
Abstract
Redox flow batteries (RFBs) that employ sustainable, abundant, and structure-tunable redox-active species are of great interest for large-scale energy storage. As a vital class of redox-active species, metal coordination complexes (MCCs) possessing the properties of both the organic ligands and transition metal ion centers are attracting increasing attention due to the advantages of multielectron charge transfer, high structural tailorability, and reduced material crossover. Herein, we present a critical overview of RFBs that employ MCCs as redox-active materials in both aqueous and nonaqueous mediums. The progress is comprehensively summarized, including the design strategies, solubility characteristics, electrochemical properties, and battery cycling performance of MCCs. Emphasis is placed on the ligand selection and modification strategies used to tune the critical properties of MCCs, including their redox potential, solubility, cycling stability, and electron transfer redox reactions, to achieve stable cycled RFBs with a high energy density. Furthermore, we discuss the current challenges and perspectives related to the development of MCC-based RFBs for large-scale energy storage implementations.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
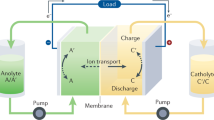
The adoption of renewable energy sources, such as solar and wind energy, provides an important opportunity to address environmental issues associated with fossil fuels while satisfying the increasing energy demand [1]. However, due to the intermittent nature of these renewable energy sources, energy storage devices are necessary to integrate these sources into smart electricity grids [2]. Among various energy storage technologies, redox flow batteries (RFBs) hold greater potential for long-duration large-scale energy storage applications, owing to their attractive features of decoupled power and energy, high safety, long lifetime, and good scalability [3,4,5,6,7,8,9,10,11]. In an RFB, the electroactive species are stored as a solution in an outer tank and pumped through the electrode interface, where chemical/electrochemical energy conversion occurs (Fig. 1). RFBs are broadly classified as aqueous and nonaqueous types, depending on the medium. One of the most important components of RFBs is the electroactive material. Traditional electroactive materials for RFBs include inorganic metal ions that have been actively studied (e.g., vanadium RFBs, Zn-Br2 and Fe-Cr RFBs) [4, 12,13,14,15,16,17,18,19,20]. There has been considerable interest in the development of abundant, sustainable, and highly structurally tailorable organic electroactive materials for RFBs [21,22,23,24,25,26,27,28,29,30,31,32,33]. Several comprehensive reviews have summarized the progress related to these materials [15, 27, 34,35,36,37,38,39,40,41,42,43,44,45,46].

Schematic illustration of an RFB system
Another class of redox-active species with excellent potential for achieving high energy density and long battery lifetime includes metal coordination complexes (MCCs), which possess the properties of both metal ions and organic ligands [37, 47,48,49,50]. By varying the organic ligand scaffold and metals, it is feasible to tune the properties of these complexes, including the solubility and redox potential. MCCs can undergo multielectron transfer redox reactions due to the various oxidation states of the metal center. The crossover of the active species through the membrane is reduced owing to the larger molecular size of MCCs. The development of RFBs that adopt MCCs as redox-active species has been partially examined in several reviews [37, 38, 40, 47, 49, 51,52,53]. For instance, Yu et al. [51, 52] summarized the molecular engineering of organometallic molecules for organic redox flow batteries (ORFBs). Liu et al. [38] discussed the relationship between the physicochemical properties of redox-active species and battery performance. Davis et al. [53] presented a comparative review of metal-based charge carriers in nonaqueous RFBs (NARFBs). Fu et al. [49] reviewed the application of organometallic complexes in rechargeable battery systems such as Li, Na, Zn, Li-S, Li-O2, and RFBs. Notably, in 2019, Toghill et al. [47] reviewed the performance of MCCs for nonaqueous flow batteries. While more progress has been achieved in recent years, there appears to be no account comprehensively summarizing the broad field of MCC-based aqueous and nonaqueous RFBs.
In this review, we present a comprehensive review of the development of RFBs that employ MCCs as redox-active materials in both aqueous and nonaqueous mediums. The main goal is to reveal the structure-performance relationship of MCC systems. Since the electrochemical properties of MCCs are considerably influenced by the ligand, the ligand development is a primary focus in this field. Hence, emphasis is placed on introducing the ligand selection and modification strategies used to tune the critical properties of MCCs, including the redox potential, solubility, cycling stability, and reversible electron transfer redox reactions to achieve stable cycled RFBs with a high energy density. We begin by presenting the structure and performance metrics of an RFB. Then, the progress of MCCs in RFBs, including the solubility, redox potential, and battery cycling performance, is accordingly summarized according to different ligand types. The key challenges and research perspectives of MCC-based RFBs are also discussed.
2 The Architecture and Performance Metrics of Redox Flow Batteries
An RFB typically consists of external electrolyte storage tanks, pipes, pumps, and electrochemical cells, as shown in Fig. 1. The cell consists of two electrode compartments and a separator. The electrodes serve as active sites for redox reactions of dissolved redox-active species, while the separator enables the movement of charge carriers (ions) and prevents the crossover of electroactive species between half-cells. Electrolytes are pumped from the outer tanks and flow through the electrochemical cell to undergo electrochemical reactions [10, 52, 54].
The performance of an RFB can be described by parameters such as energy density, power density, Coulombic efficiency (CE), voltage efficiency (VE), energy efficiency (EE), and capacity retention (CR) and utilization [35]. The energy density is recognized as the primary and the most important parameter, as described in Eq. (1):
where n represents the number of electrons transferred in the reaction, C is the lower concentration of either the catholyte or anolyte, F is Faraday’s constant, V is the working voltage of the flow battery, and μv is the volume factor [μv = 1 + (lower electrolyte concentration)/(higher electrolyte concentration)]. This equation indicates that the energy density of an RFB is determined by the solubility of redox-active species, the cell voltage, and the number of electrons involved. Another important battery performance metric is power density, which is the amount of power per unit area that a battery can supply. It can be calculated by using Eq. (2), where I is the discharge current, V is the output potential, and S is the active surface area of the separator.
CE, VE, and EE are also vital parameters for evaluating RFB performance. CE is the ratio between the discharge capacity and the charge capacity, which can be affected by the crossover of redox species and irreversible side redox reactions. VE is the ratio between the average discharge and charge voltages. The mean discharge voltage can be reduced due to a variety of overpotentials, including ohmic polarization, activation polarization, and concentration polarization. EE equals CE times VE, which is the ratio between the discharge energy and the charge energy. CR, which reflects the cycling stability of a battery, is expressed as either a total CR percentage after an extended cycling test or an average maintained capacity percentage per cycle. Capacity utilization is the ratio between the practically achieved capacity and the theoretical capacity of a battery and is mainly affected by the current density, battery resistance, and the state of charge. Notably, the battery performance parameters in an RFB are predominantly decided by the redox-active materials. To obtain an RFB with high performance (e.g., high energy density, efficiency, and power density), the redox-active species must have a high solubility, reasonable redox potential, and sufficient stability in the redox reactions.
3 Metal Coordination Complexes for RFBs
3.1 General Properties and Classification of Metal Coordination Complexes
A metal complex is composed of a central metal atom or ion that is bonded to ligands, which share electrons with the metal center. The redox reactions of MCCs depend on the electronic nature of the metallic center and ligands. In general, the redox process of a transition metal coordination complex with innocent ligands involves only the oxidation or reduction of the metal center, while complexes bearing noninnocent ligands (e.g., dithiolenes) can undergo both metal-based and ligand-based redox reactions [55, 56]. Metal coordination complexes are commonly classified into metal-ligand complexes and metallocenes based on the ligand types [38, 51, 52]. Both metal ligand complexes and metallocenes can be categorized into aqueous and nonaqueous systems depending on the solvent. In the following sections, we describe the complex structure in detail and provide the main performance metrics, including the solubility, redox potential, and battery performance, for each type of MCCs.
Interest in metal complexes for RFBs stems from their high structural tunability, multielectron configurations and superior stability in various redox states, and reduced species crossover [19, 24, 47]. The judicious choice of metals and incorporated ligands is important for optimizing the redox potential and solubility of these complexes. In 1982, Matsuda et al. reported an [Ru(bpy)3]2+ (bpy = bipyridine) complex as a redox-active species in an NARFB for the first time [57]. Since then, MCCs with various metal ion centers and ligands have been extensively studied for RFBs. For the sake of easy presentation, we group these metal complexes according to ligands in the following discussion of the main performance metrics, including solubility, redox potential, and battery performance. Figure 2 shows typical ligands, including metallocene, bipyridine, tridentate N-donor ligands, acetylacetonate (acac), dithiolene, aminopolycarboxylic acid (APCA), triethanolamine (tea), and other organic ligands.

Typical metal coordination complexes in RFBs
3.1.1 Complexes with Bipyridine Ligands
Bipyridine ligands are among the earliest studied MCCs for RFBs due to their commercial availability [58,59,60,61,62]. Scheme 1 shows the structures of the studied complexes bearing pyridine derivatives, and Table 1 summarizes their redox potential, solubility, and battery performance. Importantly, bipyridine complexes have been explored mostly for nonaqueous systems using organic solvents such as acetonitrile (MeCN) and propylene carbonate (PC), with reported solubility ranging from 0.2 to 0.8 mol L−1. The electrochemical properties of [M(bpy)3]n+ have been studied in detail for complexes with metals such as Ru, Co, Fe, Cr, and Ni. These complexes typically exhibit multielectron redox reactions. The typical redox features of the complexes are shown in Fig. 3. The chromium bipyridine complex [Cr(bpy)3]2+ gives rise to up to six redox waves from − 2.81 to − 0.56 V. Complexes of the [M(bpy)3]n+ type have been explored as catholytes, anolytes, and bipolar species. Fe-bipyridine complexes are commonly used as catholytes, while Ni-, Cr-, and Co-bipyridine complexes are usually used as anolytes. For instance, Mun and coworkers studied the charge-discharge properties of a flow battery employing tris(2,2ʹ-bipyridine)iron(II) tetrafluoroborate [Fe(bpy)3(BF4)2] (the 1a in Table 1) and tris(2,2ʹ-bipyridine)nickel(II) tetrafluoroborate [Ni(bpy)3(BF4)2] (the 2a in Table 1) as the catholyte and anolyte, respectively [63]. A CV experiment revealed a one-electron oxidation process of FeII/FeIII and a two-electron reduction of NiII/Ni0 at 0.65 and − 1.66 V (vs. Ag/Ag+), respectively. The demonstrated cell had a cell voltage of 2.2 V. Co(II) polypyridine complexes (the 3a–e in Table 1) can produce open-circuit voltages close to 2 V when paired with Fe(II) polypyridine complexes (the 1e–g in Table 1) [60]. As these bipyridine complexes usually exhibit multielectron redox reactions with a large redox potential gap, they can be adopted as single active species in both the anode and cathode to construct symmetric flow cells. For example, Fe(bpy)3(BF4)2 (the 1a in Table 1) and Ru(bpy)3(BF4)2 (the 2d in Table 1) produce open-circuit cell voltages of 2.4 and 2.6 V, respectively.

