
Abstract
Transition metal oxides (TMOs) are considered as promising anode materials for lithium-ion batteries in comparison with conventional graphite anode. However, TMO anodes suffer severe volume expansion during charge/discharge process. In this respect, a porous Fe2O3 nanorod-decorated hollow carbon nanofiber (HNF) anode is designed via a combined electrospinning and hydrothermal method followed by proper annealing. FeOOH/PAN was prepared as precursors and sacrificial templates, and porous hollow Fe2O3@carbon nanofiber (HNF-450) composite is formed at 450 °C in air. As anode materials for lithium-ion batteries, HNF-450 exhibits outstanding rate performance and cycling stability with a reversible discharge capacity of 1398 mAh g−1 after 100 cycles at a current density of 100 mA g−1. Specific capacities 1682, 1515, 1293, 987, and 687 mAh g−1 of HNF-450 are achieved at multiple current densities of 200, 300, 500, 1000, and 2000 mA g−1, respectively. When coupled with commercial LiCoO2 cathode, the full cell delivered an outstanding initial charge/discharge capacity of 614/437 mAh g−1 and stability at different current densities. The improved electrochemical performance is mainly attributed to the free space provided by the unique porous hollow structure, which effectively alleviates the volume expansion and facilitates the exposure of more active sites during the lithiation/delithiation process.
Graphical abstract
Porous Fe2O3 nanorod-decorated hollow carbon nanofibers exhibit outstanding rate performance and cycling stability with a high reversible discharge capacity.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The dominated lithium-ion batteries (LIBs) as the advanced energy storage systems for portable electronic devices and electrical vehicles have achieved extraordinary progress in practical applications with considerable capacity performance [1,2,3]. Among the major components of the LIBs, the electrode material is the key to capacity performance. Currently, graphite-based materials are still predominantly used as anode materials for commercial LIBs. However, graphite anode suffers a low theoretical capacity (372 mAh g−1) and thus hardly to meet the explosive demand in growing green energy because of the increasing concerns on environmental protection [4,5,6,7].
Therefore, it is pressing to develop new anode materials with high capacity performance for LIBs, and fortunately, FexOy would be a promising alternative due to their high theoretical capacity with the merits of low cost, non-flammability, and environmental friendliness [8, 9]. In previous studies [10,11,12,13,14], Fe2O3 anode delivered a high specific capacity of 1007 mAh g−1; however, volume expansion happening during the process of charge and discharge causes pulverization and failure of contact with the current collector. In this regard, extensive efforts have been devoted to design varieties of nanostructured composites to alleviate the volume expansion, such as 0–3D nanoparticles, nanowires, nanorods, and nanoboxes [15,16,17,18,19]. The nanomodification of the materials and reduction of grain size are beneficial to improve the specific surface area (SSA), increase the active sites, and shorten the diffusion distance of ionic electrons [20], while the composite of C-based materials can effectively buffer the volume change of the material during cycling [21]. Notably, hollow structures with a high SSA and internal space are more effective in mitigating the volume change effect during lithiation/delithiation than others and consequently boost the electrochemical performance [22,23,24].
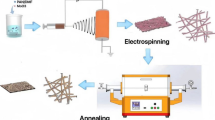
In this study, porous hollow Fe2O3@carbon nanofibers (HNF) are prepared via a facile electrospinning, hydrothermal reaction followed by heat treatment (Scheme 1a). Such a well-designed HNF combines the advantages of vertically aligned hierarchical structure and sizeable SSA. Upon applied as anode for LIBs, the HNF-450 delivers a high initial capacity of 2067 mAh g−1 at 100 mA g−1 and cycling stability with a stabilized capacity of 1398 mAh g−1 after 100 cycles owing to its unique 3D porous hollow nanofiber network for the alleviated volume expansion and improved Li ion storage.

Schematic description for the synthesis process hollow Fe2O3@carbon nanofibers (a); formation mechanism of hollow Fe2O3@carbon nanofibers (b)
2 Experimental
2.1 Material synthesis
Preparation of FeOOH/PAN nanofibers (FeOOH/PAN NF): Typically, 1.4 g polyacrylonitrile (PAN, Mw = 150,000, Sinopharm) was added into 13 mL N, N-Dimethylformamide (DMF, 99.5%, Sigma-Aldrich) and stirred at room temperature for 10 h. After a homogeneous solution was obtained, it was injected into a 20 mL syringe with a needle of 22 G and syringed on an electrospinning device. A high voltage of 19 kV was applied for electrospinning at a flow rate of 0.9 mL/h with a distance of 15 cm from needle to a collector. The PAN film was obtained after electrospinning for 12 h. Then, 36 mmol FeCl3·6H2O (99%, Macklin) was dissolved in 180 mL deionized (DI) water under magnetic stirring at room temperature for 20 min and then transferred into a Teflon-lined autoclave. The PAN film was then added and reacted for 12 h at 100 °C. After the reaction, the obtained products were washed twice with deionized water and dried in a vacuum oven at 60 °C for 12 h to obtain the FeOOH/PAN NF film.
