
Abstract
Tomato bacterial canker, caused by Clavibacter michiganensis subsp. michiganensis (Cmm) (Li et al. 2018), is a common destructive disease worldwide. The bacterium colonizes plant’s vascular system causing characteristic symptoms such as wilting, cankers, brown and black discoloration of vessels, white spots on the fruit and leaves. In the present work, isolates were collected from infected tomato plants of 11 Greek Prefectures during the years 2003-2018, and the genetic variability was investigated. After initial identification, 93 selected Cmm strains were subjected to phylogenetic analyses based on the sequencing of multiple loci (Multi-Locus Sequencing Analysis, MLSA) utilizing four housekeeping genes (atpD, ppk, kdpA and sdhA). MLSA highlighted 4.1% variability in nucleotide sequences and classified the 93 Cmm strains into 38 haplotypes. Nevertheless, the phylogenetic grouping of the strains was not in accordance with the geographical distribution, a fact that probably advocates the introduction of the disease from infected propagating material. However, strains isolated from the same area in a different year were placed in the same group which means that the pathogen can remain active between growing seasons. The study provides important epidemiological information on the disease outbreaks and the spread of Cmm in Greece.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Clavibacter michiganensis subsp. michiganensis (Cmm) is a seed-born Gram-positive bacterium that causes the bacterial canker of tomatoes (Tsiantos 1987). It was first isolated in Michigan, USA, in 1910 (Smith 1910, Davis et al. 1984) and has since spread to many parts of the world where tomato is cultivated. The disease reduces the yield and the marketability of the product, causing destruction of the crop if not contained. Bacterial canker is a serious threat to tomato producing countries worldwide, having caused catastrophic outbreaks over the years with severe economic losses (Smith 1910, Kleitman et al. 2008, Blank et al. 2016). The first appearance of bacterial canker in Greece was recorded by Zachos and Georgopoulos (1957). Since then, the disease has been observed in most parts of the country where outdoor or greenhouse tomato crops are established. In the last few years the outbreaks of the disease have become more frequent due to the import of seeds and the intensification of crops, which favor the growth of the bacterium. From 2003 to 2018, twelve bacterial canker outbreaks were recorded in Greece, during which significant economic losses were documented.
Cmm colonizes plants through wounds or natural openings, reaches the vascular system, where it grows, and can be transmitted systematically to every plant tissue, causing plant rot and death (Carlton et al. 1998). The infection of the tomato plant causes symptoms that depend on the age or the environmental conditions, such as discoloration of the stem and canker, leaf-wilting that may reach up to hemiplegia, or the pathogen-characteristic spots on the fruits called “bird’s eye” (de León et al. 2011, Gleason et al. 1993). The bacterium easily spreads through the commercial routes of infected seeds, covering large geographic regions worldwide in short time (Fatmi et al. 1991, Quesada-Ocampo et al. 2012). Additionally, Cmm may spread through infected residues that survive in the soil, where the bacterium may survive for 2 to 3 years (Fatmi and Schaad 2002, Hadas et al. 2005) or through irrigation and cultivation practices (Gitaitis et al. 1991, Carlton et al. 1998, Xu et al. 2010). Unfortunately, there is neither a resistant cultivar (Umesha 2006) nor an effective chemical or biological control to date (Umesha 2006, de León et al. 2008).
Cmm is a regulated pathogen listed in the A2 list of the European and Mediterranean Plant Protection Organization (EPPO) since 1982 (Jensen 1982) due to its inability to be controlled and the serious financial losses it produces. So far, prevention is the only way to control the disease through the application of good agricultural practices and the utilization of healthy seeds or seedlings (de León et al. 2011).
Several DNA fingerprinting methods have been utilized to exploit the genetic diversity of Cmm strains isolated in different geographic regions or growing seasons. Among them, repetitive element polymerase chain reaction (rep-PCR) using REP, ERIC or BOX primers was very popular as it had the ability to separate strains at the subspecific level, as was shown by Louws et al. (1998). Since then, rep-PCR has often been used for genetic diversity studies, as in isolations from Northwestern Iran (Nazari et al. 2007), Israel (Kleitman et al. 2008), Japan (Kawaguchi et al. 2010, Kawaguchi and Tanina 2014), Turkey (Basım and Basım 2018), Southern Italy (Ialacci et al. 2016), Canary Islands (De León et al. 2009), and Argentina (Wassermann et al. 2017). Apart from rep-PCR, DNA fingerprinting methods used for Cmm phylogenetics were Random Amplification of Polymorphic DNA (RAPD) for strains in Lithuania (Burokiene et al. 2005) and Canary Islands (De León et al. 2009), Inter Simple Sequence Repeats (ISSR) for strains in Japan (Kawaguchi and Tanina 2014) and Turkey (Baysal et al. 2011), Amplified Fragment Length Polymorphism (AFLP) for strains in Canary Islands (De León et al. 2009) and Southern Italy (Ialacci et al. 2016), and Multiple Locus Variable number of tandem repeat Analysis (MLVA) for strains in Belgium (Zaluga et al. 2013).
The above-mentioned DNA fingerprinting methods have been widely used to study the genetic diversity of Cmm. Most of them have shown a satisfactory level of resolution and have provided important information on the structure of the studied populations. Nevertheless, many of these methods are time-consuming, relatively expensive, and present low interlaboratory accuracy and reproducibility. Multi Locus Sequence Typing and Multi Locus Sequence Analysis (MLST and MLSA respectively) are two different approaches to study of Cmm genetic diversity that overcome the abovementioned limitations (Maiden et al. 1998).
The genes that are exploited in a MLSA approach are not regulated, meaning that there is a plethora of genes to choose from. Jacques et al. (2012) working on isolated strains from different regions worldwide and Croce et al. (2016) working on isolated strains from Uruguay studied the genetic correlation between Cmm strains performing MLSA and MLST analysis based on six housekeeping genes (atpD, dnaK, gyrB, ppK, recA and rpoB). Similarly, Osdaghi et al. (2018) and Ansari et al. (2019) made genetic diversity studies on strains isolated from Iran but utilized five instead of six genes as was also the case for strains from Chile (Valenzuela et al. 2018). In an analysis with a different selection of housekeeping genes than those that were mentioned above (kdpA, sdhA, ligA, gyrB and bipA), Milijašević-Marčić et al. (2012) studied the genetic variability of Cmm strains isolated in Serbia. Similarly, Tancos et al. (2015) added one more housekeeping (dnaK) and three pathogenic (celA, tomA and nagA) genes to study New York strains by MLSA analysis, while Sen et al. (2018) added three pathogenic genes (ppaA, chpC and tomA) on isolates from Turkey. In Michigan, USA, 96 Cmm strains were analyzed utilizing six genes related to pathogenesis (celA, pat-1, ABC transporter, mop, perforin, and phosphatase C). The same strains were also studied simultaneously by rep-PCR (Quesada-Ocampo et al. 2012). Beyond the population analysis that detected 36 haplotypes, the population structure seemed to follow the geographical distribution of the strains.
All the abovementioned analyses did not conclude to any geographical or temporal isolate distribution while the primary infection was consistently coming from infected plant residues left in the soil rather than from contaminated seeds (de León et al. 2011).