Structures of selected bipyridine complexes

Redox potentials of [M(bpy)3]n+ complexes in nonaqueous electrolytes
It is viable to modify bipyridine ligands by anchoring functional substituents to optimize the redox potentials and electrochemical stability of metal complexes [64]. Sanford et al. found that incorporating perfluorinated substituents or long aliphatic chains onto bipyridine led to the formation of complexes with irreversible redox behavior and decreased electrochemical stability. Anderson and coworkers demonstrated that the redox potentials of Fe(bpyR)3(BF4)2 complexes can be tuned by varying the substituents (R = –CF3, –CO2Me, –Br, –H, –tBu, –Me, –OMe, –NH2). For example, the redox potentials of Fe(bpy-OMe)3(BF4)2 and Fe(bpy-CO2Me)3(BF4)2 are 0.49 and 1.08 V, respectively. Subsequent symmetric and asymmetric RFB tests have indicated that the unsubstituted complex Fe(bpy)3(BF4)2 exhibits better battery performance in terms of efficiency and CR than the complexes bearing –OMe, –CF3, and –CO2Me substituents (the 1h–j in Table 1) [61].
In addition to their electrochemical properties, the solubility of bipyridine complexes is also a crucial factor for RFBs. To optimize the solubility of bipyridine complexes, Sanford et al. conducted a systematic investigation of the solubility and electrochemical properties of a series of [Cr(bpyʹ)3]3+ (e.g., the 4a–d in Table 1) and [Cr(bpyʹ)3]0 complexes of bipyridine ligands with different substituents [64]. Incorporating alkoxy substituents into bipyridine enhances its solubility. Depending on the ligands, the solubility values of the Cr3+ complexes range from 0.05 to 0.71 mol L−1, while those of the Cr0 complexes range from < 0.001 5 to 210 mmol L−1. Neutral Cr0 compounds are generally less soluble than the corresponding cationic Cr3+ species. In a symmetric H-cell test, the complex 4d in Table 1 exhibited two single electron transfer redox processes in the charging segment, implying its ability to show multielectron transfer redox reactions.
In addition to the substituents on the bipyridine ligand, the counter anions of bipyridine complexes can also affect solubility. For example, Lee et al. evaluated the effect of counter anions, including BF4−, bis[(trifluoromethyl)sulfonyl]azanide (TFSI−), and bis(fluorosulfonyl)imide (FSI−), on the solubility of Fe-bipyridine and Ni-bipyridine complexes and found that bulky counter anions increased the solubility of the metal complexes [59]. Fe(bpy)3(TFSI)2 (the 1b in Table 1) exhibited the highest solubility of 0.75 mol L−1 in propylene carbonate (PC), while Fe(bpy)3(BF4)2 (the 1a in Table 1) and Fe(bpy)3(FSI)2 (the 1c in Table 1) showed lower solubility values of 0.71 and 0.53 mol L−1, respectively. The complex 1b in Table 1 was used to construct a symmetric H-cell with a cell voltage of 2.3 V.
As analogs of bipyridine complexes, phenanthroline complexes have similar electrochemical properties and are potentially useful for NARFBs [65]. An asymmetric NARFB with a cell voltage of 1.45 V was successfully assembled by using [Fe(phen)3]2+/3+ (the 5a in Table 1) and [Co(phen)3]1+/2+ (the 5b in Table 1) as the catholyte and anolyte, respectively [66, 67].
There have been only a few reports on ARFBs that use water-soluble bipyridine complexes as redox-active materials. For example, Chen et al. reported asymmetric ARFBs with an [Fe(bpy)3]SO4 complex (the 1k in Table 1) as the catholyte and methyl viologen (MV) as the anolyte [71]. The complex exhibited a solubility of 0.1 mol L−1 in NaCl aqueous solution, and the flow battery had a voltage of 1.4 V, with a CE of 99.8% and a CR value greater than 99.9% per cycle within 215 cycles. Remarkably, Li et al. developed asymmetric iron complexes bearing 2,2′-bipyridine-4,4′-dicarboxylic (Dcbpy) acid and cyanide ligands (Fig. 4) [72]. The Na4[FeII(Dcbpy)3] complex bearing a bipyridine ligand with a carboxylic substituent (the 1l in Table 1) had a solubility of 0.26 mol L−1 in H2O and a high redox potential of 0.95 V vs. Ag/AgCl. By introducing the –CN ligand to break the symmetry of the complex, the asymmetric complex M4[FeII(Dcbpy)2(CN)2] exhibited an increased solubility of 1.22 mol L−1, which was approximately 4.2 times higher than that of M4[FeII(Dcbpy)3]. As shown in Fig. 4, the fabricated RFB employing 0.1 mol L−1 Na4[FeII(Dcbpy)2(CN)2] as the catholyte and 1,1′-bis(3-sulfonatopropyl)-4,4′-bipyridinium (SPr-Bpy) as the anolyte delivered a cell voltage of 1.2 V and excellent cycling performance for 6 000 cycles, with a capacity fading rate of 0.001 58% per cycle.

Reproduced with permission from Ref. [72]. Copyright © 2021, Springer Nature
a Synthesis of the asymmetric Na4[FeII(Dcbpy)2(CN)2] and Na4[FeII(Dcbpy)(CN)4] complexes. b, c Ratability test and discharge capacity, CE, VE, and EE of 0.1 mol L−1 SPr-Bpy anolyte and 0.1 mol L−1 Na4[FeII(Dcbpy)2(CN)2] catholyte under different current densities. d, e Representative charge/discharge profiles of extended cycles and charge/discharge capacity, CE, VE, EE, and long-term cycles for flow cells with 0.1 mol L−1 SPr-Bpy as the anolyte and 0.1 mol L−1 Na4[FeII(Dcbpy)2(CN)2] as the catholyte.
3.1.2 Complexes with Tridentate N-Donor Ligands
Complexes with tridentate N-donor ligands are attractive electroactive materials because of their high stability resulting from the chelating effect. Numerous tridentate N-donor ligands have been used to construct MCCs for RFBs. Common examples of such ligands include derivatives of terpyridine ligand [73], diazole-pyridine [74], pyrazole-pyridine mixed ligand [68], bipyridylimino isoindoline (BPI) [75], N-(quinoline-8-yl)pyridine-2-sulfonamido [76], and pyridine-2,6-diimine [77, 78]. Scheme 2 shows the structures of selected examples of such complexes. Table 2 presents the redox potential, solubility, and cycling properties of these complexes.

Examples of complexes with tridentate N-donor ligands
Terpyridine complexes of Fe [73], Co [74], and Ni [79] also exhibit multielectron redox properties and have been commonly applied in NARFBs, similar to bipyridine complexes. For instance, in 2017, Toghill’s group synthesized a series of cobalt(II) complexes with tridentate terpyridine, diazole-pyridine, and pyrazole-pyridine ligands (the 6a, 7b, 8b, 9 in Table 2) [74]. They found that these cobalt complexes displayed high tunable solubility varying from 0.18 to 0.51 mol L−1 and a tunable potential difference from 1.07 to 1.91 V. Charge-discharge tests in an H-type cell delivered a high CE of 89.7%–99.8%, VE of 70.3%–81.0%, and EE of 63.1%–80.8%. Yang et al. [68] also investigated the battery performance of the 7a, 8a, and 8b in Table 2 bearing pyrazole-pyridine mixed ligands. Notably, the complex 8a in Table 2 exhibited the stablest CR of about 98% after 300 cycles in a symmetric coin cell, reflecting the stable cycling of the Co complexes.
McKenzie et al. developed a cobalt-N-(quinoline-8-yl)pyridine-2-sulfonamido complex (the 10 in Table 2), which exhibited much more negative potentials for the CoII/CoI and CoII/CoIII redox processes than the terpyridine analog. By using this complex as a bipolar species in an H-cell setup, a large Ecell of 1.75 V was achieved [76]. Sevov and their coworkers attempted to develop BPI-based complexes (the 11a–f in Table 2) for NARFBs [75]. It was revealed that MCCs with metals such as Mn and Fe are more polar and can exhibit higher solubility than those of Zn and Ni in polar solvents such as CH3CN. In bulk electrolysis, the two-electron-reduced forms of Ni (the 11e in Table 2), Co (the 11d in Table 2), and Fe (the 11c in Table 2) complexes exhibited no detectable loss of stored capacity over 50 charge/discharge cycles to 100% SOC. In contrast, complexes of early transition metals showed very rapid capacity fade. The nickel complex offered good stability at an electrolyte concentration of 0.1 mol L−1 without detectable precipitation or degradation in the charged state for days.
Bis(2,6-diimine-pyridine)-based complexes (the 12 and 13 in Table 2) have been adopted as electroactive materials for both aqueous and nonaqueous RFBs, despite their poor cycling stability [77, 78, 80]. Wang et al. demonstrated a 0.93 V symmetric ARFB with the water-soluble bis(pyridine-2,6-diimine) cobalt(II) complex 13 in Table 2 [80]. By limiting the charge capacity to 41% of the theoretical value, the battery with 1 mmol L−1 active species delivered moderate efficiencies (VE = 73.2%, EE = 73.5%, and CE = 99.0%) and a CR rate of 89% after 100 cycles. Herbert demonstrated a symmetric H-cell with a bis(pyridine-2,6-diimine) iron(II) complex (the 12a in Table 2), with a cell voltage of 2.49 V and a CE of 99.9% [77].
3.1.3 Complexes with Acetylacetonate and Related Ligands
Transition metal complexes with acetylacetonate (acac) ligands constitute another class of MCCs that have been extensively studied for RFBs. Typical molecular structures are shown in Scheme 3. Acetylacetonate complexes, in general, exhibit multiple electron transfer processes across a broad voltage window, making acetylacetonate complexes promising bipolar species for symmetric NARFBs. For instance, the complex V(acac)3 (the 14b in Table 3) shows two redox couples at − 1.75 and 0.45 V. In 2009, Chakrabarti demonstrated a symmetric H-cell with a cell voltage of 1.77 V by using a ruthenium complex Ru(acac)3 as the redox-active species [81]. Subsequently, the electrochemical and charge-discharge properties of acac complexes of V, Mn, Fe, and Cr were investigated [82,83,84,85,86,87,88]. In particular, vanadium acetylacetonate [V(acac)] complexes have been widely studied in areas such as electrochemistry on different electrode materials [89], battery performance with various organic solvent mediums [90], and cycling performance in batteries with different membranes [91]. However, the decay of the capacity was fast, mainly due to the poor chemical stability of the vanadium acetylacetonate in the catholyte during battery cycling.

Structures of selected complexes with acetylacetonate and related ligands
Suttil et al. [92] conducted a systematic study of a series of V and Cr complexes bearing substituted acac ligands (Fig. 5) to investigate the impact of the chemical structures on the electrochemical properties, solubility, and battery performance. Their study revealed that the solubility of the complexes in acetonitrile ranges over four orders of magnitude (from 5.7 × 10−5 to 1.92 mol L−1) depending on the substituents of the acac ligands. Incorporation of polar functional groups, such as ester, ether, and amine to acac ligands, significantly improved the solubility of the complex to up to 1.92 mol L−1 with minimal impact on the electrochemical properties. In a related study, Kucharyson et al. [93] developed descriptors to predict the standard potentials, solubility, and cycling stability by using density functional theory calculations. Their study suggested that the experimentally determined stability was related to the percentage of the highest occupied molecular orbital (HOMO) or lowest unoccupied molecular orbital (LUMO).