Preparation of hollow Fe2O3@carbon nanofibers (HNF): The HNF was obtained by pyrolysis of the as-prepared FeOOH/PAN NF in two steps. For the first stage, the as-prepared FeOOH/PAN NF was annealed at 600 °C with a ramp of 5 °C min−1 under nitrogen protection for 1 h and then cooled down to room temperature. In the second stage, the above samples were pyrolyzed at 400, 450, and 600 °C with a heating rate of 5 °C min−1 in the air for 1 h, and labeled as HNF-400, HNF-450, and HNF-600, respectively. It should be noted that the two-step calcination is the key to form the HNF. As illustrated in Scheme 1b, the thermal vibration causes the adjacent hydroxyl groups in FeOOH crystals to detach and combine into water diffusing from inside to outside in a dry N2 atmosphere. Meanwhile, Fe3+ and O2− in the crystals migrated and rearranged with more produced defects as a consequence of porous Fe2O3 nanorods formed on the carbon nanofibers (CNFs) derived from PAN nanofibers, and ultimately γ-Fe2O3@CNF was produced. Followed by heat treatment at 450 °C, migration and rearrangement of both Fe3+ and O2− were further driven; more nanorods with intense pores were produced, and γ-Fe2O3 was transformed into α-Fe2O3. Simultaneously, the CNFs were partially decomposed into CO2 and CO gas overflow during the calcination process, producing a hollow structure for the fibers. Eventually, Fe2O3@carbon (HNF) electrode featured with a hollow structure was designed, which would improve the contact with the electrolyte due to its enhanced SSA, and effectively buffer the suffering volume expansion during the charging and discharging process.
2.2 Material characterization
The crystallographic phase of samples was characterized by X-ray diffraction measurements (XRD, Cu Kα, γ = 1.5406 Å, KD2 PHASER, BRUKER AXS) at a scanning rate of 2° min−1 in a 2θ range from 5 to 90°. The SSA and porosity were determined by a Micromeritics TriStar surface area and pore analyzer (BET, BJH, Tristar 3020, USA). The morphology and structure of the HNF were observed by a scanning electron microscope (SEM, Hitachi S4800), a transmission electron microscopy (TEM, JEOL JEM-2100), and high-resolution transmission electron microscopy (HRTEM). X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB 250XI) was used to identify the chemical state and electronic structure of the composites. Energy dispersive spectrometer (EDS, Bruker Quantax 400) was conducted to examine the elemental composition of the as-prepared samples.
2.3 Electrochemical characterizations
The electrochemical properties of the prepared samples were investigated by assembled CR2032 coin cell with a lithium foil as the counter electrode. The working electrode consists of active substance, carbon black, and polyvinylidene fluoride (PVDF, Mw = 800,000, Sinopharm) with a mass ratio of 7:2:1. The mixture was ground and dispersed into proper N-methyl-2-pyrrolidone (NMP, 99%, Sigma-Aldrich) under stirring for 1 h to form a slurry. The slurry was then coated onto a copper foil and dried in a vacuum oven at 60 °C for 8 h to remove the extra solvent. The loading mass of active material was 0.5 mg cm−2 for each electrode. The cells were assembled in a glove box with high purity argon gas (H2O ≤ 0.5 ppm, O2 ≤ 0.5 ppm) with LiPF6 (1 M, KJ Group) in ethylene carbonate (EC)/diethyl carbonate (DEC)/ethyl methyl carbonate (1:1:1 v/v/v) as the electrolyte and Celgard 2400 microporous polyethylene film as the separator. The assembled cells were rested for 12 h before the test. As for the full cell, the prepared HNF-450 served as anode and coupled with a commercial LiCoO2 (Macklin) cathode to fabricated LiCoO2//HNF-450 full cells with the same procedures of the half-cell. In order to reduce the initial irreversible capacity, the anode material was pre-lithiated before assembling the full cell. Electrochemical measurements, including cyclic voltammetry (CV) at a scanning rate of 0.1 mV s−1 in the range from 3.00–0.01 V and electrochemical impedance spectroscopy (EIS) at a frequency range of 0.01 to 100 kHz with an amplification voltage of 10 mV, were performed on an electrochemical working station (CHI660E, Shanghai, China). The galvanostatic charge–discharge (GCD), cycling stability, and rate performance were performed on a battery tester (LAND Battery program, Shenzhen, China) in a voltage range of 0.01–3.00 V.
