
Abstract
Background
The neuropeptide oxytocin (OT) is crucial in several conditions, such as lactation, parturition, mother-infant interaction, and psychosocial function. Moreover, OT may be involved in the regulation of eating behaviors.
Methods
This review briefly summarizes data concerning the role of OT in eating behaviors. Appropriate keywords and medical subject headings were identified and searched for in PubMed/MEDLINE. References of original articles and reviews were screened, examined, and selected.
Results
Hypothalamic OT-secreting neurons project to different cerebral areas controlling eating behaviors, such as the amygdala, area postrema, nucleus of the solitary tract, and dorsal motor nucleus of the vagus nerve. Intracerebral/ventricular OT administration decreases food intake and body weight in wild and genetically obese rats. OT may alter food intake and the quality of meals, especially carbohydrates and sweets, in humans.
Discussion
OT may play a role in the pathophysiology of eating disorders with potential therapeutic perspectives. In obese patients and those with certain eating disorders, such as bulimia nervosa or binge/compulsive eating, OT may reduce appetite and caloric consumption. Conversely, OT administered to patients with anorexia nervosa may paradoxically stimulate appetite, possibly by lowering anxiety which usually complicates the management of these patients. Nevertheless, OT administration (e.g., intranasal route) is not always associated with clinical benefit, probably because intranasally administered OT fails to achieve therapeutic intracerebral levels of the hormone.
Conclusion
OT administration could play a therapeutic role in managing eating disorders and disordered eating. However, specific studies are needed to clarify this issue with regard to dose-finding and route and administration time.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Hypothalamic nuclei play a role in eating behaviors, stimulating or inhibiting food intake. Appetite is triggered by nuclei located in the lateral hypothalamus, also known as the “hunger center,” while it is suppressed by neurons located in the ventromedial nucleus of the hypothalamus (VMH) (“satiety center”) [1,2,3,4,5].
Oxytocin (OT), a neurohypophyseal hormone, is synthesized by hypothalamic magnocellular neurosecretory neurons (MCN) of the supraoptic (SON) and paraventricular nuclei neurons (PVN) [6, 7]. It is pivotal in inducing parturition and lactation and in regulating social behaviors. Also, OT is synthesized in other areas of the forebrain, such as the amygdala, hippocampus, septum, striatum, and bed nucleus of the terminal stria and is also detectable in the cerebrospinal fluid [6, 7].
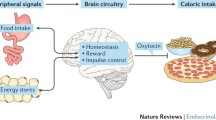
The role of OT in regulating eating behaviors has yet to be understood entirely. In this review, we have summarized the neuroanatomical and biochemical OT-ergic pathways synapsing on brain areas involved in regulating eating behaviors in animal models and humans (Fig. 1). PubMed/MEDLINE was searched for references of original articles and reviews. Appropriate keywords and medical subject headings terms were identified and included the following: “oxytocin,” “eating disorders,” “eating behaviors,” “hypothalamus,” “hypothalamic ventromedial nucleus,” “amygdale,” “area postrema,” “nucleus of the solitary tract,” and “dorsal motor nucleus of the vagus nerve.” References were screened according to a hierarchical strategy by title and abstract, and full text. Original papers and reviews were screened, selected when appropriate, and discussed in detail.

Simplified depiction of the relationship between OT-ergic hypothalamic PVN and SON neurons and brain sites involved in regulating eating behaviors (blue arrows). OT-ergic projections from PVN synapse within the amygdala (AMY), area postrema (AP), nucleus of the solitary tract (NTS), and dorsal motor nucleus of vagus nerve (DMV) playing a role in anorexigenic response, taste aversion, gastric emptying (red arrows)
Overview of physiological aspects of oxytocin
The stimulation of the cervix uteri and nipples stimulates the synthesis of a precursor protein encoded by the OT gene to synthesize OT and its carrier protein neurophysin 1 [8, 9]. These peripheral signals originating from cervix uteri and nipple receptors are transmitted to the SON and PVN OT-ergic neurons via the spinal cord, the dorsal lateral fasciculus, the medial forebrain bundle, and the mammillary peduncle [10,11,12]. The glial cells, mainly astrocytes, stimulate the morphological plasticity of the OT-ergic neurons during lactation and parturition to facilitate the synthesis and transport of OT [10, 13].
The neurokinin 3 receptor is associated with the nuclear chaperone protein importin β1, which induces the internalization of the OT precursor in the Golgi complex and rough endoplasmic reticulum [10, 11].
Once synthesis has occurred, OT and its carrier protein neurophysin 1 are conveyed to the neurohypophysis where they are stored in secretory vesicles and released into the systemic circulation [10, 13, 14].
The OT receptor (OTR) is a Gq/11 protein-linked receptor, and OT-binding sites have been localized in the brain. Efferent pathways arising from the hypothalamic OT-ergic neurons project to brain areas containing a high concentration of OTR, including the olfactory bulb, bed nucleus of the stria terminalis, nucleus accumbens septi, hypothalamic suprachiasmatic, arcuate and VM nuclei, amygdala, hippocampus, septum, the nucleus of the solitary tract (NTS) of the oblongata medulla, cingulate cortex, and spinal cord [15]. OTR is also expressed in peripheral tissue such as the kidney, heart, thymus, pancreas, and adipose tissue [16].
Neuroanatomical organization of regions regulating food intake, appetite, satiety, and reward
Hypothalamic OT-ergic neurons display widespread projections throughout the brain, mainly to the amygdala, contributing to the satiety process by causing the sensation of fullness [17]. Other brain areas typically regulating eating behaviors receive afferent projections from hypothalamic OT-ergic neurons, including the area postrema, the NTS, and the dorsal motor nucleus of the vagus nerve [18, 19].
