
Abstract
In this work, we introduce a low-cost open-source flow system that includes a dual syringe pump with implemented pressure sensor and back pressure regulator. The entire system can be built for around 500 €. Commercially available flow systems can be very expensive with equipment starting at, but often greatly exceeding, 10,000 €. This high price of entry makes such technology prohibitively expensive for many research groups. Such systems stand to benefit the emerging academic pharmaceutical field by providing the experience and availability of reliable and affordable solutions. To implement accessible flow chemistry at research facilities, the systems must be made affordable. In addition, space in research laboratories is usually limited and commercially available flow systems can be very bulky. Having a compact and individually adjustable system is thus beneficial, with 3D printing technology offering the solution. Our compact 3D-printed system meets the needs of many applications in flow chemistry research as well as educational requirements for universities. As a proof of concept, we conceptualized, developed, and tested a custom flow system that can be used to synthesize [18F]2-fluoro-2-desoxy-d-glucose ([18F]FDG), the most commonly used PET-tracer. This system was designed to perform the typical functions and operations required in radiotracer production i.e. radiofluorination, dilution, SPE-trapping, deprotection, and SPE-elution. With this proof-of-concept in hand, the system can be easily customized to produce other radiopharmaceuticals.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Continuous flow chemistry has undergone rapid growth over the last two decades [1,2,3,4]. Flow chemistry offers the unique benefits of improved mixing and heating efficiency as well as easy scale-up while reducing reaction time and solvent consumption [5,6,7,8]. Due to its beneficial properties, many applications for research and industrial organic synthesis have been developed. It can be combined with almost every technology, like photochemistry [9], catalysis [10], or electrochemistry [11]. One of the drawbacks is the prerequisite for expensive proprietary equipment that is not always affordable or obtainable. The combination of 3D printing and flow chemistry has emerged in recent years and could solve the hurdle of expensive equipment. Both, microreactors and various pump systems have already been described using 3D printing [12,13,14,15,16,17,18,19]. It can also be a useful tool for other sciences, printing various scientific devices such as microscopes, centrifuges, or 3D models for educational purposes [20].
One application in which the full potential of flow chemistry is utilized is the synthesis of radiopharmaceuticals and PET-tracers [21,22,23]. This is mostly reliant on automation using expensive and proprietary equipment that is unobtainable to many researchers. To address this need, we developed an open-source 3D printed microfluidic system that can be produced for around 500 €. The system includes a dual syringe pump, a back pressure regulator (BPR) as well as a pressure sensor (PS). In combination with our 3D-printed microreactors, which were published in our previous work [14], the system can be customized to perform crucial radiosynthetic steps such as fluorination, dilution, and SPE (solid phase extraction) trapping/eluting. This system can be customized to rapidly produce a wide range of tracers and prosthetic radioligands with high molar activity and activity concentration, all within a small footprint. In this work, we evaluated this new system by synthesizing [18F]2-fluoro-2-desoxy-d-glucose ([18F]FDG), the gold standard of PET radiotracers (Fig. 1).

Synthesis of [18F]FDG
[18F]FDG is the most common and approved radiopharmaceutical for positron emission tomography (PET) and is used for non-invasive in vivo measurements of glucose metabolism [24]. Tumor cells, because of the Warburg effect, exhibit characteristically high glucose metabolism and can thus be differentiated from other normal tissue in PET by their high tracer uptake [25,26,27]. Lung cancer, brain cancer, lymphoma, melanoma as well as breast cancer are examples of cancer cells with higher glucose metabolism [28, 29]. [18F]FDG is a structural analog of glucose and recognized by cells accordingly. After being transported via GLUT-1 and GLUT-3 into cells, it is phosphorylated by hexokinase to [18F]FDG-6-phosphate. This cannot be further metabolized and is thus trapped in the cell [30, 31]. Being deficient of neutrons, the radioactive 18F- isotope strives for a more stable energy state, which is achieved through a β+-decay. In this process a proton (p) decays into a neutron (n), emitting a positron (e+) and an electron neutrino (ν–) [32]. The reaction can be described by the following equation.
The nucleon number in the nucleus remains the same, while the atomic number is decreased by the value of one. In the case of radioactive β+-decay of 18F, the stable oxygen isotope 18O is formed. Positrons are antiparticles of electrons and differ only by their sign. They dissipate kinetic energy by inelastic interactions with the tissue in the body and recombine with an electron present in the environment, forming a positronium exotic atom. In subsequent annihilation, the mass of the two particles converts into a pair of anti-parallel 511 keV gamma photons. These photons are simultaneously detected within a ring of scintillator detectors and a tomographical map of tracer accumulation (PET image) can be generated [33].