3 Results and discussion
3.1 Physical properties
XRD was performed to explore the crystallographic structure and phase composition of the composites. A series of peaks located at 11.9°, 16.9°, 26.8°, 34.2°, 35.3° were presented for FeOOH/PAN NF, which were attributed to (1 1 0), (2 0 0), (1 3 0), (4 0 0), and (2 1 1) planes (JCPDS No.75–1594) [25] (Fig. 1a), and no obvious diffraction peaks were detected for PAN as a polymer. Figure 1b shows the XRD pattern of γ-Fe2O3@CNF with diffraction peaks at 30.4°, 35.6°, 57.2°, and 62.9° that can be indexed to (2 2 0), (3 1 1), (5 1 1), and (4 4 0) planes of γ-Fe2O3 (JCPDS No.39–1346), respectively [26]. The characteristic peak for amorphous carbon was not obvious because of the suppression of the strong diffraction peaks of γ-Fe2O3. In Fig. 1c, diffraction peaks at 24.2°, 33.2°, 35.6°, 40.9°, 49.5°, 54.9°, and 62.5° were showed for HNF-400, corresponding to (0 1 2), (1 0 4), (1 1 0), (1 1 3), (0 2 4), (1 1 6), and (2 1 4) planes of α-Fe2O3 (JCPDS card no. 33–0664) [27, 28]. Meanwhile, the diffraction peaks indexed to γ-Fe2O3 (JCPDS card No.39–1346) at 30.3°, 35.6°, 43.3°, 54.9°, and 57.3° appeared, indicating the presence of γ-Fe2O3. With the increasing calcination temperature in the air from 400 to 600 °C, the intensity of the typical diffraction peak at 30.3° of γ-Fe2O3 for HNF-400/450/500/600 was gradually becoming weak and disappeared ultimately, demonstrating the phase transition from γ-Fe2O3 to α-Fe2O3. Besides, the phase transition was further recognized by the color change from brown to red-brown before and after heat treatment for HNF.

XRD patterns of FeOOH/PAN (a), γ-Fe2O3@CNF (b), and varied-phase HNF (c); nitrogen isothermal adsorption/desorption curves and pore size distribution of HNF-400, HNF-450, and HNF-600 (d)
Both pore size distribution and SSA of the HNF-400/450/600 were examined by nitrogen adsorption/desorption isotherm analysis. As shown in Fig. 1d, a typical type-IV isotherm curves at medium relative pressure points out the presence of mesoporous structures were presented for all samples. According to the Brunauter-Emmett-Teller (BET) method, the calculated SSAs of HNF-400, HNF-450, and HNF-600 were 37.7 m2g−1, 46.17 m2g−1, and 20.8 m2g−1, respectively. Mesopores with pore size of 2–3 nm existed for HNF-400/450/600 as displayed in the pore size distribution (inset of Fig. 1d). Such a large SSA of HNF-450 was originated from the hollow structure of the composite while a lower SSA for HNF-400 and HNF-600 was led by the bridging collapse or incomplete hollowing at an overheated or insufficient calcination temperature.
The morphologies of all prepared samples were revealed by SEM. Figure 2a shows SEM images of PAN nanofibers with smooth surfaces and uniform diameter distribution of 300–400 nm. FeOOH nanorods with tens of nanometers in length and nanometers in diameter were radially grown on PAN nanofibers in a high-density as shown in Fig. 2b. To explore the effect of different calcination temperatures and time on the morphology of the prepared samples, SEM of HNF-400, HNF-450, and HNF-600 was conducted. As shown in Fig. 2c and d, HNF-400 displayed a hierarchical structure overall, and Fe2O3 nanorods were vertically anchored on nanofibers. With increasing temperature at 450 °C, Fe3+ and O2− migration and rearrangement were accelerated and more defects were formed, delivering more abundant and intensive pores for HNF-450 (Fig. 2e and f), which was also confirmed by BET results. Then at an extended treatment time of 6 h, HNF-450–6 suffered severe fracture and low porosity and the loaded Fe2O3 nanorods evolved into flakes though a general nanofiber structure was maintained (Fig. 2g and h). At 600 °C, the size of the Fe2O3 nanorods became smaller with cured pores and relatively smooth surface for HNF-600 because of the completed Fe3+ and O2− migration and rearrangement led to a stabilized structure with closed pores (Fig. 2i and j).

