI (2020) The complete genome of a divergent Grapevine virus I isolate naturally infecting grapevine in Greece. Arch Virol 165:3003–3006. https://doi.org/10.1007/s00705-020-04762-9
Maliogka VI, Martelli GP, Fuchs M, Katis NI (2015a) Control of viruses infecting grapevine. Adv Virus Res 91:175–227. https://doi.org/10.1016/bs.aivir.2014.11.002
Maliogka VI, Olmos A, Pappi PG et al (2015b) A novel grapevine badnavirus is associated with the Roditis leaf discoloration disease. Virus Res 203:47–55. https://doi.org/10.1016/j.virusres.2015.03.003
Maree HJ, Blouin AG, Diaz–Lara A, Mostert I, Al Rwahnih M, Candresse T (2020) Status of the current vitivirus taxonomy. Arch Virol 165:451–458. https://doi.org/10.1007/s00705-019-04500-w
Martelli GP (2014) Directory of virus and virus-like diseases of the grapevine and their agents. J Plant Pathol 96:1–88. https://doi.org/10.4454/JPP.V96I1SUP
Martelli GP, Minafra A, Saldarelli P (1997) Vitivirus, a new genus of plant viruses. Arch Virol 142:1929–1932
Martelli GP, Adams MJ, Keuze JF, Dolja VV (2007) Family Flexiviridae: a case study in virion and genome plasticity. Annu Rev Phytopathol 45:73–100. https://doi.org/10.1146/annurev.phyto.45.062806.094401
Massé D, Filloux D, Candresse T et al (2022) Identification of a novel vitivirus from pineapple in Reunion Island. Arch Virol 167:2355–2357. https://doi.org/10.1007/s00705-022-05512-9
Minafra A, Mawassi M, Goszczynski D, Saldarelli P (2017) Grapevine vitiviruses. In: Meng B, Martelli GP, Golino DA, Fuchs M (eds) Grapevine viruses: molecular biology, diagnostics and management. Cham, Switzerland, pp 229–256
Molenaar N, Burger JT, Maree HJ (2015) Detection of a divergent variant of grapevine virus F by next-generation sequencing. Arch Virol 160:2125–2127. https://doi.org/10.1007/s00705-015-2466-3
Nakaune R, Toda S, Mochizuki M, Nakano M (2008) Identification and characterization of a new vitivirus from grapevine. Arch Virol 153:1827. https://doi.org/10.1007/s00705-008-0188-5
Oláh R, Deák T, Turcsán M, Szénási M, Bordé Á, Szegedi E (2017) Use of an intron containing grapevine gene as internal control for validation of cDNA synthesis in virus detection by RT-PCR. Eur J Plant Pathol 149:765–770. https://doi.org/10.1007/s10658-017-1218-5
Panailidou P, Lotos L, Olmos A et al (2019) First Report of Grapevine Virus E and Grapevine Virus F in Grapevine in Greece. Plant Dis 103:1440. https://doi.org/10.1094/PDIS-11-18-2108-PDN
Panailidou P, Galeou A, Beris D et al (2023) Identification and genetic diversity of grapevine virus L in Greece. Arch Virol 168:127. https://doi.org/10.1007/s00705-023-05756-z
Rasool S, Naz S, Rowhani A, Diaz-Lara A, Golino DA, Farrar KD, Al Rwahnih M (2019) Survey of grapevine pathogens in Pakistan. J Plant Pathol 101:725–732. https://doi.org/10.1007/s42161-019-00263-0
Read DA, Thompson GD, Swanevelder D, Pietersen G (2021) Detection and diversity of grapevine virus L from a Vitis cultivar collection in Stellenbosch, South Africa. Eur J Plant Pathol 161:1007–1011. https://doi.org/10.1007/s10658-021-02380-y
Read DA, Thompson GD, Cordeur NL, Swanevelder D, Pietersen G (2022) Genomic characterization of grapevine viruses N and O: novel vitiviruses from South Africa. Arch Virol 167:611–614. https://doi.org/10.1007/s00705-021-05333-2
Rowhani A, Daubert S, Arnold K, Golino D, Uyemoto JK (2018) Synergy between grapevine vitiviruses and grapevine leafroll viruses. Eur Eur J Plant Pathol 151:919–925. https://doi.org/10.1007/s10658-018-1426-7