The synthesis of [18F]FDG has already been excessively well described in the literature [23, 34, 35] and the GMP tracer is routinely produced in multi-patient doses from commercially available kits designed for expensive automated radiosynthesis devices. The requirements and considerations for our custom system address several of the factors that play a role in typical diagnostic radiopharmaceutical production: (1) Due to the half-life of the 18F-Isotope (109.5 min) [36], the total reaction time, including all purification, should be as short as possible. (2) Handling radioactive material should be as safe as possible. The transition to highly adaptable automated systems decreases the radiation exposure of the operator. (3) The reactions should be efficient and reproducible. The tracer precursors are expensive, and the amount used for each synthesis should be minimized. More so, the production of radioactive and chemical by-products should be minimized as their presence would require time-consuming purification by solid phase extraction (SPE) or HPLC. (4) Furthermore, the system should be affordable and easy to handle. The most common radiosynthetic route to [18F]FDG is via the SN2 nucleophilic substitution of tetraacetyl mannose triflate, followed by C18 SPE immobilization, base hydrolysis, and elution. An alumina SPE cartridge is often included in series to remove chemical impurities such as [18F]fluoride, tetrabutylammonium bicarbonate (TBAHCO3), or Kryptofix®. The synthesis of [18F]FDG for clinical use by common fully automated synthesizers requires over 30 min and achieves a radiochemical purity of over 95%. With the benchmarks established, we set out to test our open-source system [37].
Results and discussion
Development of the pump system
The flexible open-source microfluidic system we describe allows for the development of semi-automated flow processes. Parameters such as reaction conditions and flow rates were investigated and optimized. To fulfill the respective requirements, essential basic elements are needed. The most important task is to guarantee a continuous flow during the experiment. For this purpose, we built a syringe pump with two syringe holders that can be separately controlled. By using syringes as solvent reservoirs, a wide range of flow rates can be reliably covered.
Apart from the electronics and the mechanical parts, such as screws and rods, almost all components were 3D-printed. Figure 2 shows the CAD drawing and the assembled pump. We printed two versions of the pump, one in PLA (polylactic acid) and one in PETG (polyethylene terephthalate glycol copolymer), but also more chemical resistant materials like Nylon or any other non-flexible material should be possible. The 3D-printed parts were assembled using threaded inserts ensuring a firm fixation. The moving parts were guided by two rods and various ball and plain bearings. To transfer the motor movement to a linear movement, a trapezoidal thread spindle was used, which ensures a better power transmission than a standard V-threaded rod.
For controlling the pump, an Arduino Mega 2560 in combination with a programmable Nextion display was used. A RAMPS shield and TMC 2208 drivers were used to control the stepper motors. One drawback of using stepper motors is that pulsation of the flow can occur at low flow rates. This is caused when the pauses between steps are too long. For this reason, we chose geared stepper motors with a ratio of 27:1, resulting in two advantages. First, by increasing the number of steps required for one rotation, a constant flow could be ensured even at low flow rates. Second, the geared motor pump is much more powerful than the stepper motor and can easily generate pressures up to 10 bar, depending on the used syringes. One disadvantage is that the maximum pump speed is reduced. If needed, the gear motor can be easily changed to a smaller gear ratio, and the variable “gear-ratio” could be simply changed in the Arduino software (line 39: int gear-ratio = 27). Of course, a smaller gear ratio leads not only to a faster pump speed but also a decrease in power and maximum pressure.

Exploded view CAD drawing (a) and photograph (b) of the syringe pump
The system can be used with Hamilton glass syringes as well as commercially available disposable plastic syringes. To determine flow accuracy and reliability, the syringes were filled with water at r.t. and atmospheric pressure, the flow rate was set and the amount of collected water was weighed after equilibration of the system at the end of the collection time. Next, flow rates from 0.1 to 10% of the syringe volume were investigated. A modest deviation of 2.5%, which could not be decoupled from experimental error, was determined.
From the data of the measurements, deviations for each examined flow rate can be created. If the deviation is over 2.5% the syringe/flowrate combination was deemed unsuitable for the pump. The results for the suggested flow rates are summarized in Table 1.