SEM images of PAN nanofibers (a); FeOOH/PAN composite nanofibers (b); HNF-400 (c, d); HNF-450 (e, f); HNF-450 (6 h) (g, h); and HNF-600 (i, j)
The elemental composition and valence structure of HNF-450 were characterized by XPS. Figure 3a shows the full XPS spectra of the composite, indicating the presence of Fe, O, and C elements. In the high-resolution spectra of Fe 2p (Fig. 3b), two sharp peaks located at 724.8 eV and 711.5 eV were displayed, referring to Fe 2p1/2 and Fe 2p3/2, respectively [15, 29]. Two broad satellite peaks at the high binding-energy side, which were about 8 eV higher than the characteristic peaks, pointed out there was no Fe2+. The peaks located at 529.5 eV, 530.8 eV, and 532.3 eV were attributed to O2−, C-O, and C = O [29], respectively, while the sharp peak at 529.5 eV belonged to oxygen in Fe2O3 (Fig. 3c). For the C 1 s spectra of HNF-450 (Fig. 3d), three peaks at 284.3 eV, 285.78 eV, and 287.9 eV were ascribed to the C–C graphite-like structures of CNFs, C-O, and C = O [30], respectively.

XPS survey spectrum of HNF-450 (a), high-resolution spectra of Fe 2p (b), O 1 s (c), and C 1 s (d)
The mass ratio of carbon in the composites was determined by TGA and DTA tests. As shown in Fig. 4a, the weight loss of FeOOH/PAN precursor was less than 6.6% before 200 °C due to the evaporation of crystal water. Then, PAN underwent a cyclization process from 200 to 280 °C with a weight loss of 4.1%. Afterwards, the FeOOH was transformed into γ-Fe2O3, and PAN was carbonized with an over 29.4% weight loss when heating to 600 °C. The carbon phase of γ-Fe2O3/CNFs was decomposed by oxidation at around 800 °C [31, 32]. Meanwhile, γ-Fe2O3 was in situ converted to α-Fe2O3 with a mass retention of 43.2% (Fig. 4b). Therefore, the carbon content in HNF-450 was calculated to be 4.8%, which was consistent with the XPS results.

TGA and DTA curves of FeOOH/PAN nanofibers in N2 (a) and γ-Fe2O3@CNFs in air (b)
The TEM was performed to further analyze the hollow interior of the HNF-400, HNF-450, and HNF-600 nanofibers. As shown in Fig. 5a, the porous Fe2O3 nanorods were uniformly distributed on the fiber surface, perpendicular to the fiber axially, with an overall discontinuous hollow shape. As the heat treatment process proceeds, the gas overflow was accelerated and clear through channels with a diameter of about 100 nm appear inside HNF-450 (Fig. 5b). As for HNF-600 (Fig. 5c), its continuous hollow channels were maintained, but the surface-loaded nanorods become clear and smooth. In the HR-TEM, Fe2O3 crystals with clear lattice space of 0.368 nm and 0.252 nm were observed for HNF-450, corresponding to the (012) lattice plane of α-Fe2O3, and the (311) lattice plane of γ-Fe2O3 are shown in Fig. 5d. Meanwhile, elements of Fe, O, and C were detected in the EDS spectrum of HNF-450 (Fig. 5e), reflecting the expected composition of the sample and aligning with the elemental mapping results (Fig. 5f).

TEM images of HNF-400 (a), HNF-450 (b), and HNF-600 (c); HR-TEM images of HNF-450 (d); EDS spectroscopy of HNF-450 (e); and EDS elemental mapping of Fe, O, and C for HNF-450 (f)
3.2 Electrochemical performance
The CV measurements were performed between 0.01 and 3.00 V at a scanning rate of 0.1 mV s−1 to explore the electrochemical reaction process of HNF-400, HNF-450, and HNF-600. In the first cycle, a cathodic peak at 0.67 V related to the reduction reaction from Fe3+ to Fe with the formation of SEI film and Li2O was identified for HNF-450 electrode (Fig. 6b) and expressed as Li2Fe2O3 (cubic) + 4Li+ + 4e− → 2Fe + 3Li2O. The voltage plateau at 0.91 V was attributed to the phase transformation from LixFe2O3 (hexagonal) into Li2Fe2O3 (cubic), which was easier to be detected due to its incomplete phase transition from γ-Fe2O3 to α-Fe2O3 for HNF-400. In the anodic sweeping, two broad peaks at about 1.70 and 1.77 V ascribed to the oxidation reaction from Fe0 to Fe2+, and Fe3+ (2Fe + 3Li2O ↔ Fe2O3 + 6Li+ + 6e−) was displayed [33, 34]. From the third cycle onwards, the cathodic peak gradually shifted from 0.67 to 0.87 V because of the activation of the electrode materials and fine-tuning of the structures, while the irreversible reaction during the lithiation process delivered a weakening of the peak intensity. For the CV curves of HNF-400 (Fig. 6a) and HNF-600 (Fig. 6c), similar profiles as that of HNF-450 were presented, indicating the identical oxidation/reduction processes were experienced.

