
Abstract
Rechargeable all-solid-state batteries (ASSBs) are considered to be the next generation of devices for electrochemical energy storage. The development of solid-state electrolytes (SSEs) is one of the most crucial subjects in the field of energy storage chemistry. The newly emerging halide SSEs have recently been intensively studied for application in ASSBs due to their favorable combination of high ionic conductivity, exceptional chemical and electrochemical stability, and superior mechanical deformability. In this review, a critical overview of the development, synthesis, chemical stability and remaining challenges of halide SSEs is given. The design strategies for optimizing the ionic conductivity of halide SSEs, such as element substitution and crystal structure design, are summarized in detail. Moreover, the associated chemical stability issues in terms of solvent compatibility, humid air stability and corresponding degradation mechanisms are discussed. In particular, advanced in situ/operando characterization techniques applied to halide-based ASSBs are highlighted. In addition, a comprehensive understanding of the interface issues, cost issues, and scalable processing challenges faced by halide-based ASSBs for practical application is provided. Finally, future perspectives on how to design high-performance electrode/electrolyte materials are given, which are instructive for guiding the development of halide-based ASSBs for energy conversion and storage.
Graphical Abstract
In this review, a critical overview is given on the development, synthesis, chemical stability and remaining challenges facing for halide SSEs. The design strategies for optimizing ionic conductivity of halide SSEs like elements substitution, crystal structures design are summarized in detail. Future perspectives are given on how to design high-performance electrode/electrolyte materials.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Driven by the ongoing advancement of the new energy revolution worldwide, the development of high-energy and high-power energy storage devices simultaneously exhibiting enhanced safety features is urgently required to satisfy the stringent demands of grid-scale and utility-scale stationary storage [1,2,3]. Conventional lithium-ion batteries with organic liquid electrolytes have been typically applied as the dominant energy storage devices for the mobile electronics industry and electric vehicles [4,5,6]. However, the thermal runaway risks caused by flammable organic electrolytes as well as the low energy densities (up to 300 Wh kg−1) profoundly limited by the nature of the material chemistry have seriously impeded the further development of traditional lithium-ion batteries with liquid electrolytes [7,8,9,10]. All-solid-state batteries (ASSBs) are prevailingly acknowledged as the most promising candidate for next-generation energy storage, primarily benefiting from their high energy density and high safety through the incorporation of nonflammable solid-state electrolytes (SSEs) and high-voltage cathode materials (e.g., LiCoO2 and Li[Ni,Co,Mn]O2) or high-capacity electrodes (e.g., a lithium metal anode and a S cathode) [11,12,13,14,15]. Notably, the appreciable merits of ASSBs originate from the nature of SSEs in terms of high single-ion conductivity, outstanding thermal stability and simplified packaging design [16,17,18].
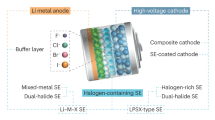
To date, several promising SSEs involving oxide, sulfide, and halide electrolytes with high ionic conductivity over 1 mS cm−1 at room temperature (RT) have received extensive attention for energy storage in ASSBs [19,20,21,22]. A comparison of various properties among the three families of ceramic solid electrolytes is shown in Fig. 1. The oxides exhibit acceptable chemical stability and good electrochemical oxidation stability; however, their mechanical rigidity results in poor interfacial contact with electrode materials, requiring high-temperature processes or infiltration of liquid electrolytes for battery assembly [23,24,25,26]. In contrast, sulfides deliver enhanced properties in terms of higher ionic conductivity and excellent mechanical deformability, whereas they are vulnerable to moisture in the ambient atmosphere, suffering from detrimental hydrolysis reactions along with severe reduction of the ionic conductivity [27,28,29,30]. Furthermore, the intrinsic poor oxidation stability of sulfides critically restricts the direct utilization of high-voltage cathode active materials (CAMs) [31, 32]. Currently, emerging halide SSEs are a research hotspot since their superior properties integrate the merits of oxides and sulfides to some extent, and thus, they possess high ionic conductivity (> 10−3 mS cm−1), reliable deformability and superior electrochemical oxidation stability (up to 6 V vs. Li+/Li) and can even be realized through soluble water-medium synthetic routes [21, 33,34,35].

Radar plots comparing the performances among three typical families of ceramic solid electrolytes
A schematic timeline of the development of halide SSEs from 1930 to the present with the representative key milestones is shown in Fig. 2. Halide-related materials can be dated back to 1930 [36], when lithium halides (LiX, X = F, Cl, Br, I) with Li+ conducting behavior were first studied. After that, LiI SSEs were further developed and applied in thin-film-type ASSBs at the end of the 1960s [37], and subsequently, ternary halides such as Li1.52Mn1.24Cl4 and Li2TiCl4 were reported in 1984 and 1988 [38, 39]. Unfortunately, both of their ionic conductivities at RT were relatively low at ~ 10−5 S cm−1, and as a result, the development of halide SSEs was severely delayed for a long time [40,41,42,43]. In 2018, Asano et al. [44] achieved a momentous breakthrough in the field of halide SSEs; specifically, Li3YCl6 and Li3YBr6 with high ionic conductivities of (0.03–1.7) × 10−3 S cm−1 at RT were successfully synthesized through a high-energy ball milling and annealing process, which are several orders of magnitude higher than those of previously reported halide electrolytes. Shortly afterward, a series of halide superionic conductors, such as Li3ErCl6 [45], Li3−xM1−xZrxCl6 (M = Y, Er, In, Yb) [46,47,48] and Li3HoBr6 [49], were gradually developed by similar approaches. In particular, Sun’s group synthesized Li3InCl6 by employing a facile water-mediated strategy in 2019 [33] and then dexterously developed a universal ammonium-assisted methodology to synthesize various halide SSEs in 2021 [50], which enabled manufacture of halide SSEs on a large scale for commercial implementation.