Reproduced with permission from Ref. [92]. Copyright © 2015, Royal Society Chemistry
a Chemical structures of selected functionalized CrIII and VIII acetylacetonate complexes. b CV of representative CrIII and VIII acetylacetonate complexes in 0.1 mol L−1 TBABF4/MeCN. c Solubility of selected CrIII and VIII acetylacetonate complexes in MeCN.
In addition to acac complexes of V, Mn, Fe, Ru, and Cr, the charge-discharge characteristics of the cobalt(II) complex (the 16 in Table 3) bearing an acetylacetonate-like bis(acetylacetone)ethylenediamine ligand have been evaluated in an H-type nonaqueous symmetric cell by Li et al. This cell delivered a cell voltage of 2.0 V and a CE of 90.2%.
3.1.4 Complexes with APCA Ligands
Aminopolycarboxylic acid (APCA) reagents such as ethylenediaminetetraacetic (EDTA) and diethylenetriaminepentaacetic acid (DTPA) constitute an important class of industrial chelating agents that can form water-soluble di- and trivalent metal complexes for use in ARFBs (Scheme 4 and Table 4) [94]. The electrochemical redox properties of transition metal complexes of APCA have been well documented, especially those of Cr, Fe, Mn, and Ce complexes.

Structures of selected APCA metal complexes
APCA complexes have been explored as redox-active materials for RFBs. In 2002, Bae et al. [95] reported the application of chromium-EDTA complexes (the 17b in Table 4) in ARFB. After complexation with an EDTA chelating agent, the Cr(III)/Cr(II) redox couple exhibited an enhanced reaction rate, and the complex underwent a reversible Cr(III)/Cr(II) redox reaction at − 1.24 V vs. SCE. The battery performance in an H-type glass cell confirmed that the oxidized chromium-EDTA species was stable. Later, in 2011, Bae et al. [96] further studied the cycling performance of Cr(III)-EDTA. However, the EE was less than 5% because of the slow kinetics of the Cr(V)-EDTA/Cr(III)-EDTA redox couple.
To improve the limited battery performance caused by the slow kinetics of the Cr(V)-EDTA/Cr(III)-EDTA redox couple, Marshak et al. [97] proposed the use of the PDTA-chromium complex 18 in Table 4 (PDTA = 1,3-propylenediaminetetraacetic acid) for RFB applications. As shown in Scheme 4, this complex has a nearly octahedral geometry with no vacancy for H2O coordination, a more negative reduction potential, and improved electrode kinetics on a glassy carbon electrode. In a flow battery test, a battery with Cr-PDTA (1 mol L−1) as the anolyte and Fe(CN)64− as the catholyte delivered a 1.62 V cell voltage, nearly 100% CE, and 78.1% EE. When paired with the Br2/Br− redox couple, the resulting Cr-PDTA RFB possessed a high voltage of 2.13 V and showed negligible H2 generation or capacity loss. The isolated reduced Cr(II)-PDTA species had the same pseudo-octahedral structure as Cr(III)-PDTA without the coordination of water [98]. Importantly, no chemical decomposition of the Cr(III) or Cr(II) species was detected upon exposure to atmospheric O2. In addition to Cr-APCA complexes, Fe-APCA electrolytes have also been explored. A fabricated flow battery with the Fe-EDTA complex 17a in Table 4 as the anolyte and Br− as the catholyte delivered an open-circuit voltage of 1.04 V and an EE of approximately 80% at a current density of 10 mA cm−2 [99]. However, it was noted that the capacity loss was rapid because the 7-coordinated species [Fe(H2O)-EDTA]1− formed a dimer species in mildly alkaline conditions, thus decreasing the redox reversibility.
To address the issue of capacity loss caused by the formation of Fe(OH)3 or an oxo-bridged dimer complex, Waters et al. proposed using a DTPA ligand that contains 5 carboxylate anions and 3 neutral nitrogen atoms that can bind metals with up to 8 coordination bonds. The steric effect induced by the ligand prevented the binding of both H2O and OH−, hindering the formation of Fe(OH)3 or an oxo-bridged dimer complex [100]. A flow battery with a high concentration of 1.15 mol L−1 K2FeDTPA (the 19a in Table 4) as the catholyte and Cr-DTPA as the anolyte demonstrated an average CE of 100.1% ± 0.1% and a discharge capacity of 28.49 Ah L−1 after 40 cycles, corresponding to a capacity loss of 0.596% per day (0.029% per cycle). The postmortem analysis showed that no decomposition or membrane crossover of Fe-DTPA occurred, indicating good cycling stability of Fe-DTPA species. The analogous K2VDTPA exhibited a redox potential as low as − 1.2 V vs. Ag/AgCl and a solubility greater than 1.3 mol L−1. When paired with the Fe-DTPA posolyte, the maximum discharge energy density of the near neutral RFB reached 12.5 Wh L−1 [100]. The charge/discharge properties of the iron complex 21 in Table 4 bearing an ethylene-bis-(o-hydroxyphenylglycine) ligand have been examined in bulk electrolysis tests with a low electrolyte concentration [101]. Ce and Mn complexes with aminopolycarboxylate ligands such as Ce-EDTA (the 17c in Table 4) [102, 103], Ce-DTPA (the 20 in Table 4) [104], and EDTA-Mn (the 17d in Table 4) [105] have been employed as catholytes in RFB applications due to their high redox potentials.
3.1.5 Complexes with Dithiolene Ligands
Dithiolene ligands are well known for their noninnocent behavior [56]. The application of noninnocent dithiolene-based complexes in NARFBs can be traced back to 2014 [107]. Cappillino et al. reported the use of tris(mnt)V(IV) (the 22 in Table 5) (mnt2− = maleonitriledithiolate, Scheme 5) in a nonflowing cell. In the CV test, [V(mnt)3]2− exhibited three reversible redox couples corresponding to the 4−/3−, 3−/2−, and 2−/1− redox reactions at − 1.41, − 0.227, and 0.856 V vs. SHE. The static symmetric H-cell charge/discharge experiment yielded a cell voltage of 1.09 V, 90% CE, and 20% VE.

Structures of complexes bearing dithiolene ligands and triethanolamine ligands
Hogue et al. expanded the use of dithiolate complexes in flow cell experiments and tested dithiolene complexes (TEA)[Fe(mnt)2], (TEA)2[V(mnt)3], (TEA)2[Co(mnt)2], (TEA)2[Ni(mnt)2], and (TEA)2[Cu(mnt)2] (the 23a–d in Table 5) as bipolar species for NARFBs (Table 5) [55]. These complexes exhibited moderate solubility in the range of 0.30–0.90 mol L−1 in MeCN, except for (TEA)[Fe(mnt)2], which had poor solubility of 0.03 mol L−1 in MeCN. The V, Co, Ni, and Cu complexes exhibited reversible redox events, fast diffusion coefficients and electrochemical rate constants. In the symmetric RFB test, [(TEA)2Vmnt] showed a high VE of 95% and an EE of 60% and could be stably charged/discharged for up to 100 cycles.
3.1.6 Complexes with Triethanolamine Ligands
Triethanolamine is a chelating agent that is commonly used in preparing metal complexes due to its tetradentate chelating character, commercial availability, and low cost [115]. Fe complexes, in particular, have attracted considerable research attention for their use in electrochemical deposition, hydrogen production, and flow battery applications. As summarized in Table 5 and Scheme 5, the triethanolamine complexes of Fe and Co have shown encouraging performance in AORFBs. Owing to its low redox potential (− 1.05 V vs. Ag/AgCl) and fast charge transfer rate, Fe(tea) and its derivative complexes (the 24 in Table 5) have been used as anolytes in basic aqueous mediums to assemble RFBs upon pairing with catholytes such as Br2, K4Fe(CN)6, Co(tea) (the 25a in Table 5), and Co(meta-tea) (the 25d in Table 5) [113,114,115,116,117].
The bonds between the triethanolamine ligand and Fe ions are weak and can break easily, causing the conversion of Fe ions into Fe(s) with released hydrogen gas during the cycling test. To solve this problem, Kwon et al. [117] introduced 3-[bis(2-hydroxyethyl) amino]-2-hydroxypropanesulfonic acid (DIPSOH3), which can form a stronger coordinating bond. By using a 0.5 mol L−1 Fe-DIPSO complex (the 24b in Table 5) as the anolyte and K4Fe(CN)6 as the catholyte, an RFB performed better, with a discharge capacity of 14.4 Ah L−1, a power density of 93.2 mW cm−2, a CE of 95%, and a decay rate of 0.12% per cycle over 100 cycles. The cycling stability of the same system was enhanced by adjusting the pH and the concentrations of both electrolytes [110]. Additionally, they also tested an RFB with Fe(DIPSO)/Co(DIPSO), which showed a capacity loss rate of 0.025 Ah L−1 per cycle [109].
Although the DIPSO complex 24b in Table 5 has an stronger coordinating bond, it is expensive and has a moderate redox potential. To address these issues, Kwon’s group introduced an Fe complex (the 24c in Table 5), which is more durable and cost-effective. This complex exhibited a low redox potential of − 1.11 V (vs. Ag/AgCl) and a high solubility of 1.33 mol L−1 in 1 mol L−1 KOH solution. The fabricated RFB (on pairing with K4[Fe(CN)6]) showed an average capacity of 11.7 Ah L−1, an EE of 72.8%, a CE of 99.8%, and a CR rate of 99.9% within 250 cycles.
3.1.7 Complexes with Other Ligands
A considerable number of other MCCs have also been studied for RFBs, such as dipicolinic acid-chromium complexes [118], azamacrocyclic iron complexes [119], imino-pyridine iron [120], bis-trimetaphosphate cobalt and vanadium complexes [121], bis-hydroxyiminodiacetate vanadium [122], nickel(II)-1,4,8,11-tetrathiacyclotetradecane [123], Ni(II)-chelated tetradentate azamacrocyclic complexes [124], nickel bispicolinamide complexes [125, 126], and cerium-containing complexes [127]. The structures, redox potentials, solubility, and cycling properties of these complexes are presented in Scheme 6 and Table 6.