Cyclic voltammetry curves of HNF-400 (a), HNF-450 (b), and HNF-600 (c); galvanostatic charge–discharge curves of HNF-400 (d), HNF-450 (e), and HNF-600 (f)
The galvanostatic discharge–charge curves at the 1st, 2nd, 3rd, 5th, and 10th cycle over a potential range from 0.01 to 3 V at a current density of 100 mA g−1 for HNF-400, HNF-450, and HNF-600 are shown in Fig. 6d–f. A small amount of lithium was inserted into the Fe2O3 matrix from open-circuit voltage which corresponded to 2Fe + 3Li2O ↔ Fe2O3 + 6Li+ + 6e−. With the proceeded lithiation process, a phase transition from LixFe2O3 to Li2Fe2O3 occurred. Besides, a long voltage plateau at around 0.85 V was distinctly identified in the first discharge process due to the transition of Fe3+ to Fe0 and the evolution of Li2O (Fe2O3 + xLi+ + xe− → LixFe2O3) [19]. The initial capacity of HNF-400, HNF-450, and HNF-600 was identified as 1882, 2067, and 1802 mAh g−1, respectively, which was much higher than that of the theoretical one (1007 mAh g−1). Such a large capacity of HNF was attributed to the large number of spin-polarized electrons stored in the reduced Fe nanoparticles in Li2O during low-voltage discharge process [35].
The cycling performance of HNF-400, HNF-450, and HNF-600 was evaluated at a current density of 100 mA g−1 (Fig. 7a). Benefiting from the hierarchical hollow structure can buffer the volume expansion of Fe2O3 nanoparticles during the charge/discharge cycling; the HNF-450 remained an outstanding reversible capacity of 1398 mAh g−1 even after 100 cycles with the coulombic efficiency of almost 97%. In contrast, only 1236 and 902 mAh g−1 discharge capacities survived for HNF-400 and HNF-600, respectively. Figure 7b shows the rate capability of the HNF-400, HNF-450, and HNF-600. The discharge capacities of HNF-450 were 1758, 1682, 1515, 1293, 987, and 686 mAh g−1 at the current densities of 100, 200, 300, 500, 1000, and 2000 mA g−1, respectively. The discharge capacity recovered to 1748 mAh g−1 when the current density was reversed to 100 mA g−1. In strong contrast, both HNF-400 and HNF-600 generally delivered low discharge capacities of 1291, 1199, 967, 655, 411, and 279 mAh g−1 and 1071, 941, 740, 530, 344, and 231 mAh g−1 with the increasing current densities. Outstanding cycling performance at a high current density of 1.0 A g−1 was also demonstrated for HNF-450 with a reversible capacity of 814 mAh g−1 after 250 cycles (Fig. 7c). In comparison with the previously reported 1D Fe2O3/C composites [36,37,38,39,40,41], the as-prepared HNF-450 electrode exhibited much better rate performance owing to the improved migration efficiency of lithium ions through smooth diffusion channel endowed by its unique hollow nanofiber structure.

Cycling performance of HNF-400/450/600 composites at 100 mA g−1 (a); rate performance of HNF-400/450/600 composites at different current densities (b); cycling performance of HNF-450 at 1 A g−1 (c); Nyquist plots with equivalent circuit of HNF-400/450/600 electrodes (d); Nyquist plots with equivalent circuit after 1 and 100 cycles of HNF-450 (e); and SEM image of HNF-450 after 250 cycles (f)
The charge transfer resistance and ion transfer resistance were characterized by the electrochemical impedance analysis (EIS). As shown in Fig. 7d, the Nyquist plots consisted of a semicircle at the high frequency range and a spike at the low frequency region, representing the charge transfer resistance (Rct) and Li+ diffusion for HNF-400, HNF-450, and HNF-600, respectively. Of them, the HNF-450 electrode reveals a lower Rct of 330 Ω with the smallest diameter of semicircle than that of HNF-400 (457 Ω) and HNF-600 (623 Ω) in the first cycle due to its high SSA, stable SEI film, and engineered structure [19]. Fast kinetic of electrochemical reaction was deployed, and the Rct of HNF-450 dropped to 125 Ω after 100 cycles (Fig. 7e), indicating the gradual activation of the electrode materials and improved kinetics upon cycling. Moreover, the structural stability of HNF-450 electrode was deeply investigated by SEM. As shown in Fig. 7f, the fiber structure with nanorods anchored on nanofibers was well maintained even after 250 cycles for HNF-450, strongly manifesting the excellent structural stability and capability of alleviating volume expansion with cycling.
Overall, the superior electrochemical performance of HNF-450 was highly related with its unique structures and attributed to the following points: (i) the vertical arranged low-dimensional nanorods not only improved ion and electron diffusion kinetics but also facilitated the electrode activation reaction due to its large SSA; (ii) the hollow porous structure could effectively alleviate the volume expansion during charge/discharge process and offer smooth channels for ion and electron transition; (iii) the existed carbon phase significantly enhance the conductivity of the electrode materials; (iv) the annoying agglomeration of metal oxides was significantly improved counting on the cross-linked 3D network for fast ion/electron diffusion.