Regarding the pathogenicity of Cmm, the bacterium bears pathogenesis or virulence genes that encode serine proteases, chymotrypsin-like proteases, subtilases, xylanases, pectinases, endoglycanase, and a tomatinase, a type of a beta-glucosidase. The role of these genes has been determined mainly by expression and mutational studies, however, their exact function has not been clarified (Nandi et al. 2018). Pathogenic genes are mainly detected in a 129 kb low GC region called chp/tomA pathogenicity island (PAI) (Gartemann et al. 2008) and have been identified in all twelve strains that have been fully sequenced (Thapa et al. 2017). The PAI is subdivided into the chp subdomain, consisting of 16 pathogenicity genes and the tomA subdomain, containing the tomA gene encoding for tomatinase (Kaup et al. 2005, Stork et al. 2008). Usually, the bacterium carries two plasmids, the pCM1 (31-59 kb) and pCM2 (64-109 kb), bearing the celA and pat-1 genes, respectively. Tomatinase is one of the major secreted pathogenic effector proteins of Cmm which degrades the alkaloid α-tomatine in invaded tomato tissues and participates in the defense mechanism against pathogenic microorganisms. Mutational studies have shown that tomatinase is not necessary for bacterial pathogenesis, but its absence reduces the intensity of the infection (Kaup et al. 2005).
The Pat-1 (or Chp) gene belongs to a family of serine proteases and was acknowledged as the most important pathogenicity gene of pCM2 by Burger et al. (2005). However, there are wild-type strains that do not have the pCM2 plasmid without it adversely affecting their pathogenicity (Thapa et al. 2017). The Pat-1 family also includes the chpA to chpG genes, which are located in a 50 kb region of the chp/tomA PAI (Stork et al. 2008) and also have a role to Cmm pathogenicity; the suppression of the chpC, chpE, chpF, and chpG genes reduced the severity of symptoms in artificial inoculations on tomato plants (Stork et al. 2008, Chalupowicz et al. 2010). The chpC protease negatively affects the ethylene level, which appears to be part of the plant's defense against Cmm (Balaji et al. 2008, Savidor et al. 2012). The chpA, chpB, and chpD genes are considered pseudogenes because of the detected shifts or termination codons inside their open reading frames (ORF) (Stork et al. 2008).
Furthermore, the Ppa family consists of multiple chymotrypsin-like serine proteases; six genes are located on the chp/tomA PAI (ppaA to ppaE, including ppaB1/B2), four genes are dispersed in other chromosomal regions (ppaF to ppaI), and a gene is located in the pCM1 plasmid (ppaJ) (Gartemann et al. 2008). As previously described, mutational studies with the ppaA and ppaC genes showed reduced severity of symptoms in the infected plants, though without statistical significance (Chalupowicz et al. 2017).
The third group of Cmm effectors are the subtilase proteases (Siezen and Leunissen 1997) which are involved in Cmm pathogenesis. While the sbtA gene is located on the chp/tomA PAI, the sbtB and sbtC genes are located on a chromosome (Gartemann et al. 2008). When a mutated version of the sbtA gene was introduced, it gave milder symptoms (Chalupowicz et al. 2017). In addition, Cmm has enzymes that break down the cell wall of the host. Among those, a cellulase gene (celA) is located in the pCM1 plasmid and is considered a significant factor for systemic infection where the presence of celA transcripts and proteins are detected (Chalupowicz et al. 2010, Savidor et al. 2012, Chalupowicz et al. 2017). When infections on leaves are established, transcripts are detected but at lower levels (Chalupowicz et al. 2017). When the celA was introduced into the Cmm100 and CmmCASJ002 strains that did not contain the pCM1 and pCM2 plasmids, the ability to infect was restored, as observed by the wilting of the infected plants (Meletzus et al. 1993, Thapa et al. 2017).
The ability of Cmm to infect their hosts differs from strain to strain (Strider and Lucas 1970, Ialacci et al. 2016). So far, there is no strong correlation between the above-mentioned pathogenicity genes and the ability of the bacterium to infect; several studies around the world have indicated that there are strains with increased virulence that do not have any of the pathogenicity genes, and on the other hand, there are less virulent strains with all of the genes (Kleitman et al. 2008, Milijašević-Marčić et al. 2012, Tancos et al. 2015, Croce et al. 2016, Ialacci et al. 2016, Osdaghi et al. 2018). Most probably, these situations are explained either by the existence of additional unknown genes, or the loss of functionality of the pathogenicity genes, respectively (Jacques et al. 2012, Tancos et al. 2015).
Most of Cmm outbreaks in various parts of the world have originated from infected seeds and seedlings. Nevertheless, no study has been conducted so far to investigate the primary infection of outbreaks in Greece. In this research work, we describe the results from a) the study of genetic diversity of 93 Cmm strains in Greece during bacterial outbreaks in various regions of Greece from 2003 to 2018, and b) the study of gene distribution on the chp/tomA PAI and the two pCM plasmids.
Materials and methods
Bacterial isolation and growth conditions
During the period 2003-2018, 98 bacterial strains isolated (Collection of the Department of Agriculture, Hellenic Mediterranean University, HMU) and previously identified as Cmm from 11 Prefectures in Greece were selected for further analysis. All strains were isolated from infected tomatoes cultivated outdoors or in greenhouses, and no more than one strain was selected per area per year for the analyses. When different strains refer to the same Prefecture but different year (e.g., 2016 Lasithi and 2017 Lasithi), isolations were not made to samples from the same location (greenhouse or outdoor field). The isolation was achieved by dispersing the extract from plant-infected tissues onto the semi-selective medium Corynebacterium nebraskense specific medium (CNS) (Gross and Vidaver 1979, Schaad et al. 1988) and Corynebacterium nebraskense specific medium fast (SCMF) (Fatmi and Schaad 1988, EPPO 2016) and incubating at 28°C for 5 to 9 days. Isolates were preliminary identified as Cmm with Gram staining, immunofluorescence, and ELISA tests, and stored in 15% v/v glycerol at -80°C. For further experimentation, the bacterial strains were grown in the solid nutrient medium Nutrient Agar with glucose (NAG, nutrient agar 23 g L-1, NaCl 8 g L-1, glucose 2.5 g L-1) or in Lysogeny Broth (LB, tryptone 10 g L-1, NaCl 10 g L-1, yeast extract 5 g L-1, pH 7).
DNA extraction and purification
Each isolate intended for DNA extraction was grown in LB broth at 28 °C for 48 hours. Four ml of the liquid culture (108 cfu/ml) was used to purify total DNA with the DNeasy Blood and Tissue Kit (Qiagen) following the respective instructions. Quality control and quantification of isolated DNA were done with the Thermo Scientific NanoDrop 2000c spectrophotometer. DNA aliquots of 10 ng/μl were prepared and stored at -20°C.
Indirect immunofluorescence (IF) assay
The indirect immunofluorescence test was performed according to the EPPO guidelines (EPPO 2009) using the polyclonal antibodies anti-Clav 782 and anti-Clav 25 of LBBA (dilution 1/1200), the anti-Cmm polyclonal antibodies from Loewe (Cat No: 07363 Antiserum Clavibacter michiganensis subsp. michiganensis ex goat), and the secondary labeled antibody CF488A Goat anti-Rabbit IgG (H+L) (Cat No: 20012, Biotium Inc). Specimens were observed on an Olympus BH2 microscope with an epifluorescence source.