The measurements were carried out on both the PLA flow system and the PETG flow system. No significant differences were found. It was also investigated on both systems whether there were differences in the flow rate accuracy at higher pressure. For this purpose, all measurements of the Hamilton glass syringes were repeated at a pressure of 5 bar. Again, no significant differences were found. Detailed experimental data can be found in the ESI.
Development of the pressure sensor and BPR
Another important point is the continuous measurement of the pressure. Due to varying flow resistances at different flow rates, different viscosities of the running fluids, as well as lengths of the capillaries or other components used in the system, a unique basic pressure is established for the process. This pressure must be measured and displayed live so that real-time flow adjustments can be made. For this purpose, we have developed a pressure sensor (PS), which can communicate with the pump. The system was designed such that the designated maximum pressure will not be exceeded as the pump stops automatically, preventing the reactor from bursting or another system component from rupturing. In addition, by tracking the pressure live, it is possible to know when the system is in equilibrium and ready to use.
The working principle of the pressure sensor is based on the resistance change of a force-sensitive resistor (FSR). To process the resistance change, the FSR is connected to an Arduino Mega on an analog Pin with enabled internal pull-up resistor. The channel in the 3D-printed part is opened to form a cavity, allowing for the pressure of the system to be transmitted to the FSR. The cavity is sealed by a PTFE disc and damped by a silicone disc. We have used different materials and thicknesses for the discs and found that silicone with a thickness of 1 mm and PTFE with a thickness of 0.1 mm gives the best results for pressures between 0 and 6 bar.
As described in our previous publication [14], we found that a system with a guide rail gives reliably leakproof connections. The surfaces shown in Fig. 3 were sanded and the flangeless flat bottom 1/4″-28 fittings were pressed against the flat-honed surfaces.

Exploded view CAD drawing (a) and photograph (b) of the pressure sensor
Since each sensor is unique and has slight differences in measured pressure, it must be calibrated before use. The calibration can be done using a commercial manometer. Therefore, multiple data points from 0.5 to 6 bar were measured in 0.2 bar increments and converted to a calibration function. Detailed instructions for the calibration can be found in the ESI. The accuracy of our calibration was checked by multiple measurements of HPLC resistance cartridges. A maximum deviation of 0.7% at 6 bars could be determined.
The kinetics of a reaction often correlate to temperature and reactant concentration. By increasing the pressure of the system, higher reaction temperatures (above the boiling point of the solvent) can be examined. For this purpose, we have designed an externally controllable back pressure regulator (BPR). In our previous publication [14] we developed a spring-based 3D-printed BPR. We have adapted the previous version to fit our guide rail (Fig. 3), which for us is the best and most reliable system. Also, we installed a 12 V gear motor to remotely tension and release the spring (Fig. 4). The motor is controlled using an L298N motor driver, which is connected to the Arduino Mega from the sensor. This remote control makes it possible to set the desired pressure when the setup is not easily accessible during the experiment. An example of this is when the components are placed in a shielded lead hotcell during experiments with high amounts of radioactivity. For our applications (PS and BPR), the solvent touching parts were 3D-printed with PEEK (polyether ether ketone), due to its high chemical and temperature resistance, but we also evaluated PP, PVDF, and PETG. All parts are working equally well with all tested materials, so a suitable material for each application can be selected. With the selected components, pressures up to 10 bar could be reliably reached.

Exploded view CAD drawings and photographs of the motorized BPR (a, b), the manual BPR (c), and the control unit (d)
Radiofluorination
Radiochemical syntheses are generally performed behind shielding as this reduces the radiation dose and contributes to safer working. Thus, non-direct intervention in the synthesis to control or vary the reaction parameters is a major benefit to the chemist.
To determine whether the system meets the high requirements of an established synthesis, a semi-automated process for the preparation of [18F]2-fluoro-2-desoxy-d-glucose ([18F]FDG) was developed and compared to the conventional batch process. Therefore, the synthesis was investigated under the same reaction conditions for both methods. For quantification, the radio-TLC measurement and HPLC measurements are used to determine the corrected radiochemical yield (RCY) and the radiochemical purity (RCP).
[18F]HF was obtained as an aqueous solution from the cyclotron, concentrated using a preconditioned ion exchange SPE, and eluted with a solution of MeCN and TBA bicarbonate. The reaction solution was azeotropically dried with anhydrous MeCN at 100 °C under an argon stream. After several drying cycles, the dry [18F]fluoride was dissolved in 1 mL of anhydrous MeCN. This reaction solution serves as the fluorinating agent.