Furthermore, full cells with HNF-450 anode and LiCoO2 cathode were assembled (Fig. 8a). The negative and positive capacity ratio (N/P) was designed to be 1.05 to verify the application value. Figure 8b illustrates the charge/discharge profiles of LiCoO2//HNF-450 full cell at a current density of 1000 mA g−1 in a voltage range of 0.5–4.3 V. An initial charge/discharge capacity of 614/437 mAh g−1 was delivered, indicating the importance of the pre-lithiation process in reducing initial irreversible capacity. In the following 2nd, 3rd, 5th, and 10th cycles, the identified reversible capacities were 366, 315, 297, and 235 mAh g−1, respectively. In Fig. 8c, cycling performances showed that a reversible capacity of 106 mAh g−1 remained even after 600 cycles at 1000 mA g−1 with an almost 100% coulombic efficiency, demonstrating the excellent cycling stability of the LiCoO2//HNF-450 full cell. For a real application demonstration, a yellow LED bulb (2.0–2.2 V, 20 mA) was successfully lighted. Figure 8d shows the rate performance of LiCoO2//HNF-450 full cell; the capacities were 300, 173, 124, 92, 69, and 57 mAh g−1 at current densities of 500, 1000, 1500, 2000, 2500, and 3000 mA g−1, respectively. The discharge capacity recovered to 248 mAh g−1 when the current density was reversed to 500 mA g−1. Such an outstanding electrochemical performance of LiCoO2//HNF-450 full cell strongly demonstrated its promising application in practice.

The mechanism illustration (a), galvanostatic charge–discharge curves (b), cycling performance (c), and rate performance (d) of LiCoO2//HNF-450 full cells
4 Conclusions
A unique structure of hollow Fe2O3@C nanofibers decorated by porous nanorods was prepared via hydrothermal synthesis followed with a two-step calcination. Upon examined as anode materials for LIBs, the highest reversible capacity of 1398 mAh g−1 after 100 cycles at a current density of 100 mA g−1, as well as superior cycling stability and rate performance, was delivered for HNF-450. Such an excellent lithium storage performance can be attributed to the unique porous hollow structure consisting of vertically grown porous and agglomeration-free Fe2O3 nanorods, which would greatly shorten the diffusion distance of lithium ions and promote the contact with the electrolyte. Additionally, the hollow structure effectively buffered the volume change effect during repeated lithiation/delithiation while the amorphous carbon effectively reduced the resistance of the electrode. The excellent capacitance performance of HNF-450 was also demonstrated in the assembled full cells coupled with a LiCoO2 cathode. The proposed strategy can also be applied to design novel nanostructured TMO electrode materials and consequently boosted the electrochemical energy storage and conversion efficiency.
References
Zhu XZ, Xu J, Luo YP, Fu QF, Liang GS, Luo LJ, Chen YJ, Lin CF, Zhao XS (2019) MoNb12O33 as a new anode material for high-capacity, safe, rapid and durable Li+ storage: structural characteristics, electrochemical properties and working mechanisms. J Mater Chem A 7:6522–6532
Zhu XZ, Cao HJ, Li RJ, Fu QF, Liang GS, Chen YJ, Luo LJ, Lin CF, Zhao XS (2019) Zinc niobate materials: crystal structures, energy-storage capabilities and working mechanisms. J Mater Chem A 7:25537–25547
Hou CX, Wang B, Murugadoss V, Vupputuri S, Chao YF, Guo ZH, Wang CY, Du W (2020) Recent advances in Co3O4 as anode materials for high-performance lithium-ion batteries. Eng Sci 11:19–30
Hu MX, Lv Q, Lv RT (2019) Controllable synthesis of nitrogen-doped graphene oxide by tablet-sintering for efficient lithium/sodium-ion storage. ES Energy Environ 3:45–54
Fu YX, Pei XYN, Dai Y, Mo DC, Lyu S (2019) Three-dimensional graphene-like carbon prepared from CO as anode material for high-performance lithium-ion batteries. ES Energy Environ 4:66–73
Zhao XC, Yang P, Yang LJ, Cheng Y, Chen HY, Liu H, Wang G, Murugadoss V, Angaiah S, Guo ZH (2018) Enhanced electrochemical performance of Cu2+ doped TiO2 nanoparticles for lithium-ion battery. ES Mater Manuf 1:67–71