The ventromedial nucleus of the hypothalamus
The VMH, the Cajal nucleus, is a pear-shaped structure in the hypothalamic tuberal area. The neurons can synthesize OTR and express a high density of OTR on their surfaces [20, 21]. Food intake is inhibited, weight gain restricted, and energy expenditure augmented after the leptin-induced activation of steroidogenic factor 1 positive neurons in the VMH [22, 23].
Food intake, especially fasting-absorbed carbohydrates, stimulates OT release from OT-secreting neurons [24]. Moreover, several hormones may stimulate OT release after the ingestion of a meal, including leptin, cholecystokinin, and gastrointestinal incretins. The same is observed after noradrenergic stimulation of OT-secreting neurons by vagal afferences from the nucleolus of the solitary tract (NTS) [25].
It has been hypothesized that OT may be involved in regulating food intake and energy expenditure directly or by potentiating central and peripheral anorexigenic stimuli [26].
Although the VMH has a high density of OTR, it contains a few OT-ergic projections, suggesting that it could be a local target of OT [21]. To support this hypothesis, OT antagonism or silencing of the OT-induced signaling pathway in the VMH predisposes to a much-extended food intake in terms of energy intake, delayed satiation, and intake of more carbohydrates while reducing energy expenditure [27,28,29].
Extrahypothalamic structures involved in the regulation of eating behaviors
The amygdala
Innate appetite and food aversion are modulated by specific brain structures, mainly in the limbic system. The basolateral and central nuclei of the amygdala regulate the appetite in terms of the amount of ingested food and innate aversions and control qualitative predisposition toward specific food due to acquired experience [17]. In rats, the apomorphine administration before or 30 min after the ingestion of saccharine negatively affected further saccharine intake after vomiting. This response indicates that the unpleasant gastric effect of apomorphine significantly contributed to taste aversion, ultimately affecting specific food intake such as rapidly absorbed carbohydrates [30].
The amygdala determines satiation by oropharyngeal and gastric afferents; bilateral amygdala lesions lead to overeating. The suppression of food intake is mediated by cholinergic stimulation of the amygdala. Conversely, adrenergic stimuli enhance appetite and food intake in starving but not satiated animals. Therefore, the amygdala exerts two different influences on eating behaviors. First, the amygdala plays a facilitating effect in the maintenance of consuming activity induced by NA-ergic activation. Subsequently, the amygdala plays an inhibitory role leading to satiety and food intake cessation. This subsequent behavior is due to cholinergic activation, which stops NA-ergic ones [30].
OTRs are expressed on the membrane of the amygdala’s basolateral and central neurons. OT-ergic projections from the PVN nuclei to the amygdala have also been described. Experiencing gastrointestinal toxicity concomitantly to food intake is accompanied by OT release in humans and animals. Therefore, it is believed that OT interaction with cholinergic and adrenergic circuits within the amygdala may have a role in regulating eating behaviors in terms of food intake, satiety, and taste aversion or predilection [31,32,33]. Indeed, OT administration in the basolateral amygdala effectively suppresses the consumption of palatable saccharin solutions in rats. A moderate restriction of food intake was observed after the administration of OT and was attenuated by pretreatment with an OTR antagonist (L-368,899) [34]. In experimental conditions, assessing the role of OT in mediating the acquisition and retrieval of conditioned taste aversion in mice that underwent lithium-induced acute gastric toxicity, OT was found to contribute to causing taste aversion significantly. At the same time, OT antagonism partially alleviated it but did not wholly retrieve taste aversion [35].
The area postrema
Lesions of the medullary circumventricular organ in the area postrema (AP) reduce food intake and induce weight loss [36, 37]. As a chemo-sensitive organ, AP modulates the conditioned avoidance response (CAR) to toxins, such as carbonate lithium. The acquisition of CAR and conditioned palatability to oral sucrose was assessed in rats with lesions of the AP. The abolishment of OT-ergic inputs from the hypothalamic PVN induced restricted ingestion and increased aversive responses to intraoral infusion of sucrose following an intraperitoneal injection of carbonate lithium [38].
OT was found to increase intragastric pressure by vagal efferences after its administration in the fourth ventricle, specifically acting at the level of AP and NTS [39]. The peptide hormone amylin, or islet amyloid peptide, is co-secreted with insulin from the pancreatic β-cells and promotes satiety by decelerating gastric emptying. The precise mechanism by which amylin reduces food intake is mediated by the activation of NA-ergic neurons within the AP [40]. Similar effects are also induced by calcitonin, a potent amylin agonist structurally similar to amylin, as belonging to the calcitonin-like gene peptide superfamily. AP lesions affecting OT-ergic release abolish anorexic effects induced by the peripheral administration of amylin and calcitonin [41]. This could be an additive mechanism by which OT may reduce food intake and prompt satiety.
The nucleus of the solitary tract and the dorsal motor nucleus of the vagus nerve
The NTS of the dorsal medulla oblongata plays an essential role in regulating cardiovascular functions, affects the activity of hypothalamic SON and PVN neurons [42, 43], and reduces food intake and body weight [44]. Signals from the gastrointestinal tract are conveyed to the brain by vagal afferents synapsing within the medullary dorsal vagal complex, including the NTS and the dorsal motor nucleus of the vagus nerve (DMV) [45, 46]. Moreover, descending pathways from the hypothalamic SON and PVN to the NTS and DMV are involved in the beginning and termination of food intake [47].
An experimental injury of the median-caudal region of the NTS induced hypophagia with consequent body weight loss [48]. During the first 6 days following the electrolytic lesion of the NTS, rats reduced their food intake by around 80% compared to the sham controls. From the 7th day, food intake slightly recovered, but the appetite remained significantly reduced compared to baseline [37].
OT may regulate appetite, food intake, and weight gain by acting at the NTS site. An experiment in rats suggested that the administration of OT in the NTS decreased caloric intake by reducing food motivation and seeking [49]. In rats, acute intraventricular administration of OT (5 μg) 30 min before a meal consumption induced a dose-response reduction in food intake up to 72% (3rd ventricle) and 60% (4th ventricle). Chronic exposure to OT prevented excessive weight gain after exposing the rats to overfeeding with a high-fat diet, with OT-treated animals maintaining a higher leptin sensibility than vehicle controls [50].