Structures of complexes 26–35
The chromium complex 26 in Table 6 bearing a dipicolinic acid ligand has a solubility of 0.7 mol L−1 in water and has a reversible reduction wave at − 0.71 V vs. SHE. When paired with the ferrocyanide catholyte K4[Fe(CN)6], the resulting neutral asymmetric ARFB exhibited a cell voltage of 1.07 V [118]. The iron complex of the imino-pyridine ligand (the 28 in Table 6) underwent multiple reversible redox reactions over a wider voltage window [120, 128] and displayed two reversible reduction events and one oxidation event with redox potentials of − 1.79, − 1.51, and 0.78 V, respectively. The fabricated symmetric RFB exhibited a cell voltage of 2.29 V with 93% CE, 84% EE, and a capacity decay rate of 0.61% per cycle at 5 mA cm−2.
An NARFB (in MeCN) that utilizes [CoII/III(P3O9)2]4−/3− (the 29a in Table 6) as the catholyte and [VIII/II(P3O9)2]3−/4− (the 29b in Table 6) as the anolyte was reported by Stauber et al. [121]. The chelation of the trimetaphosphate ligand provided the advantages of unfavorable ligand dissociation, good oxidative stability, and minimal crossover of active species. The solubility of the metaphosphate complexes in polar organic mediums was improved by pairing with the highly lipophilic PPN+ [bis(triphenylphosphine)iminium] cation. The assembled static H-cell showed a high cell voltage of 2.4 V and a CE greater than 90% over 100 cycles. Vanadium bis-hydroxyiminodiacetate (the 30 in Table 6) has also been explored as a new electrolyte [122]. Oh’s group investigated the electrochemical and cycling performance of nickel(II)-1,4,8,11-tetraazacyclotetradecane (the 31 in Table 6) [129] and nickel(II)-1,4,8,11-tetrathiacyclotetradecane (the 32 in Table 6) [123] in symmetric RFBs. Davis et al. proposed a series of picolinamide nickel complexes (the 33 in Table 6) with a large redox potential separation between positive and negative waves as redox carriers for NARFBs [125, 126]. Cerium-containing complexes (the 34 and 35 in Table 6) have also been adopted to construct high-cell-voltage NARFBs. These results indicated that Ni and Ce complexes are promising catholytes for assembling RFBs with high cell voltages due to their high cathodic redox couples, although the battery performance, including their efficiencies and CR, is uncompetitive with that of other commonly employed complexes.
In addition to the aforementioned work on soluble complexes, there have been studies exploring the applications of polymeric coordination complexes in RFBs in the form of nanoparticles [130, 131]. For example, Carretero-González’s group synthesized 1D coordination polymers of Mn and Fe by using chloranilic acid (CA-H) as the organic ligand. These polymers formed stable slurries containing up to 100 g L−1 solid particles in aqueous mediums. The 1D iron coordination polymer {[Fe(CA)(H2O)2]H2O}n was used as anolyte and catholyte in a symmetrical cell by using a low-cost size exclusion cellulose membrane as the separator. The assembled cell delivered a reversible capacity value of 225 mAh L−1 at a current density value of 20 mA g−1 for 50 cycles (~ 12 days) at neutral pH. In addition, as a unique type of metal-oxygen cluster formed by covalently linking transition metal atoms and oxygen atoms, polyoxometalates (POMs) exhibit appealing features, including multiple electrons, reversible redox capabilities, and low permeability for RFBs [132, 133]. Three main types of structures, namely Keggin, Dawson, and Lindqvist types, have been applied in RFBs. For instance, Friedl et al. demonstrated an RFB with the Keggin-type POM materials, which utilized [SiW12O40]4− as the anolyte and [PV14O42]9− as the catholyte [132]. EIS studies revealed a high electron transfer of 1 × 10−2 cm s−1 for [SiW12O40]4−, and the assembled RFB could also be stably cycled for 14 days with a CE of 94%. Although low-temperature studies on ARFBs are limited, Lu and coworkers reported a class of heteropoly acid negolyte HPOMs with exceptionally fast redox kinetics (6.8 × 10−3 cm s−1) and a low freezing point (− 35 °C) for low-temperature RFBs [133]. The HPOM-vanadium RFB demonstrated a high EE of 81.82% at 100 mA cm−2 and stable cycling over 450 cycles at 300 mA cm−2 at room temperature. At low temperatures (− 20 °C), the RFB delivered a high capacity of 79.6 Ah L−1 at 160 mA cm−2 over 800 cycles (over 1 200 h) without capacity decay, demonstrating record stability and power density (282.4 mW cm−2) for low-temperature RFBs.
3.2 Metallocene-Based Complexes in RFBs
Metallocenes are among the most studied MCCs for RFBs, and they consist of two cyclopentadienyl (Cp) ligands bound to a metal center on opposite sides [52]. The chemical and coordination reactivity of metallocene is primarily determined by the p-orbitals in the Cp rings and d-orbitals of the metal center. The physical and electrochemical properties of metallocene complexes can be adjusted by incorporating functional groups on the Cp rings, making them suitable as electroactive materials for both nonaqueous and aqueous RFBs.
3.2.1 Metallocenes in NARFB Systems
Scheme 7 and Table 7 provide an overview of the structures and properties of typical metallocenes studied for NARFBs. Among various metallocenes, ferrocene (the 36a in Table 7, Fc) and its derivatives have garnered considerable interest for use in NARFBs due to their high electrochemical stability and reversibility, favorable redox potentials, and flexible structure tunability of physical and chemical properties [48]. In addition to ferrocene, cobaltocene (the 36b in Table 7) also shows appealing cycling properties as an anolyte in NARFBs.

Structures of selected metallocenes examined for NARFBs
The feasibility of Fc as a catholyte species was initially explored in an Li/Fc semiflow battery [134]. The ferrocene/ferrocenium redox couple displayed high electrochemical reversibility and stability in aprotic solvents, making it an appropriate charge storage catholyte. Subsequently, Hwang et al. demonstrated NARFBs with ferrocene and cobaltocene derivatives as active species. However, these metallocenes exhibited limited solubility of approximately 0.1–0.5 mol L−1 in MeCN or PC, precluding their practical applications.
To improve the solubility of ferrocene, Wang and coworkers synthesized the ferrocene derivatives 37 in Table 7 with an ionic-charged tetraalkylammonium pendant on the cyclopentadienyl ring and a counter anion, such as PF6− and TFSI−, resulting in enhanced solubility [135, 136]. Solvation chemistry studies using nuclear magnetic resonance (NMR) and density functional theory (DFT) calculations demonstrated that the ionic pendant intensified the interactions between the solvent and the complex, thereby significantly increasing the solubility. The positively charged tetraalkylammonium pendant also affected the redox potential, with the complex 37a in Table 7 showing a 0.23 V positive shift in the redox potential compared to the original ferrocene without deterioration of the redox reversibility. Intriguingly, counter anions can also significantly impact the solubility and electrochemical performance of ferrocenes [137]. Similar strategies involving functional groups on the Cp ring to adjust the redox potential, solubility, melting point, and molecular size have also been adopted by other groups. The Fc derivatives studied for NARFBs include Fc-COCH3 (the 38 in Table 7) [138], Fc-DIPP (the 39 in Table 7) [139], DMFc (the 40a in Table 7) [140,141,142], Fc-Br (the 40b in Table 7) [143], Fc-ODG (the 41 in Table 7) [144], [Fc4] (the 42 in Table 7) [145], and Fc-based polymer (the 43 in Table 7) [146].
Researchers have also developed ferrocene-based bipolar redox-active compounds by linking the ferrocene unit covalently to an anolyte component. Some examples of the reported bipolar molecules include ferrocene/fullerene (the 44 in Table 7) [147], ferrocene/phthalimide (the 45–47 in Table 7) [148,149,150], and ferrocene/anthraquinone (the 48 in Table 7) [151]. The battery performance of these compounds has been evaluated, with Xu et al. [150] reporting a series of bipolar redox-active molecules based on phthalimide and ferrocene moieties that show fast mass, charge transfer kinetics, and high solubility in MeCN. The fabricated symmetric flow battery delivered a stable cycling CR of 99.8% per cycle and an EE of 77.0% over 50 cycles at 20 mA cm−2.
3.2.2 Metallocenes in ARFB Systems
Scheme 8 and Table 8 summarize the structures and properties of typical metallocenes studied for ARFBs. Although unmodified ferrocene has minimal water solubility, it is possible to increase its solubility in aqueous solution by functionalizing the Cp ring with hydrophilic groups [152,153,154,155]. For instance, Liu and coworkers developed highly water-soluble (ferrocenylmethyl) trimethylammonium chloride (FcNCl, the 49 in Table 8) and N1-ferrocenylmethyl-N1, N1, N2, N2, N2-pentamethylpropane-1,2-diaminium dibromide (FcN2Br2, the 50 in Table 8) by introducing a strongly hydrophilic tetraammonium pendant to the cyclopentadienyl ring of ferrocene, as displayed in Fig. 6 [152]. When paired with MV anolyte, the fabricated RFB had a theoretical energy density of 45.5 Wh L−1 and delivered an CR rate of 99.99% per cycle after 700 cycles. The Aziz group synthesized bis[(3-trimethylammonio)propyl]-ferrocene dichloride (the 51 in Table 8), which exhibited a high solubility of 2 mol L−1 in H2O and realized an RFB [with 1.3 mol L−1 of the 51 in Table 8 paired with bis-(trimethylammonio)propyl viologen (BTMVP-Vi)] with an unprecedented stable CR of 99.994 3% per cycle. The ferrocene-based bipolar compound 52 in Table 8 containing a cathode ferrocene moiety and an anode bipyridinium moiety showed a solubility of 1.2 mol L−1 in water and delivered a symmetric RFB with a cell voltage of 0.7 V and impressive CR of 75% after 4 000 cycles. Chen et al. [156] developed six water-soluble ferrocene derivatives (the 53 in Table 8) functionalized with cationic tetraalkylammonium groups of varying chain lengths to investigate how the catholyte structure might impact battery lifetime. In addition, sulfonate-substituted ferrocenes such as the 54–57 in Table 8 have also shown promising cycling performance in ARFBs [157,158,159,160], as summarized in Table 8. Xu et al. [161] developed a strategy to obtain water-soluble Fc-based catholytes by simply mixing Fc derivatives and β-cyclodextrins (β-CDs) through host-guest inclusion. This approach has the potential to be applied to other families of redox-active metal complexes for AORFB applications.

Structures of metallocenes applied in ARFBs

Reproduced with permission from Ref. [152]. Copyright © 2015, American Chemical Society
a Synthetic steps of FcNCl and FcNBr2. b Cyclic voltammograms of MV, FcN, FcNCl, and FcNBr2. c Extended 700 cycling performance of the 0.5 mol L−1 FcNCl/MV AORFB at 60 mA cm−2. d Polarization and power density curves of the FcNCl/MV AORFB at 0.5 and 0.7 mol L−1.
Ferrocene derivatives with a leaving group at the α or β positions are potentially unstable at higher temperatures since they can undergo elimination and substitution reactions in a nucleophilic solvent [157, 162, 163]. Hence, Borchers et al. [164] developed a ferrocene-containing methacrylamide copolymer electrolyte (the 58 in Table 8) with a C3 spacer between the leaving group and ferrocene. A flow battery using the ferrocene-containing polymer as the catholyte and BTMAP-Vi as the anolyte demonstrated a stable cycling behavior over 100 cycles at ambient temperature and elevated temperatures (60 °C), with an average CE greater than 99.8% through all cycling experiments. Additionally, Luo and coworkers systematically studied the decay mechanism of the Fc catholytes 49a–c in Table 8 in AORFBs [155]. Both the half-cell and full-cell flow battery tests showed that the cycling stability was in the order of 49a < 49b < 49c.
4 Conclusions and Perspectives
The increasing demand for large-scale storage of renewable energy sources calls for low-cost and high-performance energy storage devices. RFBs have shown excellent promise in satisfying these requirements, particularly with the use of MCCs as energy-bearing materials. Significant progress has been made in the development of MCC-based RFBs. In aqueous systems, certain MCCs, such as ferrocenes, have demonstrated a solubility greater than 3 mol L−1 and stable cycling performance, retaining over 99% capacity retention after 500 cycles. In nonaqueous systems, batteries with a cell voltage as high as 3.4 V have been demonstrated despite most cell studies being conducted with a static H-cell configuration, and the long cycling stability was generally poor. Despite these advances, there are still critical challenges that need to be addressed in both aqueous and nonaqueous RFBs to enable practical large-scale applications.
The first challenge in MCC-based RFBs is to improve their energy density to compete with organic or inorganic RFB systems. The main reason for the lower energy density is the lack of suitable MCCs with high volumetric capacity and suitably low or high redox potential. Developing highly soluble MCCs at both charge and discharge states is crucial, and this can be achieved through strategies such as modifying ligands with hydrophilic/hydrophobic functional groups, optimizing the counterions, using asymmetric molecular structures, and introducing hydrotropes. It is also important to optimize the redox potential of MCCs to optimize the use of the stable electrochemical window of the solvents. The redox potential of MCCs is closely associated with the electron-donating or electron-accepting feature of organic ligands, as illustrated in Fig. 7 by the variation in redox potentials of iron complexes with ligands. The incorporation of electron-withdrawing and electron-donating groups on the ligand can also tune the redox potential of the complex. Effective utilization of MCCs that can undergo multielectron redox reactions could also help to maximize the energy density of RFBs.