Xie J, Lu YC (2020) A retrospective on lithium-ion batteries. Nat Commun 11:2499
Yang Y, Yuan W, Zhang XQ, Wang C, Yuan YH, Huang Y, Ye YT, Qiu ZQ, Tang Y (2020) A review on FexOy-based materials for advanced lithium-ion batteries. Renew Sust Energ Rev 127:109884
Zhang L, Wu HB, Lou XWD (2014) Iron-oxide-based advanced anode materials for lithium-ion batteries. Adv Energy Mater 4:1300958
Yu SH, Ng VMH, Wang FJ, Xiao ZH, Li CY, Kong LB, Que WX, Zhou K (2018) Synthesis and application of iron-based nanomaterials as anodes of lithium-ion batteries and supercapacitors. J Mater Chem A 6:9332–9367
Keppeler M, Shen N, Nageswaran S, Srinivasan M (2016) Synthesis of α-Fe2O3/carbon nanocomposites as high capacity electrodes for next generation lithium ion batteries: a review. J Mater Chem A 4:18223–18239
Guo WX, Sun WW, Lv LP, Kong SF, Wang Y (2017) Microwave-assisted morphology evolution of Fe-based metal-organic frameworks and their derived Fe2O3 nanostructures for Li-ion storage. ACS Nano 11:4198–4205
Huang W, Zheng XW, Shangguan HH, Xiao XX, Tang J, Sun HY, Molhave K, Ci LJ, Si PC, Zhang JD (2020) Microwave assisted crystalline and morphology evolution of flower-like Fe2O3@ iron doped K-birnessite composite and its application for lithium ion storage. Appl Surf Sci 525:146513
Narsimulu D, Nagaraju G, Sekhar SC, Ramulu B, Hussain SK, Yu JS (2020) Unveiling multi-channelled 3D porous iron oxide nanostructures with exalted capacity towards high-performance Li-ion battery applications. J Alloy Compd 846:156385
Liu L, Yang XF, Lv CX, Zhu AM, Zhu XY, Guo SJ, Chen CM, Yang DJ (2016) Seaweed-derived route to Fe2O3 hollow nanoparticles/N-doped graphene aerogels with high lithium ion storage performance. ACS Appl Mater Interfaces 8:7047–7053
Wang D, Dong H, Zhang H, Zhang Y, Xu YL, Zhao CJ, Sun YN, Zhou N (2016) Enabling a high performance of mesoporous alpha-Fe2O3 anodes by building a conformal coating of cyclized-PAN network. ACS Appl Mater Interfaces 8:19524–19532
Sun MF, Chu XF, Wang ZK, Yang HX, Yang Z, Ma JJ, Zhou B, Yang TY, Chen LZ (2021) Rosa roxburghii-like hierarchical hollow sandwich-structure C@Fe2O3@C microspheres as second nanomaterialsfor superior lithium storage. J Alloy Compd 855:157518
Park Y, Oh M, Park JS, Baek SH, Kim M, Kim S, Kim JH (2015) Electrochemically deposited Fe2O3 nanorods on carbon nanofibers for free-standing anodes of lithium-ion batteries. Carbon 94:9–17
Zheng MB, Tang H, Li LL, Hu Q, Zhang L, Xue HG, Pang H (2018) Hierarchically nanostructured transition metal oxides for lithium-ion batteries. Adv Sci 5:1700592
Xiao LD, Qi HJ, Qu KQ, Shi C, Cheng Y, Sun Z, Yuan BN, Huang ZH, Pan D, Guo ZH (2021) Layer-by-layer assembled free-standing and flexible nanocellulose/porous Co3O4 polyhedron hybrid film as supercapacitor electrodes. Adv Compos Hybrid Mater 4:306–316
Tian YR, Yang X, Nautiyal A, Zheng YY, Guo QP, Luo JJ, Zhang XY (2019) One-step microwave synthesis of MoS2/MoO3@graphite nanocomposite as an excellent electrode material for supercapacitors. Adv Compos Hybrid Mater 2:151–161
Hafez AM, Sheng JZ, Cao DX, Chen Y, Zhu HL (2019) Flexible lithium metal anode featuring ultrahigh current density stability with uniform deposition and dissolution. ES Energy Environ 5:85–93
Idrees M, Liu LQ, Batool S, Luo HB, Liang J, Xu BB, Wang S, Kong J (2019) Cobalt-doping enhancing electrochemical performance of silicon/carbon nanocomposite as highly efficient anode materials in lithium-ion batteries. Eng Sci 6:64–76
Qiu L, Guo P, Zou HY, Feng YH, Zhang XX, Pervaiz S, Wen D (2018) Extremely low thermal conductivity of graphene nanoplatelets using nanoparticle decoration. ES Energy Environ 2:66–72
Qi H, Cao LY, Li JY, Huang JF, Xu ZW, Cheng YY, Kong XG, Yanagisawa K (2016) High pseudocapacitance in FeOOH/rGO composites with superior performance for high rate anode in Li-ion battery. ACS Appl Mater Interfaces 8:35253–35263