Glucagon-like peptide 1 (GLP-1) inhibits food intake by acting on neurons within the NTS and abolishes food reward behaviors and motivation to food intake. Microinjections of native GLP-1 or the GLP-1 analog exendin-4 into the NTS suppressed food reward behaviors, thus reducing appetite and food consumption and, lastly, leading to weight loss. These effects are related to the food reward-suppressing role of GLP-1 agonists operating within the NTS [51].
Adrenalectomy reduces food intake significantly, but this response is reversed by OTR antagonists and by activating satiety-related responses in the NTS. It has been reported that OT-ergic projections from the PVN to NTS are highly upregulated after bilateral adrenalectomy, thus positively affecting satiety and consequently reducing meal size in primary adrenal insufficiency [52]. Glucocorticoid replacement therapy prompts the opposite effect.
Evidence shows that OT directly injected in the dorsal vagal complex (DVC) stimulates gastric secretion via the vagal pathway [53]. OT levels in the DVC were significantly increased in response to food intake, and OTR signaling within DVC neurons plays a counter regulator of gastrointestinal activity by stimulating satiation signals to reduce food intake [54].
Oxytocin and eating behaviors: what do we know?
OT may have a role in controlling emotion and cognition [55] and regulating food intake [56]. In normal-weight and obese animals, OT administered centrally reduces food intake and facilitates weight loss [57]. These responses were also observed when OT was injected peripherally [58]. Moreover, a pretreatment with both central and peripheral (fully permeable to the blood-brain barrier) administration of OTR antagonists reduced the attenuation of food intake after OT administration [57, 58].
Rats with lesions of the PVN, the leading site of OT secretion, exhibited more food intake and weight gain than controls [59]. In this model, the peripheral and central administration of OT reduced, in a dose-dependent manner, food intake and increased the time intercurrent of the consumption of two consecutive meals [60]. The results suggest that OT could be involved in the induction and prolongation of satiety.
Patients with neuropsychiatric diseases often exhibit eating disorders, with hyperphagia and increased meal size usually the leading determinant of weight gain. The therapeutic potential of OT was analyzed in this cluster of patients, hypothesizing that OT could have also played a role in the regulation of psychosocial functions coupled with eating behaviors. A pivotal study conducted in 16 patients with an established diagnosis of schizophrenia on a stable antipsychotic treatment with overweight or obesity (BMI > 27 kg/m2) showed that the intranasal administration of OT (24 IU) compared to placebo a few minutes before the meal consumption did not affect satiety, meal size, post-meal serum glucose, and insulin levels [61].
In a randomized clinical trial, administering OT twice daily for 3 months compared to placebo improved social behavior and reduced appetite in children (3–11 years) with Prader-Willy syndrome [62].
The encephalic functional magnetic resonance imaging revealed that the intranasal administration of OT in obese men attenuated the ventral tegmental area firing to food motivation regions such as the insula, oral somatosensory cortex, and amygdala in response to high-calorie visual food images. The results suggested that OT may exert an anorexigenic effect by dampening eating cravings activated by reward anticipation in patients with obesity [28].
In women with stress-induced eating disorders, the overall exposure to serum cortisol is usually higher than normal. This mechanism may contribute to increase the appetite and positively affect food intake. The intranasal administration of a 24 IU shot compared to the placebo (saline solution) was found to reduce the intake of sweet and fatty snakes by 15 min after administering the neurohypophyseal hormone. Interestingly, the salivary cortisol levels (assessed to test the level of stress) throughout the observation remained unchanged up to 75 min after the administration of OT [63]. The findings were consistent with the fact that OT affected eating behavior independently of the background stress level by acting with a direct mechanism.
To better investigate the efficacy, safety, and mechanisms via which OT is involved in reducing appetite, caloric intake, and body weight and affecting energy expenditure, body composition, glucose and lipid metabolism, and brain activation and control of behaviors and impulses in response to food images, an 8-week randomized, double-blind, placebo-controlled trial has been designed and is currently ongoing. The study will clarify several exciting issues about OT as a pharmacologic treatment of obesity [64]. Moreover, dysfunction in the OT-ergic mechanisms has also been reported in patients with anorexia nervosa, with specific patterns that include lower circulating levels of OT at fasting and after stimulation, lower nocturnal levels of OT, and higher peripheral OT concentration after meal ingestion [65]. Derangements of OT homeostasis in AN are close to the opposite of those observed in other eating disorders characterized by weight excess or propension to gain weight and are reversible after rehabilitation programs and weight gain in AN. Although no specific trials have been carried out, OT administration may have a particular and disease-related role in improving food intake in AN. The mechanisms potentially explaining this sui generis and paradoxical effect could be attributable to the contribution of OT administration in reducing eating-related attention and concerns, attenuating cognitive rigidity, improving emotional expression, and weakening the attitude of avoiding social situations or contexts emotionally provoking stimuli, lastly improving social behavior [66, 67].
The observation of a dimorphic action of OT in these two kinds of eating disorders suggests that OT regulates the brain circuits subserving eating behaviors. Therefore, the physiological involvement of OT in eating disorders can support its beneficial therapeutic effect in clinical practice since the administration of OT by intranasal route may bypass the blood-brain barrier [68] and reach the amygdala and brainstem structures involved in the control of eating behaviors such as the AP, NTS, and DMV.
However, the current level of evidence does not suggest a possible positive effect of intranasal OT treatment in eating disorders. This could be attributable to the fact that cerebral exposure to OT, after its intranasal administration, may not be sufficient to elicit desirable effects and probably higher doses, alternative routes, and timing of administration should be considered [69].