Redox potential comparison of iron-based complexes with anionic and neutral ligands
The second challenge faced by MCC-based RFBs is the need to improve their long-term cycling stability. Most of the reported MCCs have been demonstrated at only low concentrations (mostly lower than 1.0 mol L−1) with inferior capacity retention rates. The stability of MCCs can be optimized by increasing the chelating effect between the ligand and metal ion center, thereby enabling a stabler cycling performance. Characterizing the cycled electrolyte using UV-Vis, NMR, CV, and liquid chromatography-mass spectrometry (LC-MC) techniques can help identify the degradation mechanisms of MCCs. This information can guide structural optimization and identify suitable supporting salt and solvent systems to reduce undesirable side reactions. The cycling stability of MCC-based RFBs is not only determined by the physicochemical properties of the active species but also influenced by the membranes, electrodes, and flow channels of the battery. It is important to develop membranes with high selectivity and conductivity, electrodes with high reaction activity, and flow channels with low-pressure drops to improve the long-term stability of the battery.
Reducing the capital cost is another challenge for MCC-based RFBs. To implement large-scale commercial RFB systems, low capital costs are needed, which mainly stem from the active materials, cell stack components (such as bipolar plates, membranes, and electrodes), and other expenses. The current cost of MCC materials is relatively high and not competitive with those of pure organic or inorganic counterparts. To address this issue, earth-abundant metal elements such as Fe, Co, and Mn can be adopted as redox-active metal centers, and more cost-effective routes for ligand production should be developed. Additionally, using inexpensive flow battery components, including membranes, electrodes, and other components, is equally important to reduce the capital cost of MCC-based RFBs.
Currently, research on MCC-based RFBs is flourishing, presenting both challenges and opportunities. MCCs constitute a promising class of active species, significantly extending the selection range of chemistry used in both aqueous and nonaqueous electrolytes. It is highly recommended to design and explore robust MCCs to facilitate the development of both aqueous and nonaqueous RFBs. This review aims to inspire more research on MCCs to overcome existing challenges, making RFB an environmentally benign, reliable, and cost-effective storage/conversion technology. With continued research and development, MCC-based RFBs have the potential to play a crucial role in meeting the increasing demand for large-scale energy storage in a sustainable and cost-effective manner.
References
Dunn, B., Kamath, H., Tarascon, J.M.: Electrical energy storage for the grid: a battery of choices. Science 334, 928–935 (2011). https://doi.org/10.1126/science.1212741
Alotto, P., Guarnieri, M., Moro, F.: Redox flow batteries for the storage of renewable energy: a review. Renew. Sustain. Energy Rev. 29, 325–335 (2014). https://doi.org/10.1016/j.rser.2013.08.001
Noack, J., Roznyatovskaya, N., Herr, T., et al.: The chemistry of redox-flow batteries. Angew. Chem. Int. Ed. 54, 9776–9809 (2015). https://doi.org/10.1002/anie.201410823
Park, M., Ryu, J., Wang, W., et al.: Material design and engineering of next-generation flow-battery technologies. Nat. Rev. Mater. 2, 1–18 (2017). https://doi.org/10.1038/natrevmats.2016.80
Skyllas-Kazacos, M., Chakrabarti, M.H., Hajimolana, S.A., et al.: Progress in flow battery research and development. J. Electrochem. Soc. 158, R55 (2011). https://doi.org/10.1149/1.3599565
Wang, W., Luo, Q.T., Li, B., et al.: Recent progress in redox flow battery research and development. Adv. Funct. Mater. 23, 970–986 (2013). https://doi.org/10.1002/adfm.201200694
Ye, R.J., Henkensmeier, D., Yoon, S.J., et al.: Redox flow batteries for energy storage: a technology review. J. Electrochem. Energy Convers. Storage 15, 010801 (2018). https://doi.org/10.1115/1.4037248
Huskinson, B., Marshak, M.P., Suh, C., et al.: A metal-free organic-inorganic aqueous flow battery. Nature 505, 195–198 (2014). https://doi.org/10.1038/nature12909
Lin, K.X., Chen, Q., Gerhardt, M.R., et al.: Alkaline quinone flow battery. Science 349, 1529–1532 (2015). https://doi.org/10.1126/science.aab3033
Soloveichik, G.L.: Flow batteries: current status and trends. Chem. Rev. 115, 11533–11558 (2015). https://doi.org/10.1021/cr500720t
Bartolozzi, M.: Development of redox flow batteries. A historical bibliography. J. Power Sources 27, 219–234 (1989). https://doi.org/10.1016/0378-7753(89)80037-0
Jiang, H.R., Sun, J., Wei, L., et al.: A high power density and long cycle life vanadium redox flow battery. Energy Storage Mater. 24, 529–540 (2020). https://doi.org/10.1016/j.ensm.2019.07.005
Zeng, Y.K., Zhou, X.L., An, L., et al.: A high-performance flow-field structured iron-chromium redox flow battery. J. Power Sources 324, 738–744 (2016). https://doi.org/10.1016/j.jpowsour.2016.05.138
Wu, M.C., Zhao, T.S., Wei, L., et al.: Improved electrolyte for zinc-bromine flow batteries. J. Power Sources 384, 232–239 (2018). https://doi.org/10.1016/j.jpowsour.2018.03.006
Pan, F., Wang, Q.: Redox species of redox flow batteries: a review. Molecules 20, 20499–20517 (2015). https://doi.org/10.3390/molecules201119711
Lourenssen, K., Williams, J., Ahmadpour, F., et al.: Vanadium redox flow batteries: a comprehensive review. J. Energy Storage 25, 100844 (2019). https://doi.org/10.1016/j.est.2019.100844
Wang, W., Sprenkle, V.: Redox flow batteries go organic. Nat. Chem. 8, 204–206 (2016). https://doi.org/10.1038/nchem.2466
Kowalski, J.A., Su, L., Milshtein, J.D., et al.: Recent advances in molecular engineering of redox active organic molecules for nonaqueous flow batteries. Curr. Opin. Chem. Eng. 13, 45–52 (2016). https://doi.org/10.1016/j.coche.2016.08.002
Wei, X.L., Pan, W.X., Duan, W.T., et al.: Materials and systems for organic redox flow batteries: status and challenges. ACS Energy Lett. 2, 2187–2204 (2017). https://doi.org/10.1021/acsenergylett.7b00650
Meda, L., Oldani, F., Tozzola, G., et al.: Searching for new redox-complexes\in organic flow batteries. Solid State Ion. 317, 142–148 (2018). https://doi.org/10.1016/j.ssi.2018.01.017
Liu, B., Tang, C.W., Jiang, H.R., et al.: An aqueous organic redox flow battery employing a trifunctional electroactive compound as anolyte, catholyte and supporting electrolyte. J. Power Sources 477, 228985 (2020). https://doi.org/10.1016/j.jpowsour.2020.228985
Liu, B., Tang, C.W., Jiang, H.R., et al.: Carboxyl-functionalized TEMPO catholyte enabling high-cycling-stability and high-energy-density aqueous organic redox flow batteries. ACS Sustain. Chem. Eng. 9, 6258–6265 (2021). https://doi.org/10.1021/acssuschemeng.0c08946
Winsberg, J., Hagemann, T., Janoschka, T., et al.: Redox-flow batteries: from metals to organic redox-active materials. Angew. Chem. Int. Ed. 56, 686–711 (2017). https://doi.org/10.1002/anie.201604925
Leung, P., Shah, A.A., Sanz, L., et al.: Recent developments in organic redox flow batteries: a critical review. J. Power Sources 360, 243–283 (2017). https://doi.org/10.1016/j.jpowsour.2017.05.057
Ding, Y., Yu, G.H.: Molecular engineering enables better organic flow batteries. Chem 3, 917–919 (2017). https://doi.org/10.1016/j.chempr.2017.11.010
Fischer, P., Mazúr, P., Krakowiak, J.: Family tree for aqueous organic redox couples for redox flow battery electrolytes: a conceptual review. Molecules 27, 560 (2022). https://doi.org/10.3390/molecules27020560
Luo, J., Wang, A.P., Hu, M.W., et al.: Materials challenges of aqueous redox flow batteries. MRS Energy Sustain. 9, 1–12 (2022). https://doi.org/10.1557/s43581-022-00023-1
Fan, H., Hu, B., Li, H.B., et al.: Conjugate-driven electron density delocalization of piperidine nitroxyl radical for stable aqueous zinc hybrid flow batteries. Angew. Chem. 134, e202115908 (2022). https://doi.org/10.1002/ange.202115908
Yang, L., Hao, Y.H., Lin, J.D., et al.: POM anolyte for all-anion redox flow batteries with high capacity retention and coulombic efficiency at mild pH. Adv. Mater. 34, e2107425 (2022). https://doi.org/10.1002/adma.202107425
Pan, M.G., Gao, L.Z., Liang, J.C., et al.: Reversible redox chemistry in pyrrolidinium-based TEMPO radical and extended viologen for high-voltage and long-life aqueous redox flow batteries. Adv. Energy Mater. 12, 2103478 (2022). https://doi.org/10.1002/aenm.202103478
Hu, B., Hu, M.W., Luo, J., et al.: A stable, low permeable TEMPO catholyte for aqueous total organic redox flow batteries. Adv. Energy Mater. 12, 2102577 (2022). https://doi.org/10.1002/aenm.202102577
Feng, R.Z., Zhang, X., Murugesan, V., et al.: Reversible ketone hydrogenation and dehydrogenation for aqueous organic redox flow batteries. Science 372, 836–840 (2021). https://doi.org/10.1126/science.abd9795
Liu, B., Tang, cw, Zhang, C., et al.: Cost-effective, high-energy-density, nonaqueous nitrobenzene organic redox flow battery. Chem. Mater. 33, 978–986 (2021). https://doi.org/10.1021/acs.chemmater.0c04118
Singh, V., Kim, S., Kang, J., et al.: Aqueous organic redox flow batteries. Nano Res. 12, 1988–2001 (2019). https://doi.org/10.1007/s12274-019-2355-2
Kwabi, D.G., Ji, Y.L., Aziz, M.J.: Electrolyte lifetime in aqueous organic redox flow batteries: a critical review. Chem. Rev. 120, 6467–6489 (2020). https://doi.org/10.1021/acs.chemrev.9b00599
Zhang, C.K., Li, X.F.: Perspective on organic flow batteries for large-scale energy storage. Curr. Opin. Electrochem. 30, 100836 (2021). https://doi.org/10.1016/j.coelec.2021.100836