Schematic timeline illustrating the developments achieved to date regarding halide SSEs for ASSBs
To date, the reported halide SSEs with the formula of Li–M–X (M = metal element, X = F, Cl, Br, I) can be classified into four categories according to the different types of central metal element [21, 51] as follows. (1) Halide SSEs with group 3 elements (Sc, Y, La–Lu) commonly exhibit exceptional electrochemical oxidation stability and decent ionic conductivity, especially chloride-based halides, whereas the high cost of rare-earth materials makes scalable manufacturing challenging. (2) Halide SSEs with group 13 elements (Al, In, Ga) possess reliable air stability and good ionic conductivity, for example, Li3InCl6 delivers high ionic conductivity (1.49 mS cm−1) at RT and desirable humidity tolerance originating from the generation of a stable hydrate phase [52]. (3) Halide SSEs with divalent metal elements (e.g., Ti, V, Cr) are mostly demonstrated to show undesirable ionic conductivity at RT, several orders of magnitude lower than the other SSEs, and consequently have received less attention. (4) Halide SSEs with tetravalent metal elements (Zr, Hf) have been developed in recent years, especially Li2ZrCl6, which has a high ionic conductivity of 0.81 mS cm−1 at RT and a remarkable cost effectiveness as well as an exceptional humidity tolerance [53]. Radar plots comparing the (electro)chemical properties of the four types of halide SSEs with various central metal elements are shown in Fig. 3. Consequently, compared with oxide and sulfide solid electrolytes, emerging halide SSEs offer a wider range of options in terms of composition, structure and chemistry for designing and synthesizing prospective electrolytes with enhanced comprehensive properties.

Radar plots comparing the performances of the four types of halide SSEs with various central metal elements, including group 3, group 4, group 13 and divalent metals
In this review, we comprehensively summarize the recent developments in halide SSEs for ASSBs. Various synthesis routes of halide SSEs, including the recently developed universal ammonium-assisted method, are systematically reviewed. Furthermore, the design strategies for optimizing the ionic conductivity of halide SSEs, such as element substitution and crystal structure design, are summarized in detail. Simultaneously, the associated chemical stability issues in terms of solvent compatibility, humid air stability and corresponding degradation mechanisms are discussed. The advanced in situ/operando techniques employed to investigate the structural evolution of halide electrolytes and the underlying redox mechanisms of halide-based ASSBs under realistic operating conditions are thoroughly introduced. In addition, a comprehensive understanding of the interface, cost, and scalable processing issues of halide-based ASSBs for practical application is provided. Finally, we offer future perspectives on pursuing high-performance electrode/electrolyte materials to address the challenges in the practical application of halide-based ASSBs.
2 Synthesis
A simple and cost-effective synthesis route for manufacturing halide SSEs with high ionic conductivity plays a decisive role in realizing practical application of halide-based ASSBs. In general, synthesis methods for halide SSEs can be classified into three categories: solid-state reaction routes, liquid-phase routes and chemical vapor routes. A schematic diagram of various synthesis routes for halide SSEs is shown in Fig. 4. The ionic conductivities of various types of halide SSEs obtained from different synthesis methods are summarized in Table 1.

Schematics of common synthesis routes for halide solid electrolytes, including the mechanochemical milling route (R1), mechanical milling with the postannealing route (R2), solid-state sintering route (R3), water-mediated synthesis route (R4) and ammonium-assisted synthesis route (R5)
2.1 Solid-State Reaction Synthesis
Solid-state reaction routes are the earliest and most popular methods for synthesizing halide solid electrolytes. According to the various operating conditions, solid-state reaction routes can be further divided into three methods: mechanochemical milling, mechanical milling with postannealing and solid-state sintering. Note that the operating conditions and the processing parameters in solid-state reactions exert a great effect on halide SSEs in terms of their lattice structure, crystallinity and ionic conductivity.
2.1.1 Mechanochemical Milling
Mechanochemical milling methods commonly employ high-energy mechanical milling technology with appropriate ball milling speed and reaction time for synthesizing halide SSEs (Fig. 5a), in which the stoichiometric precursors can undergo intense shearing, friction and collision between the grinding ball and jar wall to achieve effective reactions at the atomic level [69]. Mechanochemical synthesis enables the preparation of unique amorphous variants or strong amorphization of compounds with low coherence that cannot be obtained by classical high-temperature solid-state synthesis, while rapid crystallization with subsequent quenching can significantly reduce the amorphous structure, giving rise to augmentation of long-range coherent structures in crystalline compounds (Fig. 5b) [70]. Prevalent site disorder and defect structure of halide SSEs can be obtained by mechanochemical milling, which is favorable for enhancing their ionic conductivity, and consequently, the corresponding halide SSEs could be directly applied to battery assembly without post pulverization or heat treatment [54]. In 2018, Asano et al. [44] successfully synthesized Li3YCl6 and Li3YBr6 with high RT ionic conductivities of 0.51 and 0.72 mS cm−1, respectively, through mechanochemical milling for the first time and found that the remarkably high conductivity of the as-synthesized halide SSEs benefits from the partially disordered cation arrangement and the abundant defect structures induced by mechanical milling techniques. Sebti et al. [71] reported that coexistence of a high concentration of stacking faults and Li-only defect layers in Li3YCl6 can be achieved by mechanochemical synthesis, which allows the formation of extra site linkages with lower migration barriers and dramatically promotes Li+ mobility in the microcrystalline structure of Li3YCl6. Notably, the planar defects induced by mechanochemical synthesis are metastable, which is responsible for the highly tunable ionic conductivity of Li3YCl6. Under such circumstances, heat treatment even as low as 60 °C is demonstrated to be capable of reducing the ionic conductivity of Li3YCl6 due to the gradual elimination of metastable defect structures. Schlem et al. [72] demonstrated that prevalent cation site disorder between Er/Y sites and a disordered structure, such as LiCl65− octahedral distortion, could be generated in Li3YCl6 and Li3ErCl6 by employing mechanochemical synthesis, which exhibited markedly intensified ion transport through expansion of lithium diffusion bottlenecks. The microcrystalline structure of Li3MCl6 (space group: P\(\overline{3}\)m1) can be described as a trigonal lattice formed by three types of MCl63− octahedra (Fig. 5c–e), in which Wyckoff positions 1a and 2d are occupied by M1 and M2, respectively, and M3 occupying the M2-equivalent position in the (001) plane enables the formation of M2–M3 site disorder. The authors found that the M3/M2 ratio was high in the halide Li3ErCl6 synthesized by mechanical ball milling (Fig. 5f), yielding abundant M2–M3 disorder, which could effectively reduce the Li+ migration energy barrier along the c-axis, while the corresponding site disorder significantly decreased after heat treatment even for a short time of 1 min, which accounted for the reduced ionic conductivity of Li3ErCl6 prepared by subsequent annealing and ampoule synthesis (Fig. 5g). For the synthesis of a 1 g batch of halide samples through the mechanochemical milling approach, an excess amount of rare-earth chlorides of approximately 10 wt% (wt% means the weight percentage) was applied to compensate for the loss during the pregrinding process due to the strong adhesion between the rare-earth chloride precursors and the agate mortar.