Hypothalamic injury has a wide range of etiology, including brain surgery, encephalic trauma, tumors, chemotherapy and radiation, vascular diseases (aneurysms), cerebral infections, and inflammatory and infiltrative diseases. Depending on the sites, a hypothalamic injury may hypothetically result in different clinical consequences [70]. A lesion in the middle hypothalamic region produces direct damage to some specific centers, such as the arcuate nucleus, which is responsible for the tonic release of dopamine, suppressing the prolactin secretion from lactotrophic cells in the pituitary and phasic release of the growth hormone releasing hormone, resulting in a loss of somatotropic cells pulses of growth hormone. Moreover, the dorsomedial and ventromedial nuclei are also located in the middle region of the hypothalamus and are directly involved in controlling behaviors and gastrointestinal motility (the former) and food intake (the latter). This region may be affected in some endocrine diseases, especially pituitary macro/giant adenomas with considerable suprasellar extension or in the case of primitive hypothalamic disorders (such as craniopharyngioma or infiltrative diseases), and the consequent hypothalamic damage usually results in a progressive deterioration of food intake control, aggressive behaviors, and typically mild or moderate hyperprolactinemia. Injuries in the anterior region of the hypothalamus may harm both the supraoptic and paraventricular nuclei, thus also affecting the OT synthesis [71]. The disturbance may also be accompanied by partial or extensive anterior and posterior pituitary failure, resulting in a unimodal or multimodal hormonal deficiency. Patients with craniopharyngioma, one of the most common causes of hypothalamic damage, usually exhibit lower circulating levels of OT at baseline and after stimulation [72]. Several studies have been published seeking to determine whether OT deficiency was associated with changes in social cognition [73] and eating behaviors in craniopharyngioma survivors. Anecdotal cases suggested that the intranasal administration of OT improved emotional tasks and social behaviors in young survivors of craniopharyngioma with low (case report) [74] and detectable basal levels of OT (case series) [75]. In one cross-sectional case-control study in 34 patients with craniopharyngioma and 73 controls, adverse eating behaviors and eating disorders were more frequently observed among patients with extensive (anterior and posterior) hypothalamic injury than in those with less extensive damage, and controls. Among individuals with adverse eating behaviors, lower postprandial levels of OT compared to control were also found [76], as observed in patients with obesity [71]. The intranasal administration of OT in combination with the opiate antagonist naltrexone (10 weeks of OT alone + 38 weeks of OT and naltrexone) was significantly effective in reducing the appetite, caloric intake, and hyperphagia in a 13-year-old boy with confirmed hypothalamic obesity and hyperphagia post-resection of craniopharyngioma [77]. These positive results could be attributable to the numerous metabolic effects of the neurohormone, including direct reduction of food intake by decreasing appetite in homeostatic and reward-driven conditions (hence, properly insisting on hypothalamic regions involved in the regulation of hunger and satiation), enhancement of lipolysis and energy expenditure, positive affection of body composition due to improvement of peripheral insulin sensitivity, ultimately favoring lean over fat mass building [78]. Nevertheless, a recently published randomized, double-blind, placebo-controlled, crossover pilot study (13 patients randomized; 10 concluded) did not find any relevant changes in body weight between the OT arm 16–24 IU at the three main mealtimes and placebo after 8 weeks of treatment [79]. This finding lays the basis for better-designed multicentric trials to assess the role of OT treatment (alone or in combination) in patients with hypothalamic injuries/dysfunction.
Discussion and conclusion
One piece of evidence suggests that OT and OTR may regulate eating behaviors and food intake. In neuropsychiatric disorders manifesting with altered eating behaviors, such as anorexia nervosa, OT and OTR agonists could potentially have pharmacological use [80]. In addition, OTR gene polymorphisms may also be involved in the pathogenesis of such disorders [81].
OT administration reduces food intake in patients with bulimia nervosa, thus playing a possible protective effect by limiting food overconsumption, weight gain, and purging behaviors [82].
When OT is administered before meal consumption in healthy individuals, the caloric intake remains unchanged, even if the predilection toward carbohydrates and sweets could be reduced. Conversely, overweight and obese individuals exhibit different responses to OT administration before meals, including caloric restriction, less preference for fatty snake consumption, and unaltered propension toward carbohydrates [83].
Although OT may be enumerated as another therapeutic tool to manage weight gain or induce weight loss [84] and despite anecdotal evidence suggesting that OT administration may improve social behaviors, emotional tasks, and eating behaviors, trials are needed for deeper insight into the therapeutic role of OT in patients with hypothalamic injury, such as craniopharyngioma.
Special studies are surely necessary to verify more precisely the therapeutic role of OT in certain disorders characterized by overeating, eating disorders, and disordered eating as well.