Chen, H.N., Cong, G.T., Lu, Y.C.: Recent progress in organic redox flow batteries: active materials, electrolytes and membranes. J. Energy Chem. 27, 1304–1325 (2018). https://doi.org/10.1016/j.jechem.2018.02.009
Luo, J., Hu, B., Hu, M.W., et al.: Status and prospects of organic redox flow batteries toward sustainable energy storage. ACS Energy Lett. 4, 2220–2240 (2019). https://doi.org/10.1021/acsenergylett.9b01332
Zhan, X.W., Lu, X.C., Reed, D.M., et al.: Emerging soluble organic redox materials for next-generation grid energy-storage applications. MRS Commun. 10, 215–229 (2020)
Liu, Y.H., Chen, Q.R., Zhang, X., et al.: Degradation of electrochemical active compounds in aqueous organic redox flow batteries. Curr. Opin. Electrochem. 32, 100895 (2022). https://doi.org/10.1016/j.coelec.2021.100895
Perry, M.L., Rodby, K.E., Brushett, F.R.: Untapped potential: the need and opportunity for high-voltage aqueous redox flow batteries. ACS Energy Lett. 7, 659–667 (2022). https://doi.org/10.1021/acsenergylett.1c02225
Chen, Q.R., Lv, Y.G., Yuan, Z.Z., et al.: Organic electrolytes for pH-neutral aqueous organic redox flow batteries. Adv. Funct. Mater. 32, 2108777 (2022). https://doi.org/10.1002/adfm.202108777
Zhao, Z.M., Zhang, C.K., Li, X.F.: Opportunities and challenges of organic flow battery for electrochemical energy storage technology. J. Energy Chem. 67, 621–639 (2022). https://doi.org/10.1016/j.jechem.2021.10.037
Cameron, J.M., Holc, C., Kibler, A.J., et al.: Molecular redox species for next-generation batteries. Chem. Soc. Rev. 50, 5863–5883 (2021). https://doi.org/10.1039/d0cs01507e
Li, Z.J., Lu, Y.C.: Material design of aqueous redox flow batteries: fundamental challenges and mitigation strategies. Adv. Mater. 32, 2002132 (2020). https://doi.org/10.1002/adma.202002132
Gong, K., Fang, Q.R., Gu, S., et al.: Nonaqueous redox-flow batteries: Organic solvents, supporting electrolytes, and redox pairs. Energy Environ. Sci. 8, 3515–3530 (2015). https://doi.org/10.1039/C5EE02341F
Hogue, R.W., Toghill, K.E.: Metal coordination complexes in nonaqueous redox flow batteries. Curr. Opin. Electrochem. 18, 37–45 (2019). https://doi.org/10.1016/j.coelec.2019.08.006
Ding, Y., Zhao, Y., Li, Y.T., et al.: A high-performance all-metallocene-based, non-aqueous redox flow battery. Energy Environ. Sci. 10, 491–497 (2017). https://doi.org/10.1039/C6EE02057G
Wang, D.Y., Liu, R.L., Guo, W., et al.: Recent advances of organometallic complexes for rechargeable batteries. Coord. Chem. Rev. 429, 213650 (2021). https://doi.org/10.1016/j.ccr.2020.213650
Karmakar, A., Mukundan, R., Yang, P., et al.: Screening of metal complexes and organic solvents using the COSMOSAC-LANL model to enhance the energy density in a non-aqueous redox flow cell: an insight into the solubility. Phys. Chem. Chem. Phys. 23, 21106–21129 (2021). https://doi.org/10.1039/d1cp02591k
Ding, Y., Zhang, C.K., Zhang, L.Y., et al.: Pathways to widespread applications: development of redox flow batteries based on new chemistries. Chem 5, 1964–1987 (2019). https://doi.org/10.1016/j.chempr.2019.05.010
Ding, Y., Zhang, C.K., Zhang, L.Y., et al.: Molecular engineering of organic electroactive materials for redox flow batteries. Chem. Soc. Rev. 47, 69–103 (2018). https://doi.org/10.1039/C7CS00569E
Palmer, T.C., Beamer, A., Pitt, T., et al.: A comparative review of metal-based charge carriers in nonaqueous flow batteries. Chemsuschem 14, 1214–1228 (2021). https://doi.org/10.1002/cssc.202002354
Yang, Z.G., Zhang, J.L., Kintner-Meyer, M.C.W., et al.: Electrochemical energy storage for green grid. Chem. Rev. 111, 3577–3613 (2011). https://doi.org/10.1021/cr100290v
Hogue, R.W., Armstrong, C.G., Toghill, K.E.: Dithiolene complexes of first-row transition metals for symmetric nonaqueous redox flow batteries. Chemsuschem 12, 4506–4515 (2019). https://doi.org/10.1002/cssc.201901702
Eisenberg, R., Gray, H.B.: Noninnocence in metal complexes: a dithiolene dawn. Inorg. Chem. 50, 9741–9751 (2011). https://doi.org/10.1021/ic2011748
Matsuda, Y., Tanaka, K., Okada, M., et al.: A rechargeable redox battery utilizing ruthenium complexes with non-aqueous organic electrolyte. J. Appl. Electrochem. 18, 909–914 (1988). https://doi.org/10.1007/BF01016050
Laramie, S.M., Milshtein, J.D., Breault, T.M., et al.: Performance and cost characteristics of multi-electron transfer, common ion exchange non-aqueous redox flow batteries. J. Power Sources 327, 681–692 (2016). https://doi.org/10.1016/j.jpowsour.2016.07.015
Mun, J., Oh, D.J., Park, M.S., et al.: Highly soluble tris(2,2ʹ-bipyridine) metal bis(trifluoromethanesulfonyl)imide complexes for high energy organic redox flow batteries. J. Electrochem. Soc. 165, A215–A219 (2018). https://doi.org/10.1149/2.0791802jes
Benazzi, E., Cristino, V., Caramori, S., et al.: Electrochemical characterization of polypyridine iron(II) and cobalt(II) complexes for organic redox flow batteries. Polyhedron 140, 99–108 (2018). https://doi.org/10.1016/j.poly.2017.12.001
Cammack, C.X., Pratt, H.D., Small, L.J., et al.: A higher voltage Fe(II) bipyridine complex for non-aqueous redox flow batteries. Dalton Trans. 50, 858–868 (2021). https://doi.org/10.1039/d0dt03927f
Kim, J.H., Kim, K.J., Park, M.S., et al.: Development of metal-based electrodes for non-aqueous redox flow batteries. Electrochem. Commun. 13, 997–1000 (2011). https://doi.org/10.1016/j.elecom.2011.06.022
Mun, J., Lee, M.J., Park, J.W., et al.: Non-aqueous redox flow batteries with nickel and iron tris(2,2ʹ-bipyridine) complex electrolyte. Electrochem. Solid-State Lett. 15, A80 (2012). https://doi.org/10.1149/2.033206esl
Cabrera, P.J., Yang, X.Y., Suttil, J.A., et al.: Evaluation of tris-bipyridine chromium complexes for flow battery applications: impact of bipyridine ligand structure on solubility and electrochemistry. Inorg. Chem. 54, 10214–10223 (2015). https://doi.org/10.1021/acs.inorgchem.5b01328
Dong, S., Cabral, D.M., Pringle, J.M., et al.: Exploring the electrochemical properties of mixed ligand Fe(II) complexes as redox couples. Electrochim. Acta 362, 137109 (2020). https://doi.org/10.1016/j.electacta.2020.137109
Xing, X.Q., Zhao, Y.C., Li, Y.D.: A non-aqueous redox flow battery based on tris(1,10-phenanthroline) complexes of iron(II) and cobalt(II). J. Power Sources 293, 778–783 (2015). https://doi.org/10.1016/j.jpowsour.2015.06.016
Xing, X.Q., Zhang, D.P., Li, Y.D.: A non-aqueous all-cobalt redox flow battery using 1,10-phenanthrolinecobalt(II) hexafluorophosphate as active species. J. Power Sources 279, 205–209 (2015). https://doi.org/10.1016/j.jpowsour.2015.01.011
Yang, C.Z., Nikiforidis, G., Park, J.Y., et al.: Designing redox-stable cobalt-polypyridyl complexes for redox flow batteries: spin-crossover delocalizes excess charge. Adv. Energy Mater. 8, 1702897 (2018). https://doi.org/10.1002/aenm.201702897
Kolesnichenko, C.X., Pratt, H.D., III., Small, L.J., et al.: Elucidating instabilities contributing to capacity fade in bipyridine-based materials for non-aqueous flow batteries. ChemElectroChem 9, e202101490 (2022). https://doi.org/10.1002/celc.202101490
Cabrera, P.J., Yang, X.Y., Suttil, J.A., et al.: Complexes containing redox noninnocent ligands for symmetric, multielectron transfer nonaqueous redox flow batteries. J. Phys. Chem. C 119, 15882–15889 (2015). https://doi.org/10.1021/acs.jpcc.5b03582
Ruan, W.Q., Mao, J.T., Yang, S.D., et al.: Communication: tris(bipyridyl)iron complexes for high-voltage aqueous redox flow batteries. J. Electrochem. Soc. 167, 100543 (2020). https://doi.org/10.1149/1945-7111/ab9cc8
Li, X., Gao, P.Y., Lai, Y.Y., et al.: Symmetry-breaking design of an organic iron complex catholyte for a long cyclability aqueous organic redox flow battery. Nat. Energy 6, 873–881 (2021). https://doi.org/10.1038/s41560-021-00879-6
Chen, C., Zhang, S., Zhu, Y.Z., et al.: Pyridyl group design in viologens for anolyte materials in organic redox flow batteries. RSC Adv. 8, 18762–18770 (2018). https://doi.org/10.1039/c8ra02641f
Armstrong, C.G., Toghill, K.E.: Cobalt(II) complexes with azole-pyridine type ligands for non-aqueous redox-flow batteries: tunable electrochemistry via structural modification. J. Power Sources 349, 121–129 (2017). https://doi.org/10.1016/j.jpowsour.2017.03.034
Sevov, C.S., Fisher, S.L., Thompson, L.T., et al.: Mechanism-based development of a low-potential, soluble, and cyclable multielectron anolyte for nonaqueous redox flow batteries. J. Am. Chem. Soc. 138, 15378–15384 (2016). https://doi.org/10.1021/jacs.6b07638
Skavenborg, M.L., McPherson, J.N., Pasadakis-Kavounis, A., et al.: Leveraging coordination chemistry in the design of bipolar energy storage materials for redox flow batteries. Sustain. Energy Fuels 6, 2179–2190 (2022). https://doi.org/10.1039/d1se01966j
Duarte, G.M., Braun, J.D., Giesbrecht, P.K., et al.: Redox non-innocent bis(2,6-diimine-pyridine) ligand-iron complexes as anolytes for flow battery applications. Dalton Trans. 46, 16439–16445 (2017). https://doi.org/10.1039/c7dt03915h
Braun, J.D., Gray, P.A., Sidhu, B.K., et al.: Zn-Templated synthesis of substituted (2,6-diimine)pyridine proligands and evaluation of their iron complexes as anolytes for flow battery applications. Dalton Trans. 49, 16175–16183 (2020). https://doi.org/10.1039/d0dt00543f
Elgrishi, N., Chambers, M.B., Artero, V., et al.: Terpyridine complexes of first row transition metals and electrochemical reduction of CO2 to CO. Phys. Chem. Chem. Phys. 16, 13635–13644 (2014). https://doi.org/10.1039/c4cp00451e
Wang, H., Sayed, S.Y., Zhou, Y.Q., et al.: Water-soluble pH-switchable cobalt complexes for aqueous symmetric redox flow batteries. Chem. Commun. 56, 3605–3608 (2020). https://doi.org/10.1039/d0cc00383b
Chakrabarti, M.H., Dryfe, R.A.W., Roberts, E.P.L.: Evaluation of electrolytes for redox flow battery applications. Electrochim. Acta 52, 2189–2195 (2007). https://doi.org/10.1016/j.electacta.2006.08.052