Reproduced with permission from Ref. [70]. Copyright 2021, Wiley–VCH. c Li3ErCl6 unit cell with construction units of ErCl63− octahedra that form a trigonal unit cell and face-sharing ErCl63− octahedra chains perpendicular to the (002) plane, with three possible erbium sites (Er1, Wyckoff 1a; Er2 and Er3, Wyckoff 2d). d The vacant octahedral sites in Li3ErCl6 were occupied by lithium (Wyckoff 6g, 100% occupied; Wyckoff 6h, 50% occupied). e The possible lithium pathways in Li3ErCl6 involve face-sharing LiCl65− octahedra along the c-direction and tetrahedral voids in the a–b plane. f Evolution of the Er2–Er3 site disorder determined by G(r) fits (open circles) and Rietveld refinements (open squares). g Ionic conductivity and corresponding activation energy of Li3ErCl6 as a function of annealing time. Reproduced with permission from Ref. [72]. Copyright 2019, Wiley–VCH
Mechanochemical synthesis enhances the disordered structure of halides for fast lithium-ion transport. a Schematic diagram of the mechanochemical synthesis of solid electrolytes, in which the precursors (cyan, purple) react to form the final products (red) under the impact of the ball milling medium (black spheres). b Comparison of the mechanochemical synthesis and classic high-temperature preparation for solid electrolytes with different degrees of coherence.
Regarding sodium-conducting halide solid electrolytes, the mechanochemically synthesized samples also exhibit higher ionic conductivity than the samples with a subsequent crystallization route, which deliver similar behavior to the lithium-conducting halide counterparts. Kwak et al. [67] reported that mechanochemically prepared Na2ZrCl6 possessed an ionic conductivity of 0.018 mS cm−1 at RT, which was nearly three orders of magnitude higher than that of the sample (6.9 × 10−8 S cm−1) synthesized with subsequent annealing at 400 °C, although both types of electrolytes had the same trigonal lattice (space group P\(\overline{3}\)m1). Site disorder in terms of Zr and Na can be observed in the microcrystalline structure of Na2ZrCl6 obtained from mechanochemical milling syntheses, which could induce partial occupation of sodium interstitial sites to form additional transport pathways, thus leading to a substantially improved ionic conductivity. In contrast, sodium ions 100% occupy the crystallographic Wyckoff site (6g) without undergoing any disordering for the subsequently annealed Na2ZrCl6, and simultaneously, the Na–Cl interatomic distance is considerably shortened by heat treatment, promoting strong bonding between the Na+ and Cl− ions, which could be responsible for the significantly decreased ionic conductivity of Na2ZrCl6 obtained from the subsequent crystallization route. A similar phenomenon was also found for the sodium halide Na3YCl6 and its derivatives. Wu et al. [66] demonstrated that the mechanochemical milling approach enables remarkable improvement of the ionic conductivity for annealed Na3YCl6 by nearly three orders of magnitude, and for its derivative Na2.25Y0.25Zr0.75Cl6, the corresponding ionic conductivity can also be dramatically improved from 1.3 × 10−5 to 6.1 × 10−5 S cm−1 at RT after subjecting the material to a similar processing procedure. The major disadvantage of mechanochemical synthesis lies in its high energy and time consumption, and the partial precursors may be contaminated by the grinding balls, which is not conducive to synthesizing high-purity halide SSEs.
2.1.2 Mechanical Milling with Postannealing
Typically, in the mechanical milling with postannealing route, stoichiometric raw materials are first subjected to a high-energy mechanical milling process to achieve fully mixed and low-crystallinity halide precursors with a smaller size, then pressed into pellets and sealed in a quartz tube, followed by annealing to obtain halide SSEs. Through this method, a certain amount of a halide, such as Li3MCl6 (M = In, Yb) and Li3YBr6, can deliver higher Li+ ionic conductivity than the corresponding halide electrolytes obtained from the mechanochemical milling route [52, 55, 73], primarily ascribed to the enhanced crystallinity along with evolution of the crystal structure of halide electrolytes that may effectively facilitate Li+ diffusion.