For the batch synthesis, aliquots of 100 µL of the azeotropically dried [18F]fluoride were added to 60 µL of the acetylated mannose triflate solution into a disposable Pyrex tube and quickly vortexed. The radiofluorination reactions were carried out at 80, 90, 110, and 110 °C. The reaction time was set to 5, 10, and 15 min. The quality of the fluorination of each condition was quantified via radio-TLC and HPLC. After cooling to around 50 °C, the reaction mixtures were diluted with water. The diluted solution was then pushed through a SEP-Pak Alumina N SPE cartridge, connected in series to a C18 SPE cartridge.
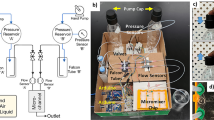
The flow synthesis system of [18F]FDG consists of our syringe pump, a 6-way valve with a built-in PEEK loop (160 µL), our pressure sensor, the remotely controllable pressure regulator, two 3D-printed PEEK reactors (900 µL and 50 µL) and two cartridges (SEP-Pak Alumina N (Waters) and Sep-Pak Plus Light C18 cartridge) (Fig. 5). Acetonitrile is used as a solvent and is continuously delivered to the system from a 10 mL syringe by the syringe pump. All components are connected using ETFE capillaries with an inner diameter of 0.5 mm and flangeless flat bottom 1/4″-28 HPLC fittings. The use of a 6-way valve allows pressure to be applied to the system at continuous flow. Reactions can thus be carried out at temperatures above the boiling point of the solvent under continuous flow. The pressure sensor is connected between the 6-way valve and the pump to avoid unnecessary radioactive contamination and unnecessary dead volume.

Schematic drawing of the flow process for [18F]FDG.
Aliquots of 100 µL of the [18F]fluoride in MeCN were premixed with 60 µL of the acetylated mannose triflate in dry MeCN. The pressure of the system was set to 2.5 bar. The reaction mixture was injected onto the loop of the 6-way valve, and into the running process by switching the 6-way valve. By changing the flow rate of the solvent, the residence or reaction time can be directly determined. Reaction times of 5, 10, and 15 min at 80, 90, 100, and 110 °C were investigated. Every condition was quantified via radio-TLC and HPLC. The results are summarized in Table 2.
Temperatures above 100 °C appear to destroy the precursor, and at temperatures below 90 °C the conversion is significantly lower. While a clear trend can be seen in the reaction times, one must also consider the half-life of the isotope of choice. Although a slightly better conversion is obtained with a residence time of 15 min, it is negligible. Under batch as well as under flow conditions, the optimum temperature was found to be 90 °C with a residence time of 10 min. To be able to make a statistical statement, repetitions of these conditions were carried out. For the batch synthesis, an RCY value of 52.3% ± 1.9% (n = 4) could be determined. For the flow system, an RCY value of 47.6% ± 1.2% (n = 5) could be determined (Table 3). Both radiochemical yields are in a similar range.
It is noticeable that considerably fewer radioactive by-products are produced under flow conditions (Fig. 6). This is a notable advantage of our flow synthesis for tracers with no included HPLC purification step such as [18F]FDG.

Radio TLC batch vs. flow after 18F-labelling
Another advantage of the flow system is that purification and deprotection can be automatically integrated into the process. For this purpose, the reaction solution is diluted 9-fold with water and passed through a Sep-Pak alumina cartridge, which is used to remove unwanted polar impurities such as [18F]fluoride. The acetylated intermediate can then be trapped on a C18 SPE cartridge that is connected afterward in series. Tests have shown that a dilution of 10% MeCN to 90% H2O is necessary. Even with 15% MeCN to 85% H2O, the fully acetylated intermediate is no longer reliably immobilized on the C18 cartridge. To verify the quality of the purification, the acetylated [18F]FDG was eluted from the C18 SPE cartridge with ethanol and examined by radio-TLC and HPLC. A radiochemical purity (RCP) of 99.1% ± 0.7% (n = 5) was observed. In comparison, the purification of the batch synthesis was performed manually. In this case, a purity of 93.5% ± 1.4% (n = 4) was determined. As expected, the purification of flow synthesis is simpler and more efficient than the manual batch process due to fewer by-products. Considering only small differences in RCY, this is an immense gain for radiochemical applications.