Liu, Q.H., Sleightholme, A.E.S., Shinkle, A.A., et al.: Non-aqueous vanadium acetylacetonate electrolyte for redox flow batteries. Electrochem. Commun. 11, 2312–2315 (2009). https://doi.org/10.1016/j.elecom.2009.10.006
Sleightholme, A.E.S., Shinkle, A.A., Liu, Q.H., et al.: Non-aqueous manganese acetylacetonate electrolyte for redox flow batteries. J. Power Sources 196, 5742–5745 (2011). https://doi.org/10.1016/j.jpowsour.2011.02.020
Bamgbopa, M.O., Shao-Horn, Y., Almheiri, S.: The potential of non-aqueous redox flow batteries as fast-charging capable energy storage solutions: demonstration with an iron-chromium acetylacetonate chemistry. J. Mater. Chem. A 5, 13457–13468 (2017). https://doi.org/10.1039/c7ta02022h
Liu, Q.H., Shinkle, A.A., Li, Y.D., et al.: Non-aqueous chromium acetylacetonate electrolyte for redox flow batteries. Electrochem. Commun. 12, 1634–1637 (2010). https://doi.org/10.1016/j.elecom.2010.09.013
Zhang, D.P., Lan, H.J., Li, Y.D.: The application of a non-aqueous bis(acetylacetone)ethylenediamine cobalt electrolyte in redox flow battery. J. Power Sources 217, 199–203 (2012). https://doi.org/10.1016/j.jpowsour.2012.06.038
Shamie, J.S., Liu, C.H., Shaw, L.L., et al.: New mechanism for the reduction of vanadyl acetylacetonate to vanadium acetylacetonate for room temperature flow batteries. Chemsuschem 10, 533–540 (2017). https://doi.org/10.1002/cssc.201601126
Shinkle, A.A., Sleightholme, A.E.S., Griffith, L.D., et al.: Degradation mechanisms in the non-aqueous vanadium acetylacetonate redox flow battery. J. Power Sources 206, 490–496 (2012). https://doi.org/10.1016/j.jpowsour.2010.12.096
Shinkle, A.A., Sleightholme, A.E.S., Thompson, L.T., et al.: Electrode kinetics in non-aqueous vanadium acetylacetonate redox flow batteries. J. Appl. Electrochem. 41, 1191–1199 (2011). https://doi.org/10.1007/s10800-011-0314-z
Herr, T., Noack, J., Fischer, P., et al.: 1,3-Dioxolane, tetrahydrofuran, acetylacetone and dimethyl sulfoxide as solvents for non-aqueous vanadium acetylacetonate redox-flow-batteries. Electrochim. Acta 113, 127–133 (2013). https://doi.org/10.1016/j.electacta.2013.09.055
Escalante-García, I.L., Wainright, J.S., Thompson, L.T., et al.: Performance of a non-aqueous vanadium acetylacetonate prototype redox flow battery: examination of separators and capacity decay. J. Electrochem. Soc. 162, A363–A372 (2014). https://doi.org/10.1149/2.0471503jes
Suttil, J.A., Kucharyson, J.F., Escalante-Garcia, I.L., et al.: Metal acetylacetonate complexes for high energy density non-aqueous redox flow batteries. J. Mater. Chem. A 3, 7929–7938 (2015). https://doi.org/10.1039/c4ta06622g
Kucharyson, J.F., Cheng, L., Tung, S.O., et al.: Predicting the potentials, solubilities and stabilities of metal-acetylacetonates for non-aqueous redox flow batteries using density functional theory calculations. J. Mater. Chem. A 5, 13700–13709 (2017). https://doi.org/10.1039/c7ta01285c
Hart, J.R.: Ethylenediaminetetraacetic acid and related chelating agents. In: Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim (2011). https://doi.org/10.1002/14356007.a10_095.pub2
Bae, C.H., Roberts, E.P.L., Dryfe, R.A.W.: Chromium redox couples for application to redox flow batteries. Electrochim. Acta 48, 279–287 (2002). https://doi.org/10.1016/S0013-4686(02)00649-7
Bae, C., Roberts, E.P.L., Chakrabarti, M.H., et al.: All-chromium redox flow battery for renewable energy storage. Int. J. Green Energy 8, 248–264 (2011). https://doi.org/10.1080/15435075.2010.549598
Robb, B.H., Farrell, J.M., Marshak, M.P.: Chelated chromium electrolyte enabling high-voltage aqueous flow batteries. Joule 3, 2503–2512 (2019). https://doi.org/10.1016/j.joule.2019.07.002
Waters, S.E., Robb, B.H., Scappaticci, S.J., et al.: Isolation and characterization of a highly reducing aqueous chromium(II) complex. Inorg. Chem. 61, 8752–8759 (2022). https://doi.org/10.1021/acs.inorgchem.2c00699
Wen, Y.H., Zhang, H.M., Qian, P., et al.: Studies on iron (Fe3+∕Fe2+)-complex/bromine (Br2/Br–) redox flow cell in sodium acetate solution. J. Electrochem. Soc. 153, A929 (2006). https://doi.org/10.1149/1.2186040
Waters, S.E., Davis, C.M., Thurston, J.R., et al.: Maximizing vanadium deployment in redox flow batteries through chelation. J. Am. Chem. Soc. 144, 17753–17757 (2022). https://doi.org/10.1021/jacs.2c07076
Schröder, P., Obendorf, D., Bechtold, T.: Electrochemistry of iron(II/III)-N,N′-ethylene-bis-(o-hydroxyphenylglycine) complexes in aqueous solution indicates potential for use in redox flow batteries. ChemElectroChem 6, 3311–3318 (2019). https://doi.org/10.1002/celc.201900658
Modiba, P., Matoetoe, M., Crouch, A.M.: Electrochemical impedance spectroscopy study of Ce(IV) with aminopolycarboxylate ligands for redox flow batteries applications. J. Power Sources 205, 1–9 (2012). https://doi.org/10.1016/j.jpowsour.2012.01.004
So, Y.M., Leung, W.H.: Recent advances in the coordination chemistry of cerium(IV) complexes. Coord. Chem. Rev. 340, 172–197 (2017). https://doi.org/10.1016/j.ccr.2016.12.009
Modiba, P., Matoetoe, M., Crouch, A.M.: Kinetics study of transition metal complexes (Ce-DTPA, Cr-DTPA and V-DTPA) for redox flow battery applications. Electrochim. Acta 94, 336–343 (2013). https://doi.org/10.1016/j.electacta.2013.01.081
Yu, X., Song, Y.X., Tang, A.: Tailoring manganese coordination environment for a highly reversible zinc-manganese flow battery. J. Power Sources 507, 230295 (2021). https://doi.org/10.1016/j.jpowsour.2021.230295
Waters, S.E., Robb, B.H., Marshak, M.P.: Effect of chelation on iron-chromium redox flow batteries. ACS Energy Lett. 5, 1758–1762 (2020). https://doi.org/10.1021/acsenergylett.0c00761
Cappillino, P.J., Pratt, H.D., III., Hudak, N.S., et al.: Energy storage: application of redox non-innocent ligands to non-aqueous flow battery electrolytes. Adv. Energy Mater. 4, 1300566 (2014). https://doi.org/10.1002/aenm.201300566
Gong, K., Xu, F., Grunewald, J.B., et al.: All-soluble all-iron aqueous redox-flow battery. ACS Energy Lett. 1, 89–93 (2016). https://doi.org/10.1021/acsenergylett.6b00049
Noh, C., Chung, Y., Kwon, Y.: Optimization of iron and cobalt based organometallic redox couples for long-term stable operation of aqueous organometallic redox flow batteries. J. Power Sources 495, 229799 (2021). https://doi.org/10.1016/j.jpowsour.2021.229799
Shin, M., Noh, C., Kwon, Y.: Stability enhancement for all-iron aqueous redox flow battery using iron-3-[bis(2-hydroxyethyl)amino]-2-hydroxypropanesulfonic acid complex and ferrocyanide as redox couple. Int. J. Energy Res. 46, 6866–6875 (2022). https://doi.org/10.1002/er.7535
Shin, M., Oh, S., Jeong, H., et al.: Aqueous redox flow battery using iron 2,2-bis(hydroxymethyl)-2,2ʹ,2ʹ-nitrilotriethanol complex and ferrocyanide as newly developed redox couple. Int. J. Energy Res. 46, 8175–8185 (2022). https://doi.org/10.1002/er.7718
Noh, C., Chung, Y., Kwon, Y.: Highly stable aqueous organometallic redox flow batteries using cobalt triisopropanolamine and iron triisopropanolamine complexes. Chem. Eng. J. 405, 126966 (2021). https://doi.org/10.1016/j.cej.2020.126966
Noh, C., Chung, Y., Kwon, Y.: Organometallic redox flow batteries using iron triethanolamine and cobalt triethanolamine complexes. J. Power Sources 466, 228333 (2020). https://doi.org/10.1016/j.jpowsour.2020.228333
Arroyo-Currás, N., Hall, J.W., Dick, J.E., et al.: An alkaline flow battery based on the coordination chemistry of iron and cobalt. J. Electrochem. Soc. 162, A378–A383 (2014). https://doi.org/10.1149/2.0461503jes
Lê, A., Floner, D., Roisnel, T., et al.: Highly soluble Fe(III)-triethanolamine complex relevant for redox flow batteries. Electrochim. Acta 301, 472–477 (2019). https://doi.org/10.1016/j.electacta.2019.02.017
Wen, Y.H., Zhang, H.M., Qian, P., et al.: A study of the Fe(III)/Fe(II)-triethanolamine complex redox couple for redox flow battery application. Electrochim. Acta 51, 3769–3775 (2006). https://doi.org/10.1016/j.electacta.2005.10.040
Shin, M., Noh, C., Chung, Y., et al.: All iron aqueous redox flow batteries using organometallic complexes consisting of iron and 3-[bis(2-hydroxyethyl)amino]-2-hydroxypropanesulfonic acid ligand and ferrocyanide as redox couple. Chem. Eng. J. 398, 125631 (2020). https://doi.org/10.1016/j.cej.2020.125631
Ruan, W.Q., Mao, J.T., Yang, S.D., et al.: Designing Cr complexes for a neutral Fe–Cr redox flow battery. Chem. Commun. 56, 3171–3174 (2020). https://doi.org/10.1039/c9cc09704j
Tsitovich, P.B., Kosswattaarachchi, A.M., Crawley, M.R., et al.: An FeIII azamacrocyclic complex as a pH-tunable catholyte and anolyte for redox-flow battery applications. Chem. A Eur. J. 23, 15327–15331 (2017). https://doi.org/10.1002/chem.201704381
Sharma, S., Andrade, G.A., Maurya, S., et al.: Iron-iminopyridine complexes as charge carriers for non-aqueous redox flow battery applications. Energy Storage Mater. 37, 576–586 (2021). https://doi.org/10.1016/j.ensm.2021.01.035
Stauber, J.M., Zhang, S.Y., Gvozdik, N., et al.: Cobalt and vanadium trimetaphosphate polyanions: synthesis, characterization, and electrochemical evaluation for non-aqueous redox-flow battery applications. J. Am. Chem. Soc. 140, 538–541 (2018). https://doi.org/10.1021/jacs.7b08751
Huang, H.B., Howland, R., Agar, E., et al.: Bioinspired, high-stability, nonaqueous redox flow battery electrolytes. J. Mater. Chem. A 5, 11586–11591 (2017). https://doi.org/10.1039/c7ta00365j
Hwang, S., Kim, H.S., Ryu, J.H., et al.: Ni(II)-chelated thio-crown complex as a single redox couple for non-aqueous flow batteries. Electrochem. Commun. 85, 36–39 (2017). https://doi.org/10.1016/j.elecom.2017.10.015