Molecular identification
Cmm strains were identified by the amplification of a 268 bp and a 132 bp DNA fragments in PCR reactions utilizing the PSA-8/R (Pastrik and Rainey 1999, EPPO 2016) and RZ_ptssk 10/11 (Berendsen et al. 2011) species specific primer pairs. All PCR reactions contained 20 ng of purified DNA as substrate and were performed on a Bio-Rad T100™ Thermal Cycler using the 2X KAPA Taq ReadyMix kit (KK1024, KAPABIOSYSTEMS) in a final volume of 25 μl. The Cmm CFBP4999 and the Dickeya solani IPO2222 were used as positive and negative reference strains, respectively. For the amplification of the 268 bp DNA fragment with the PSA-8/R primer pair (0.5 µM final primer concentration), the PCR program was: initial denaturation at 94 °C for 5 min, 35 cycles of 95 °C for 30 s for denaturation, 63 °C for 20 s for primer hybridization, and 72 °C for 45 s for product polymerization, with a final extension at 72 °C for 5 min. On the other hand, for the amplification of the 132 bp DNA fragment with the RZ_ptssk10/11 primer pair (0.3 µM final primer concentration), the PCR program was: initial denaturation at 95 °C for 10 min, 40 cycles of 95 °C for 15 s for denaturation, 60 °C for 30 s for primer hybridization, and 72 °C for 45 s for product polymerization, with a final extension at 72 °C for 10 min. The amplified fragments were visualized in 1.7% and 2% w/v agarose gels, respectively containing 0,5 μg/mL of ethidium bromide in 1X Tris-acetate-EDTA (TAE). The amplicon size was confirmed by comparison with the bands of a 100 bp DNA Ladder (NM016S, Enzyquest).
Pathogenicity tests
The pathogenicity of isolated strains was initially evaluated with infiltrations in Mirabilis jalapa leaves. Two hundred μl of a 108 cfu/ml bacterial suspension in sterile water from each Cmm strain was infiltrated with a sterile hypodermic syringe (2 replicates per strain) (Gitaitis 1990). Plants were sprayed with water and placed in a chamber with a constant temperature of 26 °C and a photoperiod of 16 h of light and 8 hours of darkness inside transparent bags. Symptoms were evaluated after 36-48 hours.
Artificial inoculations were performed on forty-day-old tomato seedlings (Solanum lycopersicum cv. Ekstasi) grown in pots with black peat mixture as substrate. Tested strains were grown for 72 hours on NAG medium to form single colonies that were used to inoculate the plant with a toothpick. The central stem was pickled between the cotyledons, 1 cm below the junction, with a sterile toothpick that was immersed in a single colony (2 plants per strain). The toothpick was left in the hole to prevent the dehydration of the wound. A toothpick soaked in clean water was used to pickle the negative control plant. The inoculated plants were kept in a chamber at 26 °C and 16:8 photoperiod. Symptoms appeared 20 days after inoculation and were recorded for another 30 days. Two independent replicates were performed, and symptoms were categorized as follows: no symptoms (-), presence of ulcers at the infection site (+), curling, yellowing, and/or wilting of one or more leaves (++), and wilting of the whole plant (+++).
Phylogenetic analysis
Ninety-three strains from the HMU collection were selected for MLSA phylogenetic analyses based on partial amplification of the atpD, kdpA, ppk and sdhA genes (Table 1). Each PCR reaction contained 20 ng of purified total DNA, a mixture of dNTPs at a final concentration of 0.2 mM each, 1.5 mM MgCl2, primers at a final concentration of 0.4 µM (atpD-F/R and ppk-F/R) or 0.5 µM (kdpA-F/R and sdhA-F/R), and 0.5 units of Taq DNA polymerase (KK1016, KAPABIOSYSTEMS) in a final volume of 25 μl. All PCR reactions were performed on a Bio-Rad T100™ Thermal Cycler. The PCR program for the amplification of the atpD and ppk DNA fragments was: initial denaturation at 94 °C for 5 min, 35 cycles with denaturation at 94 °C for 30 s, hybridization at 60 °C for 30 s and elongation at 72 °C for 1 min, final elongation at 72 °C for 10 min. On the other hand, the PCR program for the amplification of the kdpA and sdhA was: initial denaturation at 95 °C for 3 min, 35 cycles of denaturation at 95 °C for 30 s, hybridization at 58 (sdhA-F/R) or 60 °C (kdpA-F/R) for 30 s and extension at 72 °C for 1 min, final extension at 72 °C for 5 min. The products of the PCR reactions (2 μl of a 25 μl reaction) were analyzed on a 1.5% w/v agarose gel containing 0,5 μg/mL ethidium bromide in 1X TAE.
Purification and Sanger sequencing of the amplified fragments were performed by Eurofins (Eurofins Genomics Germany, GmbH Anzinger Str. 7a, 85560, Ebersberg, Germany). Processing of the raw data from the forward and reverse sequencing of the DNA fragments was done with the SnapGene software (from Insightful Science; available at snapgene.com). The consensus sequences from each gene were concatenated and aligned with the MUSCLE algorithm (Edgar 2004).
Sequence polymorphism was analyzed on single genes or on the concatenation of various combinations of gene sequences with DnaSP (Rozas et al. 2017) utilizing the Maximum Likelihood (ML) method. The subspecies insidiosus, nebraskensis, and sepedonicus of Clavibacter michiganensis were used as outgroup taxons while the Cmm strain NCPPB382 used as reference strain. Phylogenetic analysis was performed with the raxmlGUI 2.0.0-beta.14 (Stamatakis 2014, Edler et al. 2021) implementing RAxML (Randomized Axelerated Maximum Likelihood). The individual parts of the concatenated sequences were defined as separate genes (partitions) and the nucleotide substitution model GTR with Gamma distribution (GAMMA) was applied. The statistical robustness and reliability of the dendrogram topology were projected with the bootstrap test at 1000 replications. The dendrogram was visualized using the program MEGA version X (Kumar et al. 2018).
Pathogenicity gene identification
The distribution of 9 genes related to the pathogenicity of the Cmm was studied in 39 out of the 93 strains used in the phylogenetic analysis. The presence of the genes chpC, celA, ppaA, tomA, chpE, pat-1, chpG, ppaC, and sbtA was confirmed by the amplification of gene-specific DNA fragments by PCR (Table 1). The program followed was: initial denaturation at 94°C for 5 min, 35 cycles with denaturation at 94 °C for 30 s, hybridization as shown in Table 1 for 30 s and extension at 72 °C for 30 s, final extension at 72 °C for 5 min. For the amplification of the chpG the reaction program was: initial denaturation at 95 °C for 3 min, 35 cycles of denaturation at 95 °C for 30 s, hybridization as shown in Table 1 for 30 s, and extension at 72 °C for 30 s, and final extension at 72 °C for 1 min. All PCR reactions were performed on a Bio-Rad T100™ Thermal Cycler using the 2X KAPA Taq ReadyMix with dye (KK1024, KAPABIOSYSTEMS) in a final volume of 25 μl. The substrate used was 20 ng of purified total DNA and the primer concentration was as shown in Table 1. The Cmm strain CFBP4999 was used as a positive control, while the strain Dickeya solani IPO2222 was used as a negative control. The products of the PCR reactions (10 μl of a 25 μl reaction) were analyzed by agarose gel electrophoresis.