Deprotection was, in both cases, manually performed directly on the cartridge by passing 0.5 mL of 2 M NaOH through the C18 SPE cartridge. We performed a subsequent neutralization and formulation with 0.25 mL of 6 M HCl, followed by 1 mL of 1 M NaHCO3. The deprotected [18F]FDG was quantified via radioactive TLC.
The total time required for the semiautomated radiofluorination and immobilization of the acetylated intermediate on the C18 SPE cartridge was 14 min. The manual deprotection with aqueous NaOH required an additional minute. Thus, [18F]FDG was produced in under 15 min and had, according to radio-TLC, a radiochemical purity above 99%.
Experimental
Chemical reactions
If not mentioned, all reagents and solvents were purchased from commercial suppliers and used without further purification. The precursor, mannose triflate ultra-pure was purchased from ABX, Germany.
General radiochemistry
The [18F]fluoride was produced on a medical cyclotron (PETtrace 800, GE Healthcare) using the nuclear reaction of 18O(p,n)18F. Content of free [18F]fluoride as well as reaction products were analyzed by thin layer chromatography (TLC, PolyGram Sil G/UV254, Macherey–Nagel, Dueren, Germany) and analyzed on a Cyclone Plus phosphorimager (Perkin Elmer, Waltham, Massachusetts, USA). Radio-HPLC was performed using an Agilent 1260 series system (Agilent, Santa Clara, USA), with a reverse-phase analytical column (Luna C18(2), 100 Å, 250 × 4.6 mm, 5 μm; Phenomenex, Torrance, California, USA) connected to a Flow Count detection system (Model 106, Eckert & Ziegler, Berlin, Germany ) coupled to a diode detector (B-FC-3500-A, Eckert & Ziegler, Berlin, Germany). The mobile phases comprised CH3CN and water (+ 0.1% TFA) and the flow rate was 1 mL/min. The standard gradient program started with 5% CH3CN for 2 min. The CH3CN component was then increased to 100% over 15 min, where it was maintained for a further 6 min.
General procedure for fluoride preparation
Sep-Pak Plus Light QMA Carb (Waters, Waltham, MA, USA) was preconditioned with 10 mL 1 M NaHCO3, 10 mL air, 10 mL H2O, and 10 mL air. Sep-Pak Plus Light C18 SPE cartridge (Waters) was preconditioned with 10 mL ethanol and 10 mL H2O. SEP-Pak Alumina N (Waters) was preconditioned with 5 mL MeCN.
[18F]fluoride was immobilized on the QMA cartridge and eluted into a glass reactor with 0.6 mL of a premixed solution containing TBA-HCO3 (300 µL, 0.075 M, ABX, Germany) diluted with 0.3 mL dry MeCN. Azeotropical drying was carried out at 100 °C under a flow of argon and aided by multiple additions of MeCN (4 × 1 mL).
General procedure for the flow-synthesis of [18F]FDG [38, 39]
Aliquots (100 µL) of the azeotropically dried [18F]fluoride were dissolved in MeCN. 60 µL of mannose triflate solution (3 mg, 0.3 mL dry MeCN) was added to the 100 µL [18F]fluoride aliquot and quickly vortexed. The pressure of the setup was set to 2.5 bar. In a continuous flow, the premixed reaction mixture was injected through a 6-way valve into the running process. The radiofluorination was carried out at 80, 90, 110, and 110 °C. The flow rates were adjusted so that reaction times of 5, 10, and 15 min could be investigated (calculation of flow rates: Table 4). To determine the quality of the radio-fluorination, the acetylated intermediate was qualified via radio-TLC (petroleum ether/ ethyl acetate 50:50) and HPLC (tr = 14.34 min) before the deprotection took place. The process was repeated at reaction temperatures of 80, 90, 100, and 110 °C, and the optimal conditions were ascertained. To continue the process, a second syringe delivering water facilitated the dilution in the second 50 µL PEEK reactor. The diluted mixture passed through a SEP-Pak Alumina N (Waters) SPE cartridge connected in series to a Sep-Pak Plus Light C18 SPE cartridge, resulting in the immobilization of the acetylated intermediate on the latter. The deprotection was performed manually with 0.5 mL 2 M NaOH. The solution was neutralized with 0.25 mL 6 M HCl and buffered with 1 mL sat. NaHCO3 afterward. The deprotected [18F]FDG was qualified via HPLC (tr = 2.88 min) and radioactive TLC (MeCN/H2O 95:5).