Li et al. [52] investigated the effects of heat treatment conditions in terms of various temperatures and durations on the ionic conductivity of the halide Li3InCl6 synthesized by mechanical milling with postannealing and found that annealing ball-milled Li3InCl6 in the range of 260–400 °C could effectively improve the ionic conductivity, accompanied by distinctly enhanced crystallinity. Increasing the heat treatment time in the range of 260–400 °C had little influence on the ionic conductivity of Li3InCl6, and the highest ionic conductivity of 1.49 mS cm−1 at RT could be achieved by annealing at 260 °C for 2 h, which was significantly higher than that of ball-milled samples (0.83 mS cm−1). Park et al. [55] successfully synthesized Li3YbCl6 SSEs through the mechanical milling with postannealing route and demonstrated the dynamic structural evolution of Li3YbCl6, which varied with the annealing temperature. The trigonal (P\(\overline{3}\)m1) structure exhibited slightly higher RT ionic conductivity (0.19 mS cm−1) than the orthorhombic (Pnma) structure sample (0.14 mS cm−1), which were prepared by annealing ball-milled Li3YbCl6 at 400 and 500 °C, respectively. Yu et al. [73] investigated various annealing parameters, including the heat treatment temperature (250–550 °C) and duration (3–15 h), in terms of the influence on the ionic conductivity of Li3YBr6 prepared by the mechanical milling with the postannealing method (Fig. 6a–c). The halide Li3YBr6 annealed at 500 °C for 5 h exhibited the highest ionic conductivity of 3.31 mS cm−1 at RT, which is an order of magnitude higher than that of mechanical milling products (0.39 mS cm−1). The high-temperature annealing process after mechanical milling can enhance the anharmonic vibration in the lattice structure of Li3YBr6, which is favorable for appreciably enhancing the hopping rate of lithium ions between adjacent sites, thereby achieving high mobility of lithium ions in the microcrystalline structure to improve the ionic conductivity of Li3YBr6.

Reproduced with permission from Ref. [73]. Copyright 2020, Elsevier. d (d1–d4) Anisotropic thermal displacement parameters of ampoule and mechanochemically synthesized Li3YBr6 compounds. e Convolution of the effects of the disordered structures in halide compounds on the observed changes in the ionic transport. Reproduced with permission from Ref. [74]. Copyright 2021, American Chemical Society. f Illustration of the water-mediated synthesis route for Li3InCl6. Reproduced with permission from Ref. [33]. Copyright 2019, Wiley–VCH. g Schematic of solution infiltration synthesis for a Li3InCl6/glass-fiber solid-composite electrolyte film, and corresponding electrochemical test curves. Reproduced with permission from Ref. [75]. Copyright 2022, Elsevier
a XRD patterns of Li3YBr6 samples obtained from mechanical milling with postannealing at various temperatures. b Changes in the ionic conductivity of the annealed Li3YBr6 samples with the annealing temperature. c Arrhenius plots of the ionic conductivity of Li3YBr6 pellets obtained from a precursor mixture milled at 550 rpm for 32 h followed by annealing at 500 °C for various durations.
Although the additional heat treatment increases the costs and time for synthesizing halide SSEs by the mechanical milling with the postannealing route, the improved ionic conductivity endows the SSEs with application potential in ASSBs, which suggests that this method could be regarded as a potential option to manufacture halide SSEs.
2.1.3 Solid-State Sintering
The solid-state sintering route is generally regarded as a simple approach for synthesizing halide SSEs through direct thermal sintering of the stoichiometric precursors in a high-vacuum ampoule. The starting materials could be cold-pressed into pellets after manual grinding or mechanical milling at a low rotational speed for further heat treatment. Notably, the precursor ratio, sintering temperature and duration are crucial factors for successfully obtaining high-purity halide SSEs.
Liang et al. [60] optimized the composition ratio of initial precursors for synthesizing a series of Li–M–Cl (M = Tb, Dy, Ho, Y, Er, Tm) halide SSEs through direct annealing at 650 °C for 24 h, and different ratios of the starting materials could significantly affect the ionic conductivity of the halides by changing the crystal phase structure. For example, Li2.73Ho1.09Cl6 with the orthorhombic phase exhibited a fourfold increase in the ionic conductivity up to 1.3 mS cm−1 at RT compared to Li3HoCl6 with the trigonal phase. Through this method, Li3HoBr6 halide with a high RT ionic conductivity of 1.1 mS cm−1 could also be successfully synthesized through annealing at 450 °C for 12 h [64]. Kim et al. [47] investigated the effects of various annealing temperatures (350–650 °C) and durations (36–100 h) on the crystal structure and ionic conductivity of Li3YbCl6. The Li3YbCl6 with the trigonal phase annealed at 350 °C showed a markedly higher ionic conductivity (0.1 mS cm−1 at 30 ℃) than that of the orthorhombic phase (0.06 mS cm−1 at 30 °C) annealed at 650 ℃, and the metastable trigonal phase could be converted to the thermodynamically stable orthorhombic phase by increasing the annealing time from 36 to 100 h. Schlem et al. [74] demonstrated that Li3YBr6 prepared by the solid-state sintering method exhibited higher ionic conductivity than Li3YBr6 prepared by the mechanochemical milling route or mechanical milling with postannealing route, which is closely associated with the directional thermal amplitude and partial occupancies of additional sites. The structure of Li3YBr6 (space group C2/m) can be described as a monoclinic unit cell. For the ampoule-synthesized (solid-state sintering method) compound, the authors found that the larger yttrium occupancy of the lithium 4g site produced an increased electrostatic repulsion, which could promote rotation of the anisotropic thermal displacement ellipsoids away from yttrium octahedra, accompanied by a larger displacement of lithium in the a-direction (Fig. 6d1, d3), thus being pushed to the tetrahedral position. This phenomenon is highly favorable for lithium-ion transport, leading to a higher ionic conductivity. In contrast, the lower occupancy of the lithium 4g site in the mechanochemically synthesized Li3YBr6 caused displacement of lithium to octahedral sites rather than tetrahedral sites, resulting in a larger displacement of the anisotropic thermal displacement ellipsoids in the b-direction (Fig. 6 d2, d4), thereby increasing the diffusion barrier for lithium ions in the microcrystalline structure of Li3YBr6. The abundant yttrium disorder in ampoule-synthesized Li3YBr6 can effectively affect lithium polyhedral volumes and the thermal displacement amplitudes of lithium, which further facilitates the formation of optimized transport pathways for lithium ions to improve the ionic conductivity (Fig. 6e).
Although the simple solid-state sintering method is favorable for easily achieving large-scale production of halide SSEs, the underlying issues of the harsh high-vacuum environment and poor homogeneity as well as impure phases generated by elemental volatilization at high temperatures impede its widespread application for manufacturing halide electrolytes.