Change history
18 March 2024
A Correction to this paper has been published: https://doi.org/10.1007/s42000-024-00540-3
Abbreviations
- AP:
-
Area postrema
- DMV:
-
Dorsal motor nucleus of the vagus nerve
- DVC:
-
Dorsal vagal complex
- GLP-1:
-
Glucagon-like peptide 1
- MCN:
-
Magnocellular neurosecretory neurons
- NA:
-
Noradrenaline
- NTS:
-
Nucleus of the solitary tract
- OT:
-
Oxytocin
- OTR:
-
Oxytocin receptor
- PVN:
-
Paraventricular nuclei neurons
- SON:
-
Supraoptic
- VMH:
-
Ventromedial nucleus of the hypothalamus
References
Berthond HR, Munzberg H (2011) The lateral hypothalamus as integrator of metabolic and environmental needs: from electrical self-stimulation to opto-gemetics. Physiol Behav 104(1):29–39. https://doi.org/10.1016/j.physbeh.2011.04.051
Ahima RS, Antwi DA (2008) Brain regulation of appetite and satiety. Endocrinol Metab Clin N Am 37(4):811–823. https://doi.org/10.1016/j.ecl.2008.08.005
Stricker EM (2012) Neurochemical and behavioral analysis of the lateral hypothalamic syndrome: a look back. Behav Brain Res 251(2):286–288. https://doi.org/10.1016/j.bbr.2012.01.004
Stuber GD, Wise RA (2016) Lateral hypothalamus circuits for feeding and reward. Nat Neurosci 19(2):198–205. https://doi.org/10.1038/nn.4220
Elmquist JK, Elias CF, Saper CB (1999) From lesion to leptin: hypothalamic control of food intake and body weight. Neuron 22(2):221–232. https://doi.org/10.1016/s0896-6273(00)81084-3
Sofroniew MV (1983) Morphology of vasopressin and oxytocin neurons and their central and vascular projections. Prog Brain Res 60:101–114. https://doi.org/10.1016/S0079-6123(08)64378-2
Blanks AM, Thornton S (2003) The role of oxytocin in parturition. BJOG 110(20):46–51. https://doi.org/10.1016/s1470-0328(03)00024-7
Brownstein MJ, Russell JT, Gainer H (1980) Synthesis, transport and release of posterior pituitary hormones. Science 207(4429):373–378. https://doi.org/10.1126/science.6153132
Howe HE, Somponpun SJ, Sladek CD (2004) Role of neurokinin 3 receptors in supraoptic vasopressin and oxytocin neurons. J Neurosci 24(45):10103–10110. https://doi.org/10.1523/JNEUROSCI.3164-04.2004
Iovino M, Messana T, Tortora A, Giusti C, Lisco G, Giagulli VA, Guastamacchia E, De Pergola G, Triggiani V (2021) Oxytocin signaling pathway: from cell biology to clinical implications. Endocr Metab Immune Disord Drug Targets 21(1):91–110. https://doi.org/10.2174/1871530320666200520093730
Leng G, Brown CH, Russell JA (1999) Physiological pathways regulating the activity of magnocellular neurosecretory cell. Prog Neurobiol 37(6):625–655. https://doi.org/10.1016/s0301-0082(98)00072-0
Russell JA, Leng G, Douglas AJ (2003) The magnocellular oxytocin system, the fount of maternity; adaptations in pregnancy. Front Neuroendocrinol 24(1):27–61. https://doi.org/10.1016/s0091-3022(02)00104-8
Iovino M, Guastamacchia E, Giagulli VA, Licchelli B, Iovino E, Triggiani V (2014) Molecular mechanisms involved in the control of neurohypophyseal hormones secretion. Curr Pharm Des 20(42):6702–6713. https://doi.org/10.2174/1381612820666140905150730
Iovino M, Giagulli VA, Licchelli B, Iovino E, Guastamacchia E, Triggiani V (2016) Synaptic inputs of neural efferent pathways to vasopressin- and oxytocin-secreting neurons of supraoptic and paraventricular hypothalamic nuclei. Endocr Metab Immune Disord Drug Targets 16(4):276–287. https://doi.org/10.2174/1871530317666170104124229
Barberis C, Tribollet F (1996) Vasopressin and oxytocin receptors in the central nervous system. Crit Rev Neurobiol 10(1):119–154. https://doi.org/10.1615/critrevneurobiol.v10.i1.60
Kiss A, Mikkelssen JD (2005) Oxytocin--anatomy and functional assignment: a minireview. Endocr Regul 39(3):97–105
Zhang Q, Li H, Guo F (2011) Amygdala, an important regulator of food intake. Front Biol 6:82–85. https://doi.org/10.1007/s11515-011-0950-z
Iovino M, Papa M, Monteleone P, Steardo L (1988) Neuroanatomical and biochemical evidence for the involvement of the area postrema in the regulation of vasopressin release in rats. Brain Res 447(1):178–182. https://doi.org/10.1016/0006-8993(88)90982-1
Tribollet E, Barberis C, Jard S, Dubois-Dauphin M, Derifuss JJ (1992) Localization and pharmacological characterization of light affinity binding sites for vasopressin and oxytocin in the rat brain by light microscopic autoradiography. Brain Res 442(1):105–108. https://doi.org/10.1016/0006-8993(88)91437-0
Yoshimura R, Kiyama H, Kimura T, Araki T, Maeno TT, Tanizawa O (1993) Localization of oxytocin receptor messenger ribonucleic acid in the rat brain. Endocrinology 133(3):1239–1246. https://doi.org/10.1210/endo.133.3.8396014
Leng G, Ludwig M (2008) Neurotransmitters and peptides: whispered secrets and public announcement. J Physiol 586:5625–5632. https://doi.org/10.1113/jphysiol.2008.159103
Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, Coppari R, Balthasar N, Cowley MA, Chua S Jr, Elmquist JK, Lowell BB (2006) Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49(2):191–203. https://doi.org/10.1016/j.neuron.2005.12.021
Fosch A, Zagmutt S, Casals N, Rodríguez-Rodríguez R (2021) New insights of SF1 neurons in hypothalamic regulation of obesity and diabetes. Int J Mol Sci 22(12):6186. https://doi.org/10.3390/ijms22126186