Acetylated [18F]FDG intermediate RCY: 47.6% ± 1.2% (n = 5). Radiochemical purities of [18F]FDG (RCPs): 99.1% ± 0.7% (n = 5).
General procedure for the batch-synthesis of [18F]FDG
The fluoride and aliquot preparation were performed as described above. The radiofluorination reactions were performed in disposable Pyrex 7 mL tubes and were carried out at 80, 90, 110, and 110 °C. The reaction time was set to 5, 10, and 15 min. After cooling to around 50 °C, the reaction mixtures were diluted with 2 mL H2O. The diluted solution was then pushed through a SEP-Pak Alumina N SPE (Waters), connected in series to a C18 SPE cartridge. Either the acetylated intermediate was eluted with ethanol (0.5 mL) or deprotection was carried out as described above. The radiochemical yield of the acetylated intermediate was measured by radio-TLC (petroleum ether/ ethyl acetate 50:50) before the purification by alumina SPE. Purity was determined afterward. The deprotection was performed by passing 0.5 mL 2 M NaOH directly through the Sep-Pak Plus Light C18 cartridge (Waters) containing the acetylated derivative. The solution was neutralized with 0.25 mL 6 M HCl and buffered with 1 mL sat. NaHCO3 afterward. The products were qualitatively assessed as described above.
Acetylated [18F]FDG intermediate RCY: 52.3% ± 1.9% (n = 4). Radiochemical purities of [18F]FDG (RCPs): 93.5% ± 1.4% (n = 4).
Conclusion
In this work, we have developed a 3D-printed open-source microfluidic system that includes a dual syringe pump, a pressure sensor, and a remotely controlled back pressure regulator, all of which can be built for around 500 €. Owing to its high customizability, this low-cost open-source microfluidic system shows great potential in all fields of chemical research. Especially, due to its small footprint, the ever-expanding world of radiochemistry could benefit significantly. Using the [18F]FDG radiosynthesis as a benchmark, we confirmed that our system can reliably perform the functions and operations that are carried out by proprietary and expensive radiosynthesizers e.g. radiofluorination, dilution, and SPE purification. The system was thoroughly evaluated and shown to be suitable for research radiochemistry, producing results that are comparable to common batch syntheses. It can be used to synthesize radiotracers in good yield with fewer radioactive side-products compared to batch syntheses. The system is easily expandable and can be adapted to include multiple reactors and SPE purification steps, all while maintaining a footprint small enough to fit in a hotcell. The potential for this system is immense for both manual and automated radiochemistry. The automation of manual steps would reduce the overall radiation exposure to the laboratory staff while increasing reliability and reproducibility by eliminating human error. The system could also be coupled to existing radiosynthesizers, producing radioactive prosthetic ligands for more complicated, multistep radiosynthesis.
With the presented plans, descriptions, and schematics of this open-source flow system we aim to inspire other scientists to use this technology and ultimately remove some of the barriers surrounding the fascinating fields of flow- and radiochemistry.
Change history
19 April 2023
A Correction to this paper has been published: https://doi.org/10.1007/s41981-023-00269-x
References
Plutschack MB, Pieber B, Gilmore K, Seeberger PH (2017) The Hitchhiker’s guide to flow chemistry. Chem Rev 117:11796–11893. https://doi.org/10.1021/acs.chemrev.7b00183
Gutmann B, Kappe CO (2017) Forbidden chemistries — paths to a sustainable future engaging continuous processing. J Flow Chem 7:65–71. https://doi.org/10.1556/1846.2017.00009
Hughes DL (2020) Applications of flow chemistry in the pharmaceutical industry—Highlights of the recent patent literature. Org. Process Res Dev 24:1850–1860. https://doi.org/10.1021/acs.oprd.0c00156