2.2 Liquid-Phase Synthesis
Recently, wet-chemical synthesis routes employing liquid solvents as reaction media have received a strong upsurge of interest for fabricating halide solid electrolytes, which offer numerous advantages in terms of easily scalable production, effective size/morphology control and time-saving processes. Wet-chemical synthesis approaches have been successfully applied to synthesizing halides and can be classified into two strategies, water-mediated synthesis and ammonium-assisted synthesis, based on the different reaction mechanisms.
2.2.1 Water-Mediated Synthesis
Employing an ecofriendly and cost-effective water-medium approach for synthesizing halide electrolytes with high ionic conductivity and humidity tolerance is regarded as the most promising strategy for achieving commercial application of halide SSEs.
In 2019, Li et al. [33] successfully synthesized high-purity Li3InCl6 through a water-medium approach for the first time; in detail, the hydrate intermediate (Li3InCl6·nH2O) was first generated by reacting LiCl and InCl3 in an aqueous solution, followed by heat treatment in vacuum to remove water molecules (Fig. 6f). Li3InCl6 prepared at 200 ℃ in vacuum exhibited the highest ionic conductivity of 2.04 mS cm−1 at RT compared with Li3InCl6 prepared at the heat treatment temperatures of 100 and 130 ℃, and the production of high-purity Li3InCl6 can be easily scaled up to 110 g by using the water-mediated route. The authors found that the dehydrated form of Li3InCl6 could also be acquired by applying a dehydration process in an air or argon atmosphere, but the appearance of the impurity phase InOCl accompanied this process; thus, the dehydration process should be carried out under vacuum to ensure high purity. In addition, high-purity Na3InCl6 can also be successfully synthesized under similar synthetic conditions by reacting NaCl and InCl3 in an aqueous solution, followed by heat treatment at 200 ℃ in vacuum. Furthermore, Zhao et al. [75] proposed an infiltration method for preparing Li3InCl6 thin films that is compatible with the roll-to-roll manufacturing technique (Fig. 6g). Glass fibers were immersed into the precursor solution of stoichiometric LiCl and InCl3·4H2O, followed by vacuum drying at 60 and 200 °C for 4 h, and the prepared composite electrolytes were further roll pressed to obtain a free-standing and compact Li3InCl6 film with an ionic conductivity of 5.4 × 10−4 S cm−1 at RT.
However, employing the water-mediated synthesis method for halide SSEs is limited to In-based electrolytes, which is primarily ascribed to the reversible interconversion between the hydrated intermediate and dehydrated phase. Consequently, developing a general wet-chemical method for synthesizing various kinds of halide electrolytes is of great importance.
2.2.2 Ammonium-Assisted Synthesis
Recently, the universal ammonium-assisted wet-chemistry route for preparing various halide SSEs with nanoscale particles was successfully developed, which is a crucial breakthrough for realizing large-scale halide production.
Inspired by the industrial preparation of high-purity anhydrous rare-earth chlorides, Wang et al. [50] first introduced NH4Cl into an aqueous solution of YCl3 and LiCl as a coordination agent to preferentially generate the (NH4)3[YCl6] intermediate, and after the moisture was removed in vacuum at 80 °C, the acquired powders were pressed into pellets, followed by annealing at 500 °C, in which the intermediate could react with LiCl to synthesize Li3YCl6 with a high ionic conductivity of 0.345 mS cm−1 at RT. Through this method, a series of halide SSEs, such as Li3ScCl6, Li3ErCl6 and Li3YBr6 with ionic conductivities of 1.25, 0.41 and 1.09 mS cm−1 at RT, were successfully synthesized, proving that the ammonium-assisted route is a universal method to prepare various halide SSEs with decent ionic conductivity. Shi et al. [49] developed a vacuum evaporation-assisted (VEA) method for successfully synthesizing the halide Li3HoBr6 by employing a cost-effective rare-earth raw material (Ho2O3). Specifically, the mixed precursor powders of Li2CO3 and Ho2O3 were introduced to a HBr solution, followed by adding NH4Br as a coordinator, and after removing the moisture, the resulting powders were heat treated under vacuum and inert gas protection to synthesize high-quality Li3HoBr6 with a high ionic conductivity of 1.25 mS cm−1 at RT. By employing the VEA method, approximately 10 g of Li3HoBr6 with well-dispersed and nanosized particles can be prepared each time. This synthesis approach provides an important breakthrough for large-scale manufacturing of rare-earth halide SSEs with cost-effective raw materials and thus offers a critical economically viable foundation for realizing practical application of rare-earth halide-based ASSBs.
In summary, wet-chemical synthesis methods can be successfully employed to synthesize various types of high-purity halide SSEs and have shown extensive development prospects for practical applications because of the easy large-scale production, time-saving processes and homogeneity of the resulting electrolytes. However, there are still some challenging issues to be solved when synthesizing halide SSEs through liquid-phase synthesis routes. First, the synthesis process and operating conditions are generally demanding since halide SSEs are sensitive to the ambient atmosphere. Moreover, the introduction of ammonium salts induces the generation of corrosive gases, such as HCl and HBr, and thus, a special apparatus with corrosion resistance should be utilized, especially in large-scale production processes. In addition, halide SSEs synthesized by wet-chemical methods exhibit decreased ionic conductivity compared with halide SSEs synthesized by solid-state reaction routes, so an in-depth understanding of the chemical reactions involved in liquid synthesis methods is indispensable for revealing the complete reaction mechanism.
2.3 Chemical Vapor Synthesis
Chemical vapor synthesis routes are mainly employed for synthesizing thin-film halides, especially fluoride-based halides, and they can usually be categorized into thermal evaporation methods and vapor deposition methods.