Results
Isolation and identification of Cmm strains
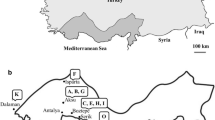
From 2003 to 2018, 12 outbreak periods of Cmm in tomato fields have been recorded in various regions of Greece, in covered as well as outdoor crops. During this time, samples of infected plants were received mainly from Crete (Lasithi, Heraklion, Chania), but also from the Prefectures of the Peloponnese (Argolis, Arcadia, Corinthia, Messinia), Central Macedonia (Imathia, Chalkidiki), Magnesia, East Macedonia and Thrace (Xanthi). From these Cmm samples, more than 150 strains were isolated and identified, which make up the Cmm collection at HMU. Colonies on NAG were mucilaginous and yellow, pale yellow, or white in color.
Ninety-eight strains from the HMU collection identified as Cmm were selected for further analyses. The identification of the isolated strains was done by collective evaluation with the hypersensitivity response reaction (HR), the 3% w/v KOH test, the indirect immunofluorescence test (IF), the two species-specific primer pairs PSA-8/R and RZ_ptssk 10/11 PCR tests, and the evaluation of artificial inoculations on tomato seedlings (Fig. 1). All strains were positive in 3% KOH (Gram positive strains), HR, IF, and PCR tests but presented variation in the reproduction of symptoms (Table 2).

Bacterial canker symptoms on tomato plants. a, typical disease symptoms in a greenhouse crop at a final stage of infection with the appearance of hydrobacteriosis in the planting line. b, unripe tomato fruit injuries presented as spots (left) or marbling (right). c, Discoloration, necrosis and separation of vascular tissue from the tomato pith. d, Pustular spots on the upper surface of the tomato leaf
The disease symptoms in artificially inoculated tomato plant seedlings appeared 20 days post-inoculation. Among the main symptoms were the canker of the stem at the point of inoculation and the upward turning of one or a few of the leaves. As the systemic infection progresses, the entire leaves may wilt and shrivel (Fig. 1). Most of the strains caused stem canker and wilting of more than one leaf or total of the plant, except for four that caused only canker (HMU4046, HMU4646, HMU4666, and HMU4698) and 6 that gave no symptoms (HMU4209, HMU4221, HMU4533, HMU4690, HMU4721, and HMU4940).
Genetic diversity analysis
The genetic diversity of 93 strains isolated from 11 Prefectures of Greece in 12 different years from 2003 to 2018 (Table 2) was studied by MLSA. The MLSA scheme was based on the concatenation of partial DNA sequences of atpD(GenBank: OP471139-OP471231), kdpA(GenBank: OP470953-OP471045), ppk (GenBank: OP471046-OP471138), and sdhA (OP470860-OP470952). The atpD and ppk genes were chosen due to the presence of increased polymorphic nucleotides in Cmm strains from Uruguay (Croce et al. 2016). Similarly, the kdpA and sdhA genes were chosen, as they proved polymorphic in strains from Serbia and New York (Milijašević-Marčić et al. 2012, Tancos et al. 2015). The diversity analysis of the single- as well as two-, three- and four-gene concatenated sequences was investigated with the DNASP software and showed relatively low genetic variability between Cmm strains (Table 3).
The highest polymorphism in individual gene sequences was observed in kdpA, with 35 polymorphic nucleotides (6.1% percentage of polymorphic nucleotides) and the highest number of haplotypes (23). The sdhA gene followed with a small difference (31 polymorphic nucleotides, 4.8% and 18 haplotypes). The atpD and ppk genes had considerably lower polymorphism; the ppk gene had a 3.5% polymorphism while the atpD had 1.4%. However, the latter yielded an increased number of haplotypes (10) than the former (8). In the sequence analysis of various gene concatenation sequences, the highest percentage of polymorphic nucleotides was observed in the kdpA-sdhA pair (5.4%, 34 haplotypes), while most haplotypes (35) arose from the kdpA-ppk and atpD-kdpA sequences (4.9% and 3.6%, respectively). When the analysis was based on a three-gene concatenation, the atpD-kdpA-sdhA combination gave 75 polymorphic nucleotides (4%) and 38 haplotypes, the same as the haplotypes yielded from the analysis of all genes together. The combinations atpD-kdpA-ppk, kdpA-ppk-sdhA, and atpD-ppk-sdhA yielded 37, 36 and 26 haplotypes, respectively. Finally, the four-gene combination (atpD-kdpA-ppk-sdhA) pointed-out 93 nucleotide polymorphisms (3.9%) and gave 38 haplotypes. Out of the 38 haplotypes, the most frequent (Hap_27), found in 13 strains, and the rarest was found in one strain.
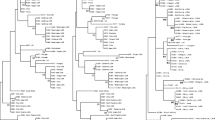
Following MLSA, a Maximum Likelihood (ML) method was utilized to generate dendrograms based on individual gene sequences of atpD (Supplementary Fig. 1), kdpA (Supplementary Fig. 2), ppk (Supplementary Fig. 3), and sdhA (Supplementary Fig. 4) as well as based on the concatenation of three-gene sequences (atpD-kdpA-sdhA, Supplementary Fig. 5) and four-gene sequences (atpD-kdpA-ppk-sdhA, Fig. 2). The atpD-kdpA-sdhA combination was selected because of the highest number of haplotypes and nucleotide polymorphisms (Table 3). No dendrograms were generated with the two-gene combinations.

Maximum Likelihood phylogenetic tree based on the concatenated sequences of atpD, kdpA, ppk and sdhA genes of Clavibacter michiganensis subsp. michiganensis strains isolated from different Prefectures of Greece between 2003 and 2018. Bootstrap values above 50% are shown in branch nodes (1000 replicates). The clades with high bootstrap values (80-100%) are highlighted with a grey shadow. Within the clades, the strains in blue-dashed rectangulars were isolated from the same region at the same year or different years
The dendrograms generated with individual genes and four-gene analysis were in-agreement with respect to their topology. In the latter, all Cmm strains were grouped with strong Bootstrap support (100%) and clearly separated from the three closely related Clavibacter michiganensis subspecies insidiosus, sepedonicus, and nebraskensis. The Cmm monophyletic group is further divided into 16 clades with strong bootstrap support (80-100%) though strains in these clades do not appear to be related either to the location or year of isolation. More specifically, isolates from the same year or county are spread across different clades. For example, strains isolated in 2016 and 2017 from the Prefecture of Lasithi and Chania are found in over five different clades, indicating the existence of different sources of infection. It is possible that those producers obtained infected seeds or seedlings from different sources where they were infected with different strains of Cmm. Although there were cases where strains isolated from the same year and from the same Prefecture had the same haplotype (HMU4505/HMU4512, HMU4548/HMU4570, HMU4678/HMU4673) it was not the rule. There were cases where strains with the same haplotype isolated from different counties and years, also with strong ootstrap support, indicating the common source of propagating material. That was also the case for the strains HMU4258 (Chalkidiki) and HMU4256 (Lasithi), which although isolated from areas outside of Crete, were grouped with strains from Crete.