Wu YZ, Zhu PB, Reddy MV, Chowdari BVR, Ramakrishna S (2014) Maghemite nanoparticles on electrospun CNFs template as prospective lithium-ion battery anode. ACS Appl Mater Interfaces 6:1951–1958
Liu M, Liu YM, Li YP, Wang K, Guo YY, Li YX, Zhao LZ (2019) Biomimetic straw-like bundle cobalt-doped Fe2O3 electrodes towards superior lithium-ion storage. Chem Eur J 25:3343–3351
Wang Z, Luan D, Madhavi S, Hu Y, Lou XW (2012) Assembling carbon-coated α-Fe2O3 hollow nanohorns on the CNT backbone for superior lithium storage capability. Energy Environ Sci 5:5252–5256
Huang P, Tao W, Wu HX, Li XG, Yin T, Zhang Q, Qi W, Gao G, Cui DX (2018) N-doped coaxial CNTs@α-Fe2O3@C nanofibers as anode material for high performance lithium ion battery. J Energy Chem 27:1453–1460
Li RR, Ke HZ, Shi C, Long ZW, Dai ZX, Qiao H, Wang KL (2021) Mesoporous RGO/NiCo2O4@carbon composite nanofibers derived from metal-organic framework compounds for lithium storage. Chem Eng J 415:128874
Hou CAX, Yang WY, Xie XB, Sun XQ, Wang J, Naik N, Pan D, Mai XM, Guo ZH, Dang F, Du W (2021) Agaric-like anodes of porous carbon decorated with MoO2 nanoparticles for stable ultralong cycling lifespan and high-rate lithium/sodium storage. J Colloid Interface Sci 596:396–407
Zhang X, Ziemer KS, Weeks BL (2019) Combustion synthesis of N-doped three-dimensional graphene networks using graphene oxide–nitrocellulose composites. Adv Compos Hybrid Mater 2:492–500
Xu YH, Jian GQ, Liu YH, Zhu YJ, Zachariah MR, Wang CS (2014) Superior electrochemical performance and structure evolution of mesoporous Fe2O3 anodes for lithium-ion batteries. Nano Energy 3:26–35
Zheng ZM, Li P, Huang JS, Liu HD, Zao Y, Hu ZL, Zhang L, Chen HX, Wang MS, Peng DL, Zhang Q (2020) High performance columnar-like Fe2O3@carbon composite anode via yolk@shell structural design. J Energy Chem 41:126–134
Li Q, Li HS, Xia QT, Hu ZQ, Zhu Y, Yan SS, Ge C, Zhang QH, Wang XX, Shang XT, Fan ST, Long YZ, Gu L, Miao GX, Yu GH, Moodera JS (2020) Extra storage capacity in transition metal oxide lithium-ion batteries revealed by in situ magnetometry. Nat Mater 20:76–83
Xu XQ, Wan YX, Liu JW, Chen YJ, Li LL, Wang XL, Xue G, Zhou DS (2017) Encapsulating iron oxide@carbon in carbon nanofibers as stable electric conductive network for lithium-ion batteries. Electrochim Acta 246:766–775
Zhu JD, Lu Y, Chen C, Ge YQ, Jasper S, Leary J, Li DW, Jiang MJ, Zhang XW (2016) Porous one-dimensional carbon/iron oxide composite for rechargeable lithium-ion batteries with high and stable capacity. J Alloy Compd 672:79–85
Guo JD, Yang Y, Yu WS, Dong XT, Wang JX, Liu GX, Wang TT (2016) Synthesis of α-Fe2O3, Fe3O4 and Fe2N magnetic hollow nanofibers as anode materials for Li-ion batteries. RSC Adv 6:111447–111456
Wu CY, Li XP, Li WS, Li B, Wang YQ, Wang YT, Xu MQ, Xing LD (2014) Fe2O3 nanorods/carbon nanofibers composite: preparation and performance as anode of high rate lithium ion battery. J Power Sources 251:85–91
Chaudhari S, Srinivasan M (2012) 1D hollow α-Fe2O3 electrospun nanofibers as high performance anode material for lithium ion batteries. J Mater Chem 22:23049–23056
Zhang B, Xu ZL, Kim JK (2014) Insitu grown graphitic carbon/Fe2O3/carbon nanofiber composites for high performance freestanding anodes in Li-ion batteries. RSC Adv 4:12298–12301
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was financially supported by the Natural Science Foundation of Jiangsu Province (BK20201343), China Postdoctoral Science Foundation (2018T110442 and 2017M610296), National Natural Science Foundation of China (21201083), and the Undergraduate Innovation Training Program of Jiangsu Province of China Grant (202110295003Z).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Long, Z., Yuan, L., Shi, C. et al. Porous Fe2O3 nanorod-decorated hollow carbon nanofibers for high-rate lithium storage. Adv Compos Hybrid Mater 5, 370–382 (2022). https://doi.org/10.1007/s42114-021-00397-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42114-021-00397-9