In 1981, Qi et al. [76] successfully prepared mLiF–nAlF3 films with different stoichiometric ratios (1/3 \(\leqslant\) m/n \(\leqslant\) 3) and a thickness of 0.8 μm by using a thermal evaporation method in a high-vacuum environment, and the ionic conductivity of the amorphous films could exceed 10−6 S cm−1 at RT. Only 3LiF–1AlF3 or Li3AlF6 can exhibit a stable state, which is primarily ascribed to the existence of the structure of vertex-sharing [AlF6]3− octahedra. Subsequently, the authors synthesized a series of mLiF–nMF3 (M = Al, Cr, Sc, Ti, V, Cr, Ga, Y and Ce) and mLiF–nMF2 (M = Mg, Ca, Ni, Cu, Zn, and Sr) thin films by thermally evaporating the precursor materials in a high-vacuum environment for 10 min [21, 77]. Notably, F-based halides with wider electrochemical windows can be used as ionic interface layers for metal oxide cathode materials to enhance the stability and energy density of full cells. For example, Xie et al. [78] successfully coated LiAlF4 on the surface of LiNi0.8Mn0.1Co0.1O2 by an atomic deposition method and found that a battery based on LiAlF4-coated cathode materials can exhibit stable cycling performance over a wide electrochemical window of 2.75–4.5 V vs. Li+/Li. In summary, chemical vapor synthesis routes are limited to preparing fluorine-based halide films and have underlying scalable production and high cost of equipment issues; thus, they may preferentially be used to meet the special requirement of fluorine-based halide films for assembling high-performance full cells.
3 Strategies for Improving the Ionic Conductivity
To date, only a few halide SSEs can experimentally show an intrinsic ionic conductivity over 1 mS cm−1 at RT, and the majority of halide SSEs deliver far less satisfactory lithium diffusion properties compared to traditional liquid electrolytes, requiring valid strategies to further improve the ionic conductivity to facilitate the application of halide SSEs in ASSBs. Substitution of elements with different valences and ionic radii is acknowledged to be a feasible strategy to improve the ionic conductivity of halide solid electrolytes by tuning the disorder in cation/anion sublattices and vacancy concentrations [55, 62, 63, 79]. Furthermore, regulating the composition of halide compounds also offers an available methodology to achieve distinct ionic conductivity by optimizing the metal/vacancy site occupations and the carrier concentrations in the microcrystal structure [57, 60, 80]. The effective strategies for improving the ionic conductivity of halide solid electrolytes are summarized in Fig. 7.

Effective strategies to enhance the ionic conductivity of halide solid electrolytes
3.1 Aliovalent Cation Substitution
The substitution of central metals in halide SSEs with aliovalent cations can be used to tune the distribution and concentration of Li+ or change the local structure framework to improve the ionic conductivity. Notably, the preferential utilization of elements with high abundance in the Earth’s crust for central metal substitution in halide electrolytes offers a promising option to realize commercial application of halides in ASSBs.
Recent studies have demonstrated that the ionic conductivity of Li3MCl6 (M = Y, In, Yb, Er) [46,47,48] and Na3MCl6 (M = Y, Er) [66, 68] can be effectively improved through central metal substitution with tetravalent Zr4+ in the structural framework of halide electrolytes. Park et al. [46] successfully synthesized new mixed-metal halides Li3−xM1−xZrxCl6 (M = Y, Er; x = 0–0.6) by substituting Y3+ (r = 90 pm) and Er3+ (r = 89 pm) with Zr4+ (r = 72 pm), reaching the corresponding highest RT ionic conductivity of 1.4 mS cm−1 for Li2.5Y0.5Zr0.5Cl6 and 1.1 mS cm−1 for Li2.5Er0.63Zr0.37Cl6. The authors reported that the gradual substitution of Zr4+ for Y3+ and Er3+ is accompanied by a structural evolution from the trigonal to orthorhombic phase through the reduction of the average transition metal ion radius (Fig. 8a–c), which is beneficial for increasing the number of additional lithium vacancies to improve the ionic conductivity. Bond valence site energy (BVSE) analysis revealed the emergence of one metastable octahedral interstitial site (“Oct.”, Fig. 8d) in the microcrystalline structure, which shares a trigonal face with the (Er1/Zr1) octahedron and yields the most favorable 1D ion transport path of a [Li3 − Oct. − Li3 − Li2] zigzag chain running along the [010] direction (Fig. 8d1, red portion). As a result, a 3D transport network for lithium ions in Li2.5Er0.5Zr0.5Cl6 can be formed by the intersection between this pathway in the ab plane and other chains of [Li2–Li3–Li1] running in the ac plane (Fig. 8d1, green and blue portions). The corresponding ion migration pathway is depicted in the bond valence energy map (Fig. 8e), viewed as the yellow isosurface of constant EBVSE(Li) for lithium in the model. In contrast, the BVSE calculations for Li3ErCl6 (Fig. 8d2) indicated that the migration energy barrier for lithium ions is approximately 1.5 times higher than that of Zr-substituted Li3ErCl6. Consequently, the additional Li sites induced by Zr substitution are a decisive factor for enhancing the ionic conductivity of halide electrolytes. Helm et al. [48] demonstrated that substitution of Zr for a central metal in Li3InCl6 can provide a series of Li3−xIn1−xZrxCl6 (0 \(\leqslant\) x \(\leqslant\) 0.5) solid solutions, which allows an increase in the RT ionic conductivity from the initial 0.47 to 1.25 mS cm−1 for Li2.4In0.6Zr0.4Cl6. This increase is primarily attributed to the incorporation of an optimal Li+/vacancy density and the induced cation site disorder capable of forming a three-dimensional Li+ diffusion pathway. Park et al. [55] synthesized a suite of Li3−xYb1−xMxCl6 (M = Hf, Zr; 0 < x < 1) materials by substituting Hf4+ and Zr4+ for Yb3+ (Fig. 8f), and the highest ionic conductivity of 1.5 mS cm−1 was achieved for Li2.6Yb0.6Hf0.4Cl6 synthesized at 400 ℃ (Fig. 8g), benefiting from the newly generated cubic close-packed (ccp) monoclinic structure with substituted halides being more favorable for Li+ diffusion than the hexagonal close-packed (hcp) orthorhombic structure of Li3YbCl6.