Consequently, there was no geographical or temporal dispersion of haplotypes since no haplotype was found in more than 5 different Prefectures or years of isolation.
Pathogenicity gene distribution in isolated strains
Most of the genes linked to Cmm's ability to cause disease are found in the chp/tomA PAI and the plasmids pCM1 and pCM2. More specifically, the chpC, chpE, chpG, ppaA, ppaC, sbtA, and tomA genes of the chp/tomA PAI and the celA and pat-1 genes in pCM1 and pCM2 were studied. Thirty-nine of the 93 isolates from the MLSA analysis were selected based on the Prefecture, the year of isolation, and the symptoms on tomato seedlings. The presence of pathogenicity genes in the genomes of the selected strains was analyzed by conventional PCR (Table 4). In addition, the pathogenicity of the strains was assessed by artificial inoculations in tomato seedlings (Table 4). The investigation of all the above-mentioned genes except celA and pat-1 gave the expected DNA band (Table 1).
Results from the pathogenicity assays were also not consistent, a fact that is probably not related to the distribution of the genes in the chp/tomA pathogenicity island or the respective plasmids. Twenty-four out of 39 strains gave the typical symptoms on one or more leaves (++) while 11 strains caused total wilting of the plant (+++). Of the rest strains, HMU4209 and HMU4721 did not cause any symptoms (-), and strains HMU4046, HMU4646 showed reduced virulence (+) causing only ulceration at the point of infection.
Reactions to detect the genes tomA, chpC, chpE, chpG, sbtA, ppaA, and ppaC gave positive signals in all tested strains (Table 4). The celA gene of pCM1 was detected in all strains except HMU4209, which did not cause any symptoms (-) in tomato seedlings. This was an indication that the lack of the celA gene may be related to the absence of pathogenicity in the strain.
The pat-1 gene was not detected in 17 strains that presented different intensities of pathogenicity. Nevertheless, this lack of PCR signal does not necessarily mean absence of the pat-1 gene and may be the result of mutations at the primer annealing sites. One of them caused no symptoms (-), two caused only an ulcer at the site of infection (+), 12 gave the typical symptoms (++) and two caused total wilting of the plant (+++). Thus, the pat-1 gene may contribute to the pathogenicity of Cmm but does not appear to be essential. Furthermore, strains lacking the pat-1 gene did not appear to be phylogenetically different from the rest of the Cmm strains in the dendrogram based on the atpD-kdpA-ppk-sdhA sequence. Most are in different branches, while only HMU4521/HMU4721, HMU4646/HMU4752, and HMU4330/HMU4337/HMU4349/HMU4375 had the same haplotype (Fig. 2).
Discussion
Tomato bacterial canker is one of the most important diseases in Greece, when it comes to growing tomatoes, causing serious losses in greenhouse and outdoor crops that can lead to destruction. The disease has spread throughout the tomato-growing regions of the world, mainly due to infected propagating material (Tsiantos 1987, Gleason et al. 1993). The lack of any chemical or biological agent to effectively combat Cmm makes the disease extremely dangerous for tomato-producing countries.
The identification of the sources of primary infections during disease outbreaks is helpful in creating a comprehensive plan to manage the importation of propagation material and the application of practices that will prevent the spread of the disease. From 2003 to 2018, twelve outbreaks of bacterial canker were recorded in Greece, during which significant economic losses were caused in tomato production. During this period, a large number of tomato plant samples were investigated, and a large number of strains were isolated. The samples derived from greenhouse and outdoor tomato crops were mainly infected during the autumn and winter; the short photoperiod favored the growth of the pathogen (EFSA_Panel_on_Plant_Health 2014). In few cases, the pathogen was isolated during the summer. From the analysis of infected plants, 98 Cmm strains were isolated and identified by phenotypic, immunological, and molecular methods as well as by pathogenicity tests.
According to EPPO classification, the Cmm is regarded as an A2 quarantine pathogen in the EU. The most effective way to prevent the import and spread of the disease is to avoid the transport of infected propagation material, the main source of inoculum, but also to ensure proper management of already infected crops to eliminate new inoculant spots (Yasuhara-Bell and Alvarez 2015).
In this work, 93 Cmm isolated strains were selected and genetically analyzed to determine the source of primary infections during the reference period. The MLSA method was utilized based on the four housekeeping genes atpD, kdpA, ppk, and sdhA, which were chosen due to the nucleotide polymorphism they present. The atpD and ppk genes showed lower polymorphism among the Cmm strains studied (1.4% and 3.5%) polymorphic loci that separated the strains into 10 and 8 haplotypes, respectively (Table 3). Those numbers were close to Uruguayan strains (1.6% and 4.3%, respectively) (Croce et al. 2016) but much lower than those derived from strains isolated from all over the world (Jacques et al. 2012). The kdpA and sdhA genes showed greater polymorphism in our study, 6.1% and 5.1%, which gave 23 and 18 haplotypes respectively. Although the kdpA gene yielded a significantly larger number of polymorphic sites compared to the study of Cmm in Serbia and New York (2.7% and 3.7%) (Milijašević-Marčić et al. 2012, Tancos et al. 2015), the sdhA gene presented slightly lower polymorphism (5.4% and 4.3%). When phylogenesis was based on the concatenated sequences of the four genes, 93 polymorphic loci (4.1%) were identified, producing 38 haplotypes. The detected polymorphism was lower than that detected when a more wide global collection of strains was analyzed (8.7%) (Jacques et al. 2012). Though, it was higher than that of Uruguayan strains (1.8%) (Croce et al. 2016).
The phylogenetic analysis of this study was done by creating a Maximum Likelihood (ML) phylogenetic tree, using the concatenated sequence consisting of the atpD, kdpA, ppk, and sdhA individual sequences (Fig. 2). Moreover, additional dendrograms were created based on the individual gene sequences (Supplementary Figs. 1-4). In all cases, the isolated strains were consistently separated from the three related subspecies (C.m. insidiosus, C.m. nebraskensis, and C.m. sepedonicus). Consequently, the concatenation of these four gene sequences is suitable for the phylogenetic analysis of Cmm strains since it increases bootstrap support and strain resolution.
Moreover, the Cmm group of strains is comprised of 16 clades with very good bootstrap support (80-100%). The separation of strains in most of these branches is not related to area or year of isolation, i.e., there are branches consisting of a) strains isolated from different areas or isolated from different years, b) strains isolated from the same area but from a different year, and c) strains isolated from the same area and the same year. More specifically, the phylogenetic analysis showed that strains isolated from the same Prefecture and the same year span more than five different branches. In these cases, the primary contamination of the crops probably came from different sources through infected seeds or seedlings supplied by the producers. The clustering of strains isolated from different regions and in different years on the same branch indicates a common initial source of infection. A possible explanation would be the import of seeds from a country in which the specific haplotype of Cmm exists and was spread in Greece in the following years. In some branches, only strains isolated from the same region and year are grouped together. This fact can be explained either by the common source of contamination through the propagating material or by the transmission of the bacterium between closely spaced fields without applying good agricultural practices.