Kim, H.S., Hwang, S., Mun, J., et al.: Counter anion effects on the energy density of Ni(II)-chelated tetradentate azamacrocyclic complex cation as single redox couple for non-aqueous flow batteries. Electrochim. Acta 308, 227–230 (2019). https://doi.org/10.1016/j.electacta.2019.04.027
Andrade, G.A., Popov, I.A., Federico, C.R., et al.: Expanding the potential of redox carriers for flow battery applications. J. Mater. Chem. A 8, 17808–17816 (2020). https://doi.org/10.1039/d0ta04511j
Chu, T., Popov, I.A., Andrade, G.A., et al.: Linked picolinamide nickel complexes as redox carriers for nonaqueous flow batteries. Chemsuschem 12, 1304–1309 (2019). https://doi.org/10.1002/cssc.201802985
Li, Y., Geysens, P., Zhang, X., et al.: Cerium-containing complexes for low-cost, non-aqueous redox flow batteries (RFBs). J. Power Sources 450, 227634 (2020). https://doi.org/10.1016/j.jpowsour.2019.227634
Lu, C.C., Bill, E., Weyhermüller, T., et al.: Neutral bis(α-iminopyridine)metal complexes of the first-row transition ions (Cr, Mn, Fe, Co, Ni, Zn) and their monocationic analogues: mixed valency involving a redox noninnocent ligand system. J. Am. Chem. Soc. 130, 3181–3197 (2008). https://doi.org/10.1021/ja710663n
Kim, H.S., Yoon, T., Jang, J., et al.: A tetradentate Ni(II) complex cation as a single redox couple for non-aqueous flow batteries. J. Power Sources 283, 300–304 (2015). https://doi.org/10.1016/j.jpowsour.2015.02.083
Montero, J., Navalpotro, P., D́Epifanio, A., et al.: Redox-active coordination polymers as bifunctional electrolytes in slurry-based aqueous batteries at neutral pH. J. Electroanal. Chem. 895, 115442 (2021). https://doi.org/10.1016/j.jelechem.2021.115442
Fernández-Benito, A., Rivera-Gálvez, F.J., Cisneros-Ruiz, P., et al.: Novel ferrocene-containing amphiphilic block copolymer nanoparticles as high-capacity charge carriers in aqueous redox flow systems. Mater. Today Chem. 27, 101271 (2023). https://doi.org/10.1016/j.mtchem.2022.101271
Friedl, J., Holland-Cunz, M.V., Cording, F., et al.: Asymmetric polyoxometalate electrolytes for advanced redox flow batteries. Energy Environ. Sci. 11, 3010–3018 (2018). https://doi.org/10.1039/c8ee00422f
Ai, F., Wang, Z.Y., Lai, N.C., et al.: Heteropoly acid negolytes for high-power-density aqueous redox flow batteries at low temperatures. Nat. Energy 7, 417–426 (2022). https://doi.org/10.1038/s41560-022-01011-y
Zhao, Y., Ding, Y., Song, J., et al.: Sustainable electrical energy storage through the ferrocene/ferrocenium redox reaction in aprotic electrolyte. Angew. Chem. Int. Ed. 53, 11036–11040 (2014). https://doi.org/10.1002/anie.201406135
Wei, X.L., Cosimbescu, L., Xu, W., et al.: Batteries: towards high-performance nonaqueous redox flow electrolyte via ionic modification of active species. Adv. Energy Mater. 5, 1400678 (2015). https://doi.org/10.1002/aenm.201400678
Hu, B., Liu, T.L.: Two electron utilization of methyl viologen anolyte in nonaqueous organic redox flow battery. J. Energy Chem. 27, 1326–1332 (2018). https://doi.org/10.1016/j.jechem.2018.02.014
Cosimbescu, L., Wei, X.L., Vijayakumar, M., et al.: Anion-tunable properties and electrochemical performance of functionalized ferrocene compounds. Sci. Rep. 5, 1–9 (2015). https://doi.org/10.1038/srep14117
Kim, H.S., Yoon, T., Kim, Y., et al.: Increase of both solubility and working voltage by acetyl substitution on ferrocene for non-aqueous flow battery. Electrochem. Commun. 69, 72–75 (2016). https://doi.org/10.1016/j.elecom.2016.06.002
Song, H., Kwon, G., Citek, C., et al.: Pyrrolinium-substituted persistent zwitterionic ferrocenate derivative enabling the application of ferrocene anolyte. ACS Appl. Mater. Interfaces 13, 46558–46565 (2021). https://doi.org/10.1021/acsami.1c11571
Cong, G.T., Zhou, Y.C., Li, Z.J., et al.: A highly concentrated catholyte enabled by a low-melting-point ferrocene derivative. ACS Energy Lett. 2, 869–875 (2017). https://doi.org/10.1021/acsenergylett.7b00115
Zhang, C.K., Qian, Y.M., Ding, Y., et al.: Biredox eutectic electrolytes derived from organic redox-active molecules: high-energy storage systems. Angew. Chem. 131, 7119–7124 (2019). https://doi.org/10.1002/ange.201902433
Zhang, C.K., Zhang, L.Y., Yu, G.H.: Eutectic electrolytes as a promising platform for next-generation electrochemical energy storage. Acc. Chem. Res. 53, 1648–1659 (2020). https://doi.org/10.1021/acs.accounts.0c00360
Hwang, B., Park, M.S., Kim, K.: Ferrocene and cobaltocene derivatives for non-aqueous redox flow batteries. Chemsuschem 8, 310–314 (2015). https://doi.org/10.1002/cssc.201403021
Chen, H., Niu, Z.H., Ye, J., et al.: Multicore ferrocene derivative as a highly soluble cathode material for nonaqueous redox flow batteries. ACS Appl. Energy Mater. 4, 855–861 (2021). https://doi.org/10.1021/acsaem.0c02733
Milton, M., Cheng, Q., Yang, Y., et al.: Molecular materials for nonaqueous flow batteries with a high Coulombic efficiency and stable cycling. Nano Lett. 17, 7859–7863 (2017). https://doi.org/10.1021/acs.nanolett.7b04131
Montoto, E.C., Nagarjuna, G., Moore, J.S., et al.: Redox active polymers for non-aqueous redox flow batteries: validation of the size-exclusion approach. J. Electrochem. Soc. 164, A1688–A1694 (2017). https://doi.org/10.1149/2.1511707jes
Friedl, J., Lebedeva, M.A., Porfyrakis, K., et al.: All-fullerene-based cells for nonaqueous redox flow batteries. J. Am. Chem. Soc. 140, 401–405 (2018). https://doi.org/10.1021/jacs.7b11041
Hwang, S., Kim, H.S., Ryu, J.H., et al.: N-ferrocenylphthalimide; a single redox couple formed by attaching a ferrocene moiety to phthalimide for non-aqueous flow batteries. J. Power Sources 395, 60–65 (2018). https://doi.org/10.1016/j.jpowsour.2018.05.053
Hwang, S., Kim, H.S., Ryu, J.H., et al.: N-(α-ferrocenyl)ethylphthalimide as a single redox couple for non-aqueous flow batteries. J. Power Sources 421, 1–5 (2019). https://doi.org/10.1016/j.jpowsour.2019.02.080
Xu, D.H., Zhang, C.J., Zhen, Y.H., et al.: Ferrocene/phthalimide ionic bipolar redox-active molecule for symmetric nonaqueous redox flow batteries. ACS Appl. Energy Mater. 4, 8045–8051 (2021). https://doi.org/10.1021/acsaem.1c01362
Zhen, Y.H., Zhang, C.J., Yuan, J.S., et al.: Ferrocene/anthraquinone based bi-redox molecule for symmetric nonaqueous redox flow battery. J. Power Sources 480, 229132 (2020). https://doi.org/10.1016/j.jpowsour.2020.229132
Hu, B., DeBruler, C., Rhodes, Z., et al.: Long-cycling aqueous organic redox flow battery (AORFB) toward sustainable and safe energy storage. J. Am. Chem. Soc. 139, 1207–1214 (2017). https://doi.org/10.1021/jacs.6b10984
Beh, E.S., De Porcellinis, D., Gracia, R.L., et al.: A neutral pH aqueous organic-organometallic redox flow battery with extremely high capacity retention. ACS Energy Lett. 2, 639–644 (2017). https://doi.org/10.1021/acsenergylett.7b00019
Zhu, Y.Z., Yang, F., Niu, Z.H., et al.: Enhanced cyclability of organic redox flow batteries enabled by an artificial bipolar molecule in neutral aqueous electrolyte. J. Power Sources 417, 83–89 (2019). https://doi.org/10.1016/j.jpowsour.2019.02.021
Luo, J., Hu, M.W., Wu, W.D., et al.: Mechanistic insights of cycling stability of ferrocene catholytes in aqueous redox flow batteries. Energy Environ. Sci. 15, 1315–1324 (2022). https://doi.org/10.1039/D1EE03251H
Chen, Q.R., Li, Y.Y., Liu, Y.H., et al.: Designer ferrocene catholyte for aqueous organic flow batteries. Chemsuschem 14, 1295–1301 (2021). https://doi.org/10.1002/cssc.202002467
Yu, J.Z., Salla, M., Zhang, H., et al.: A robust anionic sulfonated ferrocene derivative for pH-neutral aqueous flow battery. Energy Storage Mater. 29, 216–222 (2020). https://doi.org/10.1016/j.ensm.2020.04.020
Zhao, Z.L., Zhang, B.S., Schrage, B.R., et al.: Investigations into aqueous redox flow batteries based on ferrocene bisulfonate. ACS Appl. Energy Mater. 3, 10270–10277 (2020). https://doi.org/10.1021/acsaem.0c02259
Yao, Y.W., Xu, H., Tian, Z.Z., et al.: Simple-synthesized sulfonated ferrocene ammonium for aqueous redox flow batteries. ACS Appl. Energy Mater. 4, 8052–8058 (2021). https://doi.org/10.1021/acsaem.1c01363
Schrage, B.R., Zhang, B.S., Petrochko, S.C., et al.: Highly soluble imidazolium ferrocene bis(sulfonate) salts for redox flow battery applications. Inorg. Chem. 60, 10764–10771 (2021). https://doi.org/10.1021/acs.inorgchem.1c01473
Li, Y.Y., Xu, Z.A., Liu, Y.H., et al.: Functioning water-insoluble ferrocenes for aqueous organic flow battery via host-guest inclusion. Chemsuschem 14, 745–752 (2021). https://doi.org/10.1002/cssc.202002516
Chopra, R., de Kock, C., Smith, P., et al.: Ferrocene-pyrimidine conjugates: synthesis, electrochemistry, physicochemical properties and antiplasmodial activities. Eur. J. Med. Chem. 100, 1–9 (2015). https://doi.org/10.1016/j.ejmech.2015.05.043
Tappe, K., Knochel, P.: New efficient synthesis of taniaphos ligands: application in ruthenium- and rhodium-catalyzed enantioselective hydrogenations. Tetrahedron Asymmetry 15, 91–102 (2004). https://doi.org/10.1016/j.tetasy.2003.11.004
Borchers, P.S., Strumpf, M., Friebe, C., et al.: Aqueous redox flow battery suitable for high temperature applications based on a tailor-made ferrocene copolymer. Adv. Energy Mater. 10, 2001825 (2020). https://doi.org/10.1002/aenm.202001825
Acknowledgements
The work described in this paper was fully supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. T23-601/17-R). The authors thank Dr. Chun Wai Tang for valuable advice.
Funding
Open access funding provided by Hong Kong University of Science and Technology.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, B., Li, Y., Jia, G. et al. Recent Advances in Redox Flow Batteries Employing Metal Coordination Complexes as Redox-Active Species. Electrochem. Energy Rev. 7, 7 (2024). https://doi.org/10.1007/s41918-023-00205-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41918-023-00205-6