Iovino M, Messana T, Lisco G, Mariano F, Giagulli VA, Guastamacchia E, De Pergola G, Triggiani V (2022) Neuroendocrine modulation of food intake and eating behavior. Endocr Metab Immune Disord Drug Targets 22(13):1252–1262. https://doi.org/10.2174/1871530322666220127114326
Onaka T, Takayanagi Y (2019) Role of oxytocin in the control of stress and food intake. J Neuroendocrinol 31(3):e12700. https://doi.org/10.1111/jne.12700
Flanagan LM, Blackburn RE, Verbalis JG, Stricker EM (1992) Hypertonic NaCl inhibits gastric motility and food intake in rats with lesions in the rostral AV3V region. Am J Phys 263:R9–R14. https://doi.org/10.1152/ajpregu.1992.263.1.R9
Neuman J, Ludwig M, Engelmann M, Pitman QJ, Landgraf R (1993) Simultaneous microdialysis in blood and brain: oxytocin and vasopressin release in response to central and peripheral osmotic stimulation and suckling in the rat. Neuroenocrinology 58:637–645. https://doi.org/10.1159/000126604
Sabatier N, Leng G, Menzies J (2013) Oxytocin, feeding, and satiety. Front Endocrinol 4:35. https://doi.org/10.3389/fendo.2013.00035
Noble EE, Billington CJ, Kotz CM, Wang C (2014) Oxytocin in the ventromedial hypothalamic nucleus reduces feeding and acutely increases energy expenditure. Am J Phys Regul Integr Comp Phys 307(6):R737–R745. https://doi.org/10.1152/ajpregu.00118.2014
Reilly S, Bornovalova MA (2005) Conditioned taste aversion and amygdala lesions in the rat: a critical review. Neurosci Biobehav Rev 29(7):1067–1088. https://doi.org/10.1016/j.neubiorev.2005.03.025
Boccia MM, Baratti CM (2000) Involvement of central cholinergic mechanisms in the effect of oxytocin and an oxytocin receptor antagonist on retention performance in mice. Neurobiol Learn Mem 74(3):217–228. https://doi.org/10.1006/nlme.1999.3954
Campbell EJ, Holmes NM, Lingawi NW, Panayi MC, Westbrook RF (2015) Oxytocin signaling in basolateral and central amygdala nuclei differentially regulates the acquisition, expression, and extinction of context-conditioned fear in rats. Learn Mem 22(5):247–257. https://doi.org/10.1101/lm.036962.114
Ferguson JN, Aldag JM, Insel TR, Young LJ (2001) Oxytocin in the medial amygdala is essential for social recognition in the mouse. J Neurosci 21(20):8278–8285. https://doi.org/10.1523/JNEUROSCI.21-20-08278.2001
Klockars OA, Klockars A, Levine AS, Olszewski PK (2018) Oxytocin administration in the basolateral and central nuclei of amygdala moderately suppresses food intake. Neuroreport 29(6):504–510. https://doi.org/10.1097/WNR.0000000000001005
Olszewski PK, Waas JR, Brooks LL, Herisson F, Levine AS (2013) Oxytocin receptor blockade reduces acquisition but not retrieval of taste aversion and blunts responsiveness of amygdala neurons to an aversive stimulus. Peptides 50:36–41. https://doi.org/10.1016/j.peptides.2013.09.008
Hyde TM, Miselis RR (1983) Effects of area postrema/caudal medial nucleus of the solitary tract lesions on food intake and body weight. Am J Phys Regul Integr Comp Phys 244(4):R577–R587. https://doi.org/10.1152/ajpregu.1983.244.4.R577
Contreas RT, Fox E, Drugovich ML (1982) Area postrema lesions produce feeding deficits in the rat: effects of preoperative dieting an 2-deoxy-D-glucose. Physiol Behav 29(5):875–884. https://doi.org/10.1016/0031-9384(82)90338-9
Ossenkopp KP, Eckel LA (1995) Toxin-induced conditioned changes in taste reactivity and the role of the chemosensitive area postrema. Neurosci Biobehav Rev 19(1):99–108. https://doi.org/10.1016/0149-7634(94)00024-u
Kobashi M, Shimatani Y, Fujita M (2023) Oxytocin increased intragastric pressure in the forestomach of rats via the dorsal vagal complex. Physiol Behav 261:114087. https://doi.org/10.1016/j.physbeh.2023.114087
Potes CS, Turek VF, Cole RL, Vu C, Roland BL, Roth JD, Riediger T, Lutz TA (2010) Noradrenergic neurons of the area postrema mediate amylin hypophagic action. Am J Phys Regul Integr Comp Phys 299:R623–R631. https://doi.org/10.1152/ajpregu.00791.2009
Lutz TA, Senn M, Althans J, Del Prete E, Ehrensperger F, Sharrer E (1998) Lesions of the area postrema/nucleus of the solitary tract (AP/NTS) attenuates the anorectic effects of amylin and calcitonin gene-related peptide (CGRP) in rats. Peptides 19(2):309–317. https://doi.org/10.1016/s0196-9781(97)00292-1
Iovino M, Vanacore A, Steardo L (1990) Alpha 2-adrenergic stimulation within the nucleus tractus solitarius attenuates vasopressin release induced by depletion of cardiovascular volume. Pharmacol Biochem Behav 37(4):821–824. https://doi.org/10.1016/0091-3057(90)90568-3
Iovino M, Guastamacchia E, Giagulli VA, Licchelli B, Triggiani V (2012) Vasopressin secretion control: central neural pathways, neurotransmitters and effects of drugs. Curr Pharm Des 18:4714–4724. https://doi.org/10.2174/138161212802651607
Rinaman L (2010) Ascending projections from the caudal visceral nucleus of the solitary tract to brain regions involved in food intake and energy expenditure. Brain Res 13(50):18–34. https://doi.org/10.1016/j.brainres.2010.03.059
Shapiro RE, Miselis RR (1985) The central organization of the vagus nerve innervating the stomach of the rat. J Comp Neurol 238(4):473–488. https://doi.org/10.1002/cne.902380411
Rinaman L (2003) Postnatal development of hypothalamic inputs to the dorsal vagal complex in rats. Physiol Behav 79(1):65–70. https://doi.org/10.1016/s0031-9384(03)00105-7