Bogdan AR, Dombrowski AW (2019) Emerging trends in flow chemistry and applications to the pharmaceutical industry. J Med Chem 62:6422–6468. https://doi.org/10.1021/acs.jmedchem.8b01760
Wegner J, Ceylan S, Kirschning A (2011) Ten key issues in modern flow chemistry. Chem Commun 47:4583–4592. https://doi.org/10.1039/C0CC05060A
Zhang J, Wang K, Teixeira AR, Jensen KF, Luo G (2017) Design and scaling up of microchemical systems: a review. Annu Rev Chem Biomol Eng 8:285–305. https://doi.org/10.1146/annurev-chembioeng-060816-101443
Berton M, de Souza JM, Abdiaj I, McQuade DT, Snead DR (2020) Scaling continuous API synthesis from milligram to kilogram: extending the enabling benefits of micro to the plant. J Flow Chem 10:73–92. https://doi.org/10.1007/s41981-019-00060-x
Dong Z, Wen Z, Zhao F, Kuhn S, Noël T (2021) Scale-up of micro- and milli-reactors: an overview of strategies, design principles and applications. Chem Eng Sci : X 10:100097. https://doi.org/10.1016/j.cesx.2021.100097
Buglioni L, Raymenants F, Slattery A, Zondag SDA, Noël T (2022) Technological innovations in photochemistry for organic synthesis: Flow chemistry, high-throughput experimentation, scale-up, and photoelectrochemistry. Chem Rev 122:2752–2906. https://doi.org/10.1021/acs.chemrev.1c00332
Tanimu A, Jaenicke S, Alhooshani K (2017) Heterogeneous catalysis in continuous flow microreactors: a review of methods and applications. Chem Eng J 327:792–821. https://doi.org/10.1016/j.cej.2017.06.161
Pletcher D, Green RA, Brown RCD (2018) Flow electrolysis cells for the synthetic organic chemistry laboratory. Chem Rev 118:4573–4591. https://doi.org/10.1021/acs.chemrev.7b00360
Price AJN, Capel AJ, Lee RJ, Pradel P, Christie SDR (2020) An open source toolkit for 3D printed fluidics. J Flow Chem. https://doi.org/10.1007/s41981-020-00117-2
Neumaier JM, Madani A, Klein T, Ziegler T (2019) Low-budget 3D-printed equipment for continuous flow reactions. Beilstein J Org Chem 15:558–566. https://doi.org/10.3762/bjoc.15.50
Menzel F, Klein T, Ziegler T, Neumaier JM (2020) 3D-printed PEEK reactors and development of a complete continuous flow system for chemical synthesis. React Chem Eng 5:1300–1310. https://doi.org/10.1039/D0RE00206B
Parra-Cabrera C, Achille C, Kuhn S, Ameloot R (2018) 3D printing in chemical engineering and catalytic technology: structured catalysts, mixers and reactors. Chem Soc Rev 47:209–230. https://doi.org/10.1039/C7CS00631D
Alimi OA, Meijboom R (2021) Current and future trends of additive manufacturing for chemistry applications: a review. J Mater Sci 56:16824–16850. https://doi.org/10.1007/s10853-021-06362-7
Capel AJ, Rimington RP, Lewis MP, Christie SDR (2018) 3D printing for chemical, pharmaceutical and biological applications. Nat Rev Chem 2:422–436. https://doi.org/10.1038/s41570-018-0058-y
Baas S, Saggiomo V (2021) Ender3 3D printer kit transformed into open, programmable syringe pump set. HardwareX 10:e00219. https://doi.org/10.1016/j.ohx.2021.e00219
Wijnen B, Hunt EJ, Anzalone GC, Pearce JM (2014) Open-source syringe pump library. PLoS ONE 9:e107216. https://doi.org/10.1371/journal.pone.0107216
Saggiomo V (2022) A 3D printer in the lab: not only a Toy. Adv Sci 9:2202610. https://doi.org/10.1002/advs.202202610
Steel CJ, O’Brien AT, Luthra SK, Brady F (2007) Automated PET radiosyntheses using microfluidic devices. J Label Compd Radiopharm 50:308–311. https://doi.org/10.1002/jlcr.1259
Koag MC, Kim H-K, Kim AS (2014) Efficient microscale synthesis of [18F]-2-fluoro-2-deoxy-d-glucose. Chem Eng J 258:62–68. https://doi.org/10.1016/j.cej.2014.07.077
Watts P, Pascali G, Salvadori PA (2012) Positron emission tomography radiosynthesis in microreactors. J Flow Chem 2:37–42. https://doi.org/10.1556/jfc-d-12-00010