Reproduced with permission from Ref. [46]. Copyright 2020, American Chemical Society. f Schematic diagram of the phase evolution of Hf4+-substituted Li3YbCl6 synthesized by annealing at 400 or 500 °C. g Ionic conductivity and corresponding activation energies for Li3−xYb1−xHfxCl6. Reproduced with permission from Ref. [55]. Copyright 2021, Elsevier
Structural evolution and corresponding Li+ conductivity of heterovalently substituted halide SSEs. a XRD patterns of Li3−xEr1−xZrxCl6. b, c Ionic conductivity and corresponding activation energy of Li3−xEr1−xZrxCl6 and Li3−xY1−xZrxCl6. d (d1, d2) BVSE analysis of migration barriers for Li-ion migration within the Li2.5Er0.5Zr0.5Cl6 and Li3ErCl6 structures. e Li-ion migration pathway of Li2.5Er0.5Zr0.5Cl6 in the ab plane.
The newly discovered Li2ZrCl6 free of rare-earth metals has received extensive attention due to its cost-effective starting materials and good ionic conductivity as well as its high humidity tolerance. By employing the more inexpensive and earth-abundant Fe element, Kwak et al. [56] reported that the aliovalent substitution of Li2ZrCl6 with Fe3+ dramatically improved the ionic conductivity from 0.4 mS cm−1 to the maximum of 1 mS cm−1 for Li2.25Zr0.75Fe0.25Cl6, and the substitution of Zr4+ with trivalent Cr3+ and V3+ also exhibited beneficial effects, as shown in Fig. 9a, b. The Fe3+ substitution induced shrinkage of ZrCl62− octahedra and augmentation of the bond covalency, which could enlarge Li+ channels and reduce the energy landscape for favorable Li+ migration. Subsequently, the authors found that aliovalent substitution with the trivalent metals In3+ and Sc3+ was also feasible for improving the Li+ conductivity of Li2ZrCl6, and a more than two orders of magnitude improvement could be easily achieved for Li2+xZr1−xMxCl6 (M = In, Sc; 0 \(\leqslant\) x \(\leqslant\) 1.0), with a maximum of 2.1 mS cm−1 (M = In, x = 0.7) at 30 ℃ (Fig. 9c, d). This improvement is primarily attributed to the expanded anisotropic lattice volume and the increased concentration of Li+ in the (002) plane, making Li+ migration more favorable in the microcrystalline structure.

Reproduced with permission from Ref. [56]. Copyright 2021, Wiley–VCH. c, d XRD patterns and ionic conductivities with the corresponding activation energies of Li2+xZr1−xMxCl6 (M = In, Sc). Reproduced with permission from Ref. [62]. Copyright 2022, Wiley–VCH. e (e1–e4) Plots of the probability density of Na+ and Cl− in Na3YCl6 and Na2.25Y0.25Zr0.75Cl6 from AIMD simulations. f, g Ionic conductivities and XRD patterns of Na3−xY1−xZrxCl6. Reproduced with permission from Ref. [66]. Copyright 2021, Nature Publishing Group
Effect of central metal substitution on the structural evolution and ionic conductivity of representative halide SSEs. a, b XRD patterns and ionic conductivities with the corresponding activation energies of Li2+xZr1−xMxCl6 (M = Fe, V, Cr).
Regarding Na-based halide SSEs, aliovalent substitution of the central metals of Na3MCl6 (M = Y, Er) with tetravalent Zr4+ also proved to be an effective approach for improving the ionic conductivity. Wu et al. [66] reported aliovalent substitution of Y3+ in the sodium-based halide Na3YCl6 with tetravalent Zr4+, and a series of Na3–xY1–xZrxCl6 (0 < x < 0.875) materials were successfully synthesized. They demonstrated that the impurity phase appeared in the X-ray diffraction (XRD) pattern at 9.6° and 10.5° when the Zr value was increased to 0.875 (Fig. 9e), and the highest ionic conductivity of 6.6 × 10−5 S cm−1 at RT was achieved for Na2.125Y0.125Zr0.875Cl6 (Fig. 9g). Ab initio molecular dynamics (AIMD) trajectory simulations indicated that the Na+ trajectories exhibited mostly local Na+ motion with little long-range transport (Fig. 9f1) and that Cl− remained relatively static (Fig. 9f3) in Na3YCl6, while macroscopic 3D Na+ diffusion (Fig. 9f2) and substantial Cl− motion corresponding to YCl63−/ZrCl62− octahedra rotations (Fig. 9f4) were observed in Na2.125Y0.125Zr0.875Cl6. The synergistic effect between the increased carrier concentration and the polyanionic rotation was concluded to result in remarkably improved Na+ conductivity upon Zr incorporation. Similarly, Schlem et al. [68] found that Zr4+-substituted Na2.4Er0.4Zr0.6Cl6 exhibits an appreciably improved ionic conductivity of ~ 0.04 mS cm−1 compared with the value of 10−9 S cm−1 for Na3ErCl6. Accordingly, heterovalent substitution of Zr for the expensive central metal element of sodium-based halides is highly desirable not only for appreciable enhancement of the ionic conductivity but also for clear cost effectiveness to further realize practical manufacture of sodium-based halide electrolytes.
3.2 Isovalent Cation Substitution
Because the various isovalent cations have different ionic radii and chemical bonding properties, isovalent cation substitution of central metals in halide compounds can effectively optimize the parameters of the unit cell structure and carrier concentration to achieve enhancement of the ionic conductivity.