Though, this finding is consistent with data from Argentina and Turkey where rep-PCR and MLSA analysis were performed with the housekeeping genes hipA, gyrB, kdpA, ligA, and sdhA and the pathogenicity genes ppaA, chpC, and tomA (Wassermann et al. 2017, Sen et al. 2018). In contrast, a similar study in Michigan by BOX-PCR and MLSA showed a significant correlation between the genetic variability and the region of isolation of the strains (Quesada-Ocampo et al. 2012). In general, the geographical and temporal distribution of haplotypes was relatively low since no haplotype was found in more than five different Prefectures or years of isolation. This result supports the hypothesis of the introduction of the disease through the import and spread of infected propagation material.
The chpC, chpE, chpG, ppaA, ppaC, sbtA and tomA genes of the chp/tomA PAI and the celA and pat-1 genes of the pCM1 and pCM2 plasmids, respectively, are considered important for the pathogenicity of the bacterium (Nandi et al. 2018). The presence or absence of these genes was confirmed by conventional PCR in 39 Cmm strains out of the 93 analyzed by MLSA that were selected to include at least one strain from each different region and year of isolation. The pathogenicity tests revealed extreme differences in virulence, with symptoms ranging from mild to typical to extreme, causing complete wilting of the plants, while two strains showed no symptoms.
Based on the abovementioned differences in pathogenicity, it was expected that several genes that are linked with the chp/tomA PAI would be absent from the strains that showed reduced or no virulence. Nevertheless, the seven chromosomal genes of the chp/tomA PAI were detected in all 39 Cmm indicating that the intensity of the pathogenicity is not related to the genes chpC, chpE, chpG, ppaA, ppaC, sbtA, and tomA. However, we cannot support this conclusion with certainty since it may be possible that some of the genes are not fully functional. Further studies on the functionality of each gene could confirm the above conclusion. A similar study on Cmm strains in Argentina showed that the ppaA and chpC genes were absent from non-pathogenic strains, concluding that the ppaA and chpC genes are essential for the virulence of the bacterium (Kleitman et al. 2008).
The celA gene, which is located on the pCM1 plasmid, was found in all studied strains except for the non-infectious HMU4209. The absence of celA from this strain could be responsible for the lack of pathogenicity, however, we cannot support this with certainty, although studies in mutant strains lacking the celA have shown that the re-introduction of the gene restores the virulence (Meletzus et al. 1993, Thapa et al. 2017, Hwang et al. 2019). In the New York study, the celA gene was the least absent gene (Tancos et al. 2015) as is the case in this study. In the same study, of the three strains from which the celA was absent, one was fully virulent while the other two had reduced virulence. In the study with the Uruguayan and Iranian strains, only one strain lacked the celA while being pathogenic (Croce et al. 2016, Osdaghi et al. 2018). Moreover, in the global collection studied by Jacques et al. (2012), the celA was not detected in four strains, which, in contrast, retained their virulence. Consequently, it appears that the celA gene may contribute to Cmm virulence but is not always essential for it. Most probably, there are unknown virulence factors that interact with the celA gene.
Concerning the pat-1 gene on the pCM2 plasmid, it was not found in 17 of the 39 strains that were studied (Table 4), but all the other pathogenicity genes were there. The ML phylogenetic tree (Fig. 2) shows that these strains are not close to each other. This suggests that the loss of the pat-1 gene has nothing to do with their phylogenetic separation. Pathogenicity tests of these strains showed the full range of symptoms in infected plants. Most of them (12) gave the typical symptoms (++), two gave total wilting of the plant (+++), two gave simple cankers at the site of infection (+) and one plant’s symptoms were absent. These results show that pat-1 may contribute to the pathogenicity but does not seem to play an essential role in the virulence of the bacterium, and its contribution may be masked by other infectious agents. To this end, there are several studies where, although the strains are negative for the pat-1 gene, they present strong, reduced (Tancos et al. 2015, Ialacci et al. 2016, Jacques et al. 2012) or no virulence (Milijašević-Marčić et al. 2012). The abovementioned results are justified by the observed heterogeneity in the pathogenic ability of the studied strains lacking the pat-1, strengthening the hypothesis that the pat-1 contributes to Cmm virulence but is not essential for it.
In conclusion, the study of the 93 Cmm strains isolated from various areas of Greece and studied with an MLSA approach, separated the strains into 38 haplotypes, showing increased genetic variability. The analysis was based on the atpD, kdpA, ppk, and sdhA genes and appeared suitable for the clustering analysis of the Cmm population in Greece. The phylogenetic separation of the strains in the dendrogram showed that there was no regional or temporal distribution of the isolations. The increased variability has probably resulted from the introduction of the bacterium into Greece from different sources; infected seeds were possibly imported by seed distribution companies, establishing the initial contamination that was then transmitted by farmers. The wide spread of the disease was probably due to the inappropriate control of symptomatic plants and favorable weather conditions that were further spread to neighboring crops by infected plant debris, although we cannot support this since the strains were isolated from unrelated geographic areas.
Data availability
All analyzed gene sequences are freely available through Genbank (atpD = OP471139-OP471231, kdpA = OP470953-OP471045, ppk = OP471046-OP471138, sdhA = OP470860-OP470952).
References
Ansari M, Taghavi SM, Hamzehzarghani H, Valenzuela M, Siri MI, Osdaghi E (2019) Multiple introductions of tomato pathogen Clavibacter michiganensis subsp. michiganensis into Iran as revealed by a global-scale phylogeographic analysis. Appl Environ Microbiol 85:e02098–e02019
Balaji V, Mayrose M, Sherf O, Jacob-Hirsch J, Eichenlaub R, Iraki N et al (2008) Tomato transcriptional changes in response to Clavibacter michiganensis subsp. michiganensis reveal a role for ethylene in disease development. Plant Physiol 146:1797–1809
Basım H, Basım E (2018) Phenotypic and genotypic characterization of Clavibacter michiganensis subsp. michiganensis causing tomato bacterial canker and wilt disease in Turkey. Eur J Plant Pathol 151:355–369
Baysal Ö, Mercati F, İkten H, Yıldız RÇ, Carimi F, Aysan Y et al (2011) Clavibacter michiganensis subsp. michiganesis: Tracking strains using their genetic differentiations by ISSR markers in Southern Turkey. Physiol Mol Plant Pathol 75:113–119