Berthoud HR, Sutton GM, Townsend RL, Petterson LM, Zheng H (2006) Brainstem mechanisms integrating gut-derived satiety signals and descending forebrain information in the control of meal size. Physiol Behav 89(4):517–524. https://doi.org/10.1016/j.physbeh.2006.08.018
Menani JV, Colombari E, Talman WT, Johnson AK (1996) Commissural nucleus of the solitary tract lesions reduce food intake, body weight gain in rats. Brain Res 740(1-2):102–108. https://doi.org/10.1016/s0006-8993(96)00850-5
Iovino M, Messana T, De Pergola G, Iovino E, Di Cuozo F, Guastamacchia E, Giagulli VA, Triggiani V (2018) The role of neurohypophyseal hormones vasopressin and oxytocin in neuropsychiatric disorders. Endocr Metab Immune Disord Drug Targets 18(4):341–347
Spetter MS, Hallshmid M (2017) Current findings on the role of oxytocin in the regulation of food intake. Physiol Behav 176:31–39. https://doi.org/10.1016/j.physbeh.2017.03.007
Richard JE, Anerberg RH, Goteson A, Gribble FM, Reimann F, Skibicka KP (2015) Activation of the GLP-1 receptors in the nucleus of the solitary tract reduces food reward behavior and targets the mesolimbic system. PLoS One 10(3):e0119034. https://doi.org/10.1371/journal.pone.0119034
Uchoa ET, Zham S, de Cravahlo BB, Rorate R, Antunes-Rorigues J, Elias LLK (2013) Oxytocin projections to the nucleus of the solitary tract contribute to the increased meal-related satiety responses in primary adrenal insufficiency. Exp Physiol 98(10):1495–1504. https://doi.org/10.1113/expphysiol.2013.073726
McCann MJ, Rogers RC (1990) Oxytocin excites gastric-related neurons in rat dorsal vagal complex. J Physiol 428:95–108. https://doi.org/10.1113/jphysiol.1990.sp018202
Kerem L, Hadjikhani N, Holsen L, Lawson EA, Plessow F (2020) Oxytocin reduces the functional connectivity between brain regions involved in eating behavior in men with overweight and obesity. Int J Obes 44(5):980–989. https://doi.org/10.1038/s41366-019-0489-7
Ho JM, Anekonda VT, Thompson BW, Zhu M, Curry RW, Hwang BH, Morton GJ, Schwartz MW, Baskin DG, Appeleyard SM, Blevins JE (2014) Hindbrain oxytocin receptors contribute to the effects of circulating oxytocin on food intake in male rats. Endocrinology 155(8):2845–2857
Iwasaki Y, Maejima Y, Suyama S, Yoshia M, Arai T, Kataurada K, Kumari P, Nakabayshi H, Kakei M, Yaa T (2015) Peripheral oxytocin activates vagal afferent neurons to suppress feeding in normal and leptin-resistant mice. A route for ameliorating hyperphagia and obesity. Am J Physiol Regul Integr Comp Physiol 308(5):R360–R369. https://doi.org/10.1152/ajpregu.00344.2014
Leibowitz SF, Hammer NJ, Chang K (1981) Hypothalamic paraventricular nucleus lesions produce overeating and obesity in the rat. Physiol Behav 27(6):1031–1040. https://doi.org/10.1016/0031-9384(81)90366-8
Arletti R, Benelli A, Bertolini A (1989) Influence of oxytocin on feeding behavior in the rat. Peptides 10(1):89–93. https://doi.org/10.1016/0196-9781(89)90082-x
Wald HS, Chandra A, Kalluri A, Ong ZY, Hayes MR, Grill HJ (2020) NTS and VTA oxytocin reduces food motivation and food seeking. Am J Phys Regul Integr Comp Phys 319(6):R673–R683. https://doi.org/10.1152/ajpregu.00201.2020
Anekonda VT, Thompson BW, Ho JM, Roberts ZS, Edwards MM, Nguyen HK, Dodson AD, Wolden-Hanson T, Chukri DW, Herbertson AJ, Graham JL, Havel PJ, Wietecha TA, O’Brien KD, Blevins JE (2021) Hindbrain administration of oxytocin reduces food intake, weight gain and activates catecholamine neurons in the hindbrain nucleus of the solitary tract in rats. J Clin Med 10(21):5078. https://doi.org/10.3390/jcm10215078
Warren KR, Wehring HJ, Liu F, McMahon RP, Chen S, Chester C, Kelly DL (2018) Effects of intranasal oxytocin on satiety signaling in people with schizophrenia. Physiol Behav 189:86–91. https://doi.org/10.1016/j.physbeh.2018.03.008
Damen L, Grootjen LN, Juriaans AF, Donze SH, Huisman TM, Visser JA, Delhanty PJD, Hokken-Koelega ACS (2021) Oxytocin in young children with Prader-Willi syndrome: results of a randomized, double-blind, placebo-controlled, crossover trial investigating 3 months of oxytocin. Clin Endocrinol 94(5):774–785. https://doi.org/10.1111/cen.14387
Burmester V, Gibson EL, Butler G, Bailey A, Terry P (2019) Oxytocin reduces post-stress sweet snack intake in women without attenuating salivary cortisol. Physiol Behav 212:112704. https://doi.org/10.1016/j.physbeh.2019.112704
Wronski ML, Plessow F, Kerem L, Asanza E, O’Donoghue ML, Stanford FC, Bredella MA, Torriani M, Soukas AA, Kheterpal A, Eddy KT, Holmes TM, Deckersbach T, Vangel M, Holsen LM, Lawson EA (2022) A randomized, double-blind, placebo-controlled clinical trial of 8-week intranasal oxytocin administration in adults with obesity: rationale, study design, and methods. Contemp Clin Trials 122:106909. https://doi.org/10.1016/j.cct.2022.106909
Maguire S, O’Dell A, Touyz L, Russell J (2013) Oxytocin and anorexia nervosa: a review of emerging literature. Eur Eat Disord Rev 21(6):475–478. https://doi.org/10.1002/erv.2252
Olszewski PK, Klockars A, Levine AS (2016) Oxytocin: a conditional anorexigen whose effects on appetite depend on the physiological, behavioural and social contexts. J Neuroendocrinol 28(4). https://doi.org/10.1111/jne.12376
Leppanen J, Cardi V, Ng KW, Paloyelis Y, Stein D, Tchanturia K, Treasure J (2017) Effects of intranasal oxytocin on the interpretation and expression of emotions in anorexia nervosa. J Neuroendocrinol 29(3). https://doi.org/10.1111/jne.12458