Culbert PA, Adam MJ, Hurtado ET, Huser JMA, Jivan S, Lu J, Ruth TJ, Zeisler SK (1995) Automated synthesis of [18F]FDG using tetrabutylammonium bicarbonate. Appl Radiat Isot 46:887–891. https://doi.org/10.1016/0969-8043(95)00177-F
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033. https://doi.org/10.1126/science.1160809
Palsson-McDermott EM, O’Neill LA (2013) The Warburg effect then and now: from cancer to inflammatory diseases. BioEssays 35:965–973. https://doi.org/10.1002/bies.201300084
Robey IF, Stephen RM, Brown KS, Baggett BK, Gatenby RA, Gillies RJ (2008) Regulation of the Warburg effect in early-passage breast cancer cells. Neoplasia 10:745–756. https://doi.org/10.1593/neo.07724
Glaudemans AW, Signore A (2010) FDG-PET/CT in infections: the imaging method of choice? Eur. J Nucl Med Mol Imaging 37:1986–1991. https://doi.org/10.1007/s00259-010-1587-x
Gafter-Gvili A, Raibman S, Grossman A, Avni T, Paul M, Leibovici L, Tadmor B, Groshar D, Bernstine H (2015) [18F]FDG-PET/CT for the diagnosis of patients with fever of unknown origin. QJM 108:289–298. https://doi.org/10.1093/qjmed/hcu193
Wang ZG, Yu MM, Han Y, Wu FY, Yang GJ, Li DC, Liu SM (2016) Correlation of Glut-1 and Glut-3 expression with F-18 FDG uptake in pulmonary inflammatory lesions. Med (Baltim) 95:e5462. https://doi.org/10.1097/md.0000000000005462
Yu S (2006) Review of F-FDG synthesis and quality control. Biomed Imaging Interv J 2:e57. https://doi.org/10.2349/biij.2.4.e57
Turkington TG (2001) Introduction to PET instrumentation. J Nucl Med Technol 29:4–11. https://tech.snmjournals.org/content/29/1/4
Melcher CL (2000) Scintillation crystals for PET. J Nucl Med 41:1051–1055. https://jnm.snmjournals.org/content/41/6/1051
Oh SJ, Chi DY, Mosdzianowski C, Kim JY, Gil HS, Kang SH, Ryu JS, Moon DH (2005) Fully automated synthesis of [18F]fluoromisonidazole using a conventional [18F]FDG module. Nucl Med Biol 32:899–905. https://doi.org/10.1016/j.nucmedbio.2005.06.003
Decristoforo C (2007) Challenges in the small-scale preparation of radiopharmaceuticals -A European perspective. FABAD J Pharm Sci 32:131–138. http://dergi.fabad.org.tr/index/issue/abstract/32_03_5.htm
Unterweger MP, Fitzgerald R (2020) Corrigendum to “Update of NIST half-life results corrected for ionization chamber source-holder instability” [Appl. Radiat. Isot. 87 (2014) 92–94]. Appl Radiat Isot 159:108976. https://doi.org/10.1016/j.apradiso.2019.108976
Saxena P, Singh AK, Dixit M, Kheruka SC, Mahmood T, Gambhir S (2021) Establishing the [18F]-FDG production via two different automated synthesizers for routine clinical studies: our institutional experiences of 4 years. Indian J Nucl Med 36:120–124. https://doi.org/10.4103/ijnm.IJNM_137_20
Gomzina NA, Zaitsev VV, Krasikova RN (2001) Optimization of nucleophilic fluorination step in the synthesis of various compounds labelled with fluorine-18 for their use as pet radiotracers. J Label Compd Radiopharm 44:S895–S897. https://doi.org/10.1002/jlcr.25804401314
Zhou X, Li Y, Jiang X, Wang X, Chen S, Shen T, You J, Lu H, Liao H, Li Z, Cheng Z (2020) Intra-individual comparison of 18F-PSMA-1007 and 18F-FDG PET/CT in the evaluation of patients with prostate cancer. Front Oncol 10:585213. https://doi.org/10.3389/fonc.2020.585213
Acknowledgements
We thank the Karl und Anna Buck Stiftung for funding this work.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
ESM 1
3D printer setup, numbered part lists, detailed step-by-step assembly instructions end electronic setups of all components. (PDF 1.94 MB)
ESM 2
CAD files (STEP and STL), Arduino and Nextion program, and sigmoid plotting file for calibration of the pressure sensor. (ZIP 13.3 MB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Menzel, F., Cotton, J., Klein, T. et al. FOMSy: 3D-printed flexible open-source microfluidic system and flow synthesis of PET-tracer. J Flow Chem 13, 247–256 (2023). https://doi.org/10.1007/s41981-023-00267-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41981-023-00267-z