Ruiz-García AB, Bester R, Olmos A, Maree HJ (2019) Bioinformatic tools and genome analysis of Citrus tristeza virus. In: Citrus Tristeza Virus Humana. New York, NY, pp 163–178. https://doi.org/10.1007/978-1-4939-9558-5_12
Sabaghian S, Rakhshandehroo F, Zamanizadeh H, Elbeaino T (2018) First detection of grapevine virus F in Iran. J Plant Pathol 100:111–112. https://doi.org/10.1007/s42161-018-0002-5
Selmi I, Lehad A, Pacifico D, Carimi F, Mahfoudhi N (2017) First report of grapevine virus E and grapevine virus F in tunisian grapevines. J Plant Pathol 99:543
Shi BJ, Habili N, Gafny R, Symons RH (2004) Extensive variation of sequence within isolates of Grapevine virus B. Virus Genes 29:279–285. https://doi.org/10.1023/B:VIRU.0000036388.41242.c1
Shvets D, Porotikova E, Sandomirsky K, Vinogradova S (2022) Virome of Grapevine Germplasm from the Anapa Ampelographic Collection (Russia). Viruses 14:1314. https://doi.org/10.3390/v14061314
Vargas-Asencio J, Al Rwahnih M, Rowhani A, Celebi-Toprak F, Thompson JR, Fuchs M, Perry KL (2016) Limited genetic variability among american isolates of Grapevine virus E from Vitis spp. Plant Dis 100:159–163. https://doi.org/10.1094/PDIS-05-15-0556-RE
Vončina D, Almeida RPP (2018) Screening of some croatian autochthonous grapevine varieties reveals a multitude of viruses, including novel ones. Arch Virol 163:2239–2243. https://doi.org/10.1007/s00705-018-3850-6
Vončina D, Rwahnih MA, Rowhani A, Gouran M, Almeida RPP (2017) Viral diversity in autochthonous croatian grapevine cultivars. Plant Dis 101:1230–1235. https://doi.org/10.1094/PDIS-10-16-1543-RE
Yao XL, Domier LL, Qu F, Ivey MLL (2021) Grapevine virus E detected in Ohio Vineyards. Catalyst: Discovery into Practice 5:1–3. https://doi.org/10.5344/catalyst.2020.20006
Zhao L, Cao M, Huang Q et al (2020) Occurrence and molecular characterization of Actinidia virus C (AcVC), a novel vitivirus infecting kiwifruit (Actinidia spp.) in China. Plant Pathol 69:775–782. https://doi.org/10.1111/ppa.13171
Acknowledgements
We would like to thank Mr D. Dimou (Agronomist, ex. Head DAOK Argolida), professors E. Chatzivasilliou and A. Biniari (Agricultural University of Athens) and the Viticulture Department of Athens, Institute of Olive Tree, Subtropical Crops and Viticulture (ΙΟSV) (ELGO-DEMETER) in Lykovrisi Attica for providing us with grapevine samples.
Funding
This research has been financed by Greek national funds through the Public Investments Program (PIP) of the General Secretariat for Research and Technology (GSRT), under the Action “The Vineyard Roads” (project code: 2018ΣE01300000). Moreover, this research has been partially co-financed by the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – CREATE – INNOVATE (project code: T1EDK-04363).
Open access funding provided by HEAL-Link Greece.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by all authors. The first draft of the manuscript was written by Polina Panailidou, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Panailidou, P., Lotos, L., Orfanidou, C.G. et al. Prevalence and molecular characterization of grapevine virus E, F and I populations in Greek vineyards. J Plant Pathol 106, 31–43 (2024). https://doi.org/10.1007/s42161-023-01523-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42161-023-01523-w