Russell J, Maguire S, Hunt GE, Kesby A, Suraev A, Stuart J, Booth J, McGregor IS (2018) Intranasal oxytocin in the treatment of anorexia nervosa: randomized controlled trial during re-feeding. Psychoneuroendocrinology 87:83–92. https://doi.org/10.1016/j.psyneuen.2017.10.014
Ong ZY, Alhodeff AL, Grill HJ (2015) Medial nucleus tractus solitarius oxytocin receptor signaling and food intake control: the role of gastrointestinal satiation signal processing. Am J Physiol Regul Integr Comp Physiol 308(9):R800–R806. https://doi.org/10.1152/ajpregu.00534.2014
Sanchez Jimenez JG, De Jesus O (2023) Hypothalamic dysfunction. In: StatPearls. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560743/. Accessed 29 Apr 2023
Daubenbüchel AM, Hoffmann A, Eveslage M, Özyurt J, Lohle K, Reichel J, Thiel CM, Martens H, Geenen V, Müller HL (2016) Oxytocin in survivors of childhood-onset craniopharyngioma. Endocrine 54(2):524–531. https://doi.org/10.1007/s12020-016-1084-5
Gebert D, Auer MK, Stieg MR, Freitag MT, Lahne M, Fuss J, Schilbach K, Schopohl J, Stalla GK, Kopczak A (2018) De-masking oxytocin-deficiency in craniopharyngioma and assessing its link with affective function. Psychoneuroendocrinology 88:61–69. https://doi.org/10.1016/j.psyneuen.2017.11.006
Brandi ML, Gebert D, Kopczak A, Auer MK, Schilbach L (2020) Oxytocin release deficit and social cognition in craniopharyngioma patients. J Neuroendocrinol 32(5):e12842. https://doi.org/10.1111/jne.12842
Cook N, Miller J, Hart J (2016) Parent observed neuro-behavioral and pro-social improvements with oxytocin following surgical resection of craniopharyngioma. J Pediatr Endocrinol Metab 29(8):995–1000. https://doi.org/10.1515/jpem-2015-0445
Hoffmann A, Özyurt J, Lohle K, Reichel J, Thiel CM, Müller HL (2017) First experiences with neuropsychological effects of oxytocin administration in childhood-onset craniopharyngioma. Endocrine 56(1):175–185. https://doi.org/10.1007/s12020-017-1257-x
Daubenbüchel AM, Özyurt J, Boekhoff S, Warmuth-Metz M, Eveslage M, Müller HL (2019) Eating behaviour and oxytocin in patients with childhood-onset craniopharyngioma and different grades of hypothalamic involvement. Pediatr Obes 14(9):e12527. https://doi.org/10.1111/ijpo.12527
Hsu EA, Miller JL, Perez FA, Roth CL (2018) Oxytocin and naltrexone successfully treat hypothalamic obesity in a boy post-craniopharyngioma resection. J Clin Endocrinol Metab 103(2):370–375. https://doi.org/10.1210/jc.2017-02080
McCormack SE, Blevins JE, Lawson EA (2020) Metabolic effects of oxytocin. Endocr Rev 41(2):121–145. https://doi.org/10.1210/endrev/bnz012
McCormack SE, Wang Z, Wade KL, Dedio A, Cilenti N, Crowley J, Plessow F, Bamba V, Roizen JD, Jiang Y, Stylli J, Ramakrishnan A, Platt ML, Shekdar K, Fisher MJ, Vetter VL, Hocking M, Xiao R, Lawson EA (2023) A pilot randomized clinical trial of intranasal oxytocin to promote weight loss in individuals with hypothalamic obesity. J Endocr Soc 7(5):bvad037. https://doi.org/10.1210/jendso/bvad037
Kim YR, Kim JH, Kim CH, Shin JG, Treasure J (2015) Association between the oxytocin receptor gene polymorphism (rs53576) and bulimia nervosa. Eur Eat Disord Rev 23(3):171–178. https://doi.org/10.1002/erv.2354
Micali N, Crous-Bou M, Treasure J, Lawson EA (2017) Association between oxytocin receptor genotype, maternal care and eating disorder behaviors in community sample of women. Eur Eat Disord Rev 25(1):19–25. https://doi.org/10.1002/erv.2486
Kim YR, Eom JS, Yang YW, Kang J, Treasure J (2015) The impact of oxytocin on food intake and emotion recognition in patients with eating disorders: a double blind single dose within-subject cross-over design. PLoS One 10(9):e0137514. https://doi.org/10.1371/journal.pone.0137514
Dhuria SV, Hanson LR, Frey WH (2010) Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci 99:1654–1673. https://doi.org/10.1002/jps.21924
Dimitri P (2022) Treatment of acquired hypothalamic obesity: now and the future. Front Endocrinol (Lausanne) 13:846880. https://doi.org/10.3389/fendo.2022.846880
Funding
Open access funding provided by Università degli Studi di Bari Aldo Moro within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
M.I. conceived the review. M.I., T.M., G.L., S.M., and V.T. performed database searches and selected appropriate references. M.I., T.M., and G.L. drafted the manuscript. F.M., V.A.G., E.G., G.D.P., and V.T. provided minor editing. E.G., G.D.P., and V.T. provided feedback. All the authors read the text and approved the final manuscript submission.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Informed consent
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: In this article the affiliation details for Giovanni De Pergola were incorrectly given as 'National Institute of Gastroenterology "Saverio de Bellis", Research Hospital, Castellana Grotte, Bari, Italy' but should have been 'National Institute of Gastroenterology IRCCS "Saverio de Bellis", Research Hospital, Castellana Grotte, Bari, Italy'.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Iovino, M., Messana, T., Marucci, S. et al. The neurohypophyseal hormone oxytocin and eating behaviors: a narrative review. Hormones 23, 15–23 (2024). https://doi.org/10.1007/s42000-023-00505-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42000-023-00505-y