Berendsen S, Koenraadt H, Woudt B, Oosterhof J (2011) The development of a specific Real-Time TaqMan for the detection of Clavibacter michiganensis subsp. michiganensis. In: Presentedatthe American Phytopathological Society-International Plant Protection Convention Meeting, Honolulu, Hawaii
Blank L, Cohen Y, Borenstein M, Shulhani R, Lofthouse M, Sofer M et al (2016) Variables associated with severity of bacterial canker and wilt caused by Clavibacter michiganensis subsp. michiganensis in tomato greenhouses. Phytopathology 106:254–261
Burger A, Gräfen I, Engemann J, Niermann E, Pieper M, Kirchner O et al (2005) Identification of homologues to the pathogenicity factor Pat-1, a putative serine protease of Clavibacter michiganensis subsp. michiganensis. Microbiol Res 160:417–427
Burokiene D, Pulawska J, Sobiczewski P (2005) Genetic diversity of Clavibacter michiganensis subsp. michiganensis isolates from Lithuania. Phytopatol Polonica 38:79–90
Carlton W, Braun E, Gleason M (1998) Ingress of Clavibacter michiganensis subsp. michiganensis into tomato leaves through hydathodes. Phytopathology 88:525–529
Chalupowicz L, Barash I, Reuven M, Dror O, Sharabani G, Gartemann KH et al (2017) Differential contribution of Clavibacter michiganensis ssp. michiganensis virulence factors to systemic and local infection in tomato. Mol Plant Pathol 18:336–346
Chalupowicz L, Cohen-Kandli M, Dror O, Eichenlaub R, Gartemann K-H, Sessa G et al (2010) Sequential expression of bacterial virulence and plant defense genes during infection of tomato with Clavibacter michiganensis subsp. michiganensis. Phytopathology 100:252–261
Croce V, Pianzzola MJ, Durand K, González-Arcos M, Jacques M-A, Siri MI (2016) Multilocus sequence typing reveals high variability among Clavibacter michiganensis subsp. michiganensis strains affecting tomato crops in Uruguay. Eur J Plant Pathol 144:1–13
Davis MJ, Gillaspie AG Jr, Vidaver AK, Harris RW (1984) Clavibacter: a new genus containing some phytopathogenic coryneform bacteria, including Clavibacter xyli subsp. xyli sp. nov., subsp. nov. and Clavibacter xyli subsp. cynodontis subsp. nov., pathogens that cause ratoon stunting disease of sugarcane and bermudagrass stunting disease. Int J Syst Evol Microbiol 34:107–117
De León L, Rodriguez A, Llop P, López MM, Siverio F (2009) Comparative study of genetic diversity of Clavibacter michiganensis subsp. michiganensis isolates from the Canary Islands by RAPD-PCR, BOX-PCR and AFLP. Plant Pathol 58:862–871
de León L, Siverio F, López MM, Rodríguez A (2008) Comparative efficiency of chemical compounds for in vitro and in vivo activity against Clavibacter michiganensis subsp. michiganensis, the causal agent of tomato bacterial canker. Crop Protect 27:1277–1283
de León L, Siverio F, López MM, Rodríguez A (2011) Clavibacter michiganensis subsp. michiganensis, a seedborne tomato pathogen: healthy seeds are still the goal. Plant Dis 95:1328–1338
Dreier J, Bermpohl A, Eichenlaub R (1995) Southern hybridization and PCR for specific detection of phytopathogenic Clavibacter michiganensis subsp. michiganensis. Phytopathology 85:462–468
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Edler D, Klein J, Antonelli A, Silvestro D (2021) raxmlGUI 2.0: A graphical interface and toolkit for phylogenetic analyses using RAxML. Methods in Ecology and Evolution 12:373–377
EFSA_Panel_on_Plant_Health (2014) Scientific opinion on the pest categorisation of Clavibacter michiganensis subsp. michiganensis (Smith) Davis et al. EFSA J 12:3721
EPPO (2009) PM 7/97 (1): Indirect immunofluorescence test for plant pathogenic bacteria. In: EPPO Bulletin, pp 413-416
EPPO (2016) PM 7/42 (3) Clavibacter michiganensis subsp. michiganensis. In: EPPO Bulletin, pp 202-225
Fatmi M, Schaad N (1988) Semiselective agar medium for isolation of Clavibacter michiganense subsp. michiganense from tomato seed. Phytopathology 78:121–126
Fatmi M, Schaad N (2002) Survival of Clavibacter michiganensis ssp. michiganensis in infected tomato stems under natural field conditions in California, Ohio and Morocco. Plant Pathol 51:149–154
Fatmi M, Schaad N, Bolkan H (1991) Seed treatments for eradicating Clavibacter michiganensis subsp. michiganensis from naturally infected tomato seeds. Plant Dis 75:383–385
Flügel M, Becker A, Gartemann K-H, Eichenlaub R (2012) Analysis of the interaction of Clavibacter michiganensis subsp. michiganensis with its host plant tomato by genome-wide expression profiling. J Biotechnol 160:42–54
Gartemann K-H, Abt B, Bekel T, Burger A, Engemann J, Flügel M et al (2008) The genome sequence of the tomato-pathogenic actinomycete Clavibacter michiganensis subsp. michiganensis NCPPB382 reveals a large island involved in pathogenicity. J Bacteriol 190:2138–2149
Gitaitis R (1990) Induction of a hypersensitivelike reaction in four-o’clock by Clavibacter michiganensis subsp. michiganensis. Plant Dis 74:58–60
Gitaitis R, Beaver R, Voloudakis A (1991) Detection of Clavibacter michiganensis subsp. michiganensis in symptomless tomato transplants. Plant Dis 75:834–838
Gleason ML, Gitaitis RD, Ricker MD (1993) Recent progress in understanding and controlling bacterial canker of tomato in eastern North America. Plant Dis 77:1069–1076
Gross DC, Vidaver A (1979) A selective medium for isolation of Corynebacterium nebraskense from soil and plant parts. Phytopathology 69:82–87
Hadas R, Kritzman G, Klietman F, Gefen T, Manulis S (2005) Comparison of extraction procedures and determination of the detection threshold for Clavibacter michiganensis ssp. michiganensis in tomato seeds. Plant Pathol 54:643–649
Hwang IS, Oh E-J, Lee HB, Oh C-S (2019) Functional characterization of two cellulase genes in the Gram-positive pathogenic bacterium Clavibacter michiganensis for wilting in tomato. Mol Plant-Microbe Interact 32:491–501
Ialacci G, Bella P, Licciardello G, Strano C, Eichenlaub R, Gartemann KH et al (2016) Clonal populations of Clavibacter michiganensis subsp. michiganensis are responsible for the outbreaks of bacterial canker in greenhouse tomatoes in Italy. Plant Pathol 65:484–495
Jacques M-A, Durand K, Orgeur G, Balidas S, Fricot C, Bonneau S et al (2012) Phylogenetic analysis and polyphasic characterization of Clavibacter michiganensis strains isolated from tomato seeds reveal that nonpathogenic strains are distinct from C. michiganensis subsp. michiganensis. Appl Environ Microbiol 78:8388–8402
Jensen H (1982) Corynebacterium michiganense (EF Smith). EPPO Bull 12:13–18
Kaup O, Gräfen I, Zellermann E-M, Eichenlaub R, Gartemann K-H (2005) Identification of a tomatinase in the tomato-pathogenic actinomycete Clavibacter michiganensis subsp. michiganensis NCPPB382. Mol Plant-Microbe Interact 18:1090–1098
Kawaguchi A, Tanina K (2014) Genetic groups of Clavibacter michiganensis subsp. michiganensis identified by DNA fingerprinting and the effects of inoculation methods on disease development. Eur J Plant Pathol 140:399–406
Kawaguchi A, Tanina K, Inoue K (2010) Molecular typing and spread of Clavibacter michiganensis subsp. michiganensis in greenhouses in Japan. Plant Pathol 59:76–83
Kleitman F, Barash I, Burger A, Iraki N, Falah Y, Sessa G et al (2008) Characterization of a Clavibacter michiganensis subsp. michiganensis population in Israel. Eur J Plant Pathol 121:463–475
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Li X, Tambong J, Yuan KX, Chen W, Xu H, Lévesque CA et al (2018) Re-classification of Clavibacter michiganensis subspecies on the basis of whole-genome and multi-locus sequence analyses. Int J Syst Evol Microbiol 68:234
Louws FJ, Bell J, Medina-Mora CM, Smart CD, Opgenorth D, Ishimaru CA et al (1998) rep-PCR-mediated genomic fingerprinting: A rapid and effective method to identify clavibacter michiganensis. Phytopathology 88:862–868
Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R et al (1998) Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Nat Acad Sci 95:3140–3145
Meletzus D, Bermphol A, Dreier J, Eichenlaub R (1993) Evidence for plasmid-encoded virulence factors in the phytopathogenic bacterium Clavibacter michiganensis subsp. michiganensis NCPPB382. J Bacteriol 175:2131–2136
Milijašević-Marčić S, Gartemann K-H, Frohwitter J, Eichenlaub R, Todorović B, Rekanović E et al (2012) Characterization of Clavibacter michiganensis subsp. michiganensis strains from recent outbreaks of bacterial wilt and canker in Serbia. Eur J Plant Pathol 134:697–711
Nandi M, Macdonald J, Liu P, Weselowski B, Yuan ZC (2018) Clavibacter michiganensis ssp. michiganensis: bacterial canker of tomato, molecular interactions and disease management. Mol Plant Pathol 19:2036–2050
Nazari F, Niknam G, Ghasemi A, Taghavi S, Momeni H, Torabi S (2007) An investigation on strains of Clavibacter michiganensis subsp. michiganensis in north and north west of Iran. J Phytopathol 155:563–569
Osdaghi E, Ansari M, Taghavi SM, Zarei S, Koebnik R, Lamichhane JR (2018) Pathogenicity and phylogenetic analysis of Clavibacter michiganensis strains associated with tomato plants in Iran. Plant Pathol 67:957–970
Pastrik KH, Rainey FA (1999) Identification and differentiation of Clavibacter michiganensis subspecies by Polymerase Chain Reaction-based techniques. J. Phytopathol 147:687–693
Quesada-Ocampo L, Landers N, Lebeis A, Fulbright D, Hausbeck M (2012) Genetic structure of Clavibacter michiganensis subsp. michiganensis populations in Michigan commercial tomato fields. Plant Dis 96:788–796
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE et al (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302
Savidor A, Teper D, Gartemann K-H, Eichenlaub R, Chalupowicz L, Manulis-Sasson S et al (2012) The Clavibacter michiganensis subsp. michiganensis-tomato interactome reveals the perception of pathogen by the host and suggests mechanisms of infection. J Proteome Res 11:736–750
Schaad NW, Jones JB, Chun W (1988) Laboratory guide for identification of plant pathogenic bacteria. APS Press, St. Paul, MN, USA
Sen Y, Aysan Y, Mirik M, Ozdemir D, Meijer-Dekens F, van der Wolf JM et al (2018) Genetic characterization of Clavibacter michiganensis subsp. michiganensis population in Turkey. Plant Dis 102:300–308
Siezen RJ, Leunissen JAM (1997) Subtilases: The superfamily of subtilisin-like serine proteases. Protein Sci 6:501–523
Smith E (1910) A new tomato disease of economic importance. Science 31:794–796
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Stork I, Gartemann KH, Burger A, Eichenlaub R (2008) A family of serine proteases of Clavibacter michiganensis subsp. michiganensis: chpC plays a role in colonization of the host plant tomato. Mol Plant Pathol 9:599–608
Strider D, Lucas L (1970) Variation in virulence in Corynebacterium michiganense. Plant Dis Rep 54:976–978
Tancos MA, Lange HW, Smart CD (2015) Characterizing the genetic diversity of the Clavibacter michiganensis subsp. michiganensis population in New York. Phytopathology 105:169–179
Thapa SP, Pattathil S, Hahn MG, Jacques M-A, Gilbertson RL, Coaker G (2017) Genomic analysis of Clavibacter michiganensis reveals insight into virulence strategies and genetic diversity of a Gram-positive bacterial pathogen. Mol Plant-Microbe Interact 30:786–802
Tsiantos J (1987) Transmission of bacterium Corynebacterium michiganense pv. michiganense by seeds. J Phytopathol 119:142–146
Umesha S (2006) Occurrence of bacterial canker in tomato fields of Karnataka and effect of biological seed treatment on disease incidence. Crop Protect 25:375–381
Valenzuela M, Besoain X, Durand K, Cesbron S, Fuentes S, Claverías F et al (2018) Clavibacter michiganensis subsp. michiganensis strains from central Chile exhibit low genetic diversity and sequence types match strains in other parts of the world. Plant Pathol 67:1944–1954
Wassermann E, Montecchia MS, Correa OS, Damián V, Romero AM (2017) Clavibacter michiganensis subsp. michiganensis strains virulence and genetic diversity. A first study in Argentina. Eur J Plant Pathol 149:35–42
Xu X, Miller SA, Baysal-Gurel F, Gartemann K-H, Eichenlaub R, Rajashekara G (2010) Bioluminescence imaging of Clavibacter michiganensis subsp. michiganensis infection of tomato seeds and plants. Appl Environ Microbiol 76:3978–3988
Yasuhara-Bell J, Alvarez AM (2015) Seed-associated subspecies of the genus Clavibacter are clearly distinguishable from Clavibacter michiganensis subsp. michiganensis. Int J Syst Evol Microbiol 65:811–826
Zachos DG, Georgopoulos SG (1957) The bacterial canker of tomatoes in Greece. Annals of Benaki Phytopathological Institute 1:98–102
Zaluga J, Stragier P, Van Vaerenbergh J, Maes M, De Vos P (2013) Multilocus Variable-Number-Tandem-Repeats Analysis (MLVA) distinguishes a clonal complex of Clavibacter michiganensis subsp. michiganensis strains isolated from recent outbreaks of bacterial wilt and canker in Belgium. BMC Microbiol 13:1–15
Acknowledgments
This research has been co‐financed by the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH - CREATE - INNOVATE (project: VEG-PROTECT; code: T1EDK-04142). The authors would like to acknowledge the support of Hellenic Mediterranean University that being a member of HEAL-Link provided the publication of this article in Open Access mode. We would also like to thank Aglaia Antoniou, researcher at HCMR, for her valuable advice in phylogenetic analysis.
Funding
Open access funding provided by HEAL-Link Greece.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors have no competing interests to declare that are relevant to the content of this article
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
42161_2023_1375_MOESM5_ESM.tif
Maximum Likelihood phylogenetic tree based on the concatenated sequences of atpD, kdpA, and sdhA genes of Clavibacter michiganensis subsp. michiganensis strains isolated from different Prefectures of Greece between 2003 and 2018. Bootstrap values above 50% are shown in branch nodes (1000 replicates). The clades with high bootstrap values (80-100%) are highlighted with a grey shadow. Within the clades, the strains in blue-dashed rectangulars were isolated from the same region at the same year or different years.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Malliarakis, D., Pagoulatou, M.G., Mpalantinaki, E. et al. Phylogenetic diversity of Clavibacter michiganensis subsp. michiganensis isolates causing bacterial canker of tomato in Greece. J Plant Pathol 105, 1403–1419 (2023). https://doi.org/10.1007/s42161-023-01375-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42161-023-01375-4