Li et al. [81] investigated the effect of substituting In3+ for the central metal in Li3YCl6 on its structural evolution and ionic conductivity, and a series of Li3Y1−xInxCl6 (0 \(\leqslant\) x < 1) materials were synthesized by mechanical milling with postannealing at 260 °C. Li3YCl6 has an orthorhombic structure without the introduction of In3+, and the corresponding crystal structure can transform to a trigonal structure when the substitution ratio of In3+ increases to 0.1, whereas both microcrystalline structures are based on hcp anion arrangements. The gradual structural conversion from the hcp anion arrangement to the ccp anion arrangement can be traced when the substitution ratio of In3+ is over 0.2, which allows a dramatic improvement in the ionic conductivity, with a maximum of 1.42 mS cm−1 for Li3Y0.5In0.5Cl6. The In3+ substitution can be interpreted to induce conspicuous changes in the lithium substructure and the metal-Cl covalency along with the generation of the ccp anion arrangement, which are responsible for the appreciably facilitated fast Li+ transport in the microcrystalline structure of In-substituted Li3YCl6. Zhou et al. [59] reported a family of lithium mixed-metal chlorospinels Li2InxSc0.666−xCl4 (0 < x < 0.666) with a relatively low activation energy of ~ 0.33 eV as well as a high Li+ conductivity of 1.80–2.03 mS cm−1, which is much higher than that of spinel-type superionic Li2Sc2/3Cl4 (1.5 mS cm−1). Li2InxSc0.666−xCl4 exhibits a pure cubic spinel microcrystalline structure within the corresponding solid solution range of 0 \(\leqslant\) x < 0.444, whereas the impurity phases Li3InCl6 and LiCl appear in high In3+ content (x > 0.555) compounds, as determined by XRD. The highest ionic conductivity of 2.03 mS cm−1 at RT can be achieved for Li2In1/3Sc1/3Cl4 with four Li sites per unit cell, similar to the crystallite structure of Li2Sc2/3Cl4 but with different occupation, which primarily benefits from the three-dimensional Li-ion diffusion pathway induced by the highly disordered Li-ion distribution in the spinel structure. More importantly, Li2In1/3Sc1/3Cl4 with an ultralow electronic conductivity of 4.7 × 10−10 S cm−1 could be successfully applied in high-output-voltage (4.8 V vs. Li+/Li) ASSBs with the LiNi0.85Co0.1Mn0.05O2 CAM and simultaneously deliver stable cycling with very slow capacity degradation during the charge‒discharge process. Consequently, isovalent cation substitution of central metals in halide compounds can be regarded as a promising option to further enhance the ionic conductivity and stability of halide SSEs.
3.3 Anion Substitution
The central metal of halide SSEs can be incorporated with various halogen anions to crystallize different types of lattice structures, such as Li3ErCl6 (trigonal, P\(\overline{3}\)m1) and Li3ErBr6 (monoclinic, C2/m), which thus offers an efficient approach to enhance the ionic conductivity through the design of halide structures based on halogen anion substitution. Liu et al. [63] successfully synthesized a new family of halide electrolytes Li3MBr3Cl3 (M = Y, Er) by substituting Br− for halogen anions in Li3MCl6 (M = Y, Er), and the Li3YBr3Cl3 obtained by mechanochemical milling exhibited a high ionic conductivity of 1.6 mS cm−1 at RT, which is threefold higher than that of Li3YCl6. Notably, interesting coexistence of Li tetrahedral occupation (1/3 in total) and octahedral occupation (2/3 in total) in the layered structure of Li3YBr3Cl3 is achieved through substitution of Br−, along with corresponding changes in the lattice parameters, which is rarely reported in other halide electrolytes but reasonable in thermodynamics. The presence of tetrahedral Li can induce more vacancies at the octahedral sites as well as change the energy landscape by making it flat, consequently giving rise to the appreciably enhanced Li+ diffusion pathway in the microcrystalline structure of Li3YBr3Cl3. Moreover, the ionic conductivity of the Li3YBr3Cl3 compound could be further improved to 7.2 mS cm−1 at RT after a hot-pressing process at 170 °C, mainly ascribed to the Br− substitution inducing rapid 3D diffusion pathways, resulting from a mass of Li at the tetrahedral sites as well as enhancement of grain boundary contact through hot-pressing. Due to the higher ionic conductivity of Li3YBr3Cl3, a primarily improved specific capacity of the full cell fabricated with Li3Ybr3Cl3 was achieved compared to the cell with Li3YCl6. Through this method, a noteworthy enhancement of the ionic conductivity could also be obtained for Li3ErBr3Cl3, indicating that this anion substitution strategy appears to be general in terms of improving the ionic conductivity for mixed anionic halide electrolytes. Tomita et al. [43] investigated the effect of Cl− substitution of Br− in Li3InBr6 on its ionic conductivity and thermal stability, and a series of Li3InBr6−xClx (0 < x < 6) solid solutions were prepared by mechanical ball milling and subsequent annealing. They reported that Li3InBr3Cl3 exhibited the highest ionic conductivity of 1.2 × 10−4 S cm−1 at 300 K, and the phase transition temperature was lowered to approximately 295 K. The substitution of bromine with chlorine was demonstrated to allow reduction of the lattice volume and induction of partial diffusion of lithium ions, which was confirmed by 7Li NMR analysis that showed faster diffusion of lithium ions in Li3InBr3Cl3.
3.4 Heterogeneous Compositing
A heterogeneous compositing strategy can be used to enhance the ionic conductivity of halide SSEs by introducing uniformly dispersed heterogeneous materials, and it is preferentially applied to the case of halide compounds with halogen fluoride anions. Typically, the introduced heterogeneous phase could exhibit chemical affinity for the mobile ions on the surface of halide compounds and absorb the mobile cations, resulting in defect concentration and consequently enhancement of the ionic conductivity of halide solid electrolytes [82, 83].
Feinauer et al. [84] investigated the effect of introducing nanocrystalline alumina (γ-Al2O3) on the ionic conductivity of the halide β-Li3AlF6, and a series of β-Li3AlF6 + Al2O3 (2–20 wt%) composites were prepared through mechanical milling. The ionic conductivity of the halide β-Li3AlF6 can be increased up to 10 times through compositing with 2 wt% γ-Al2O3, whereas a higher content will not further enhance the ionic conductivity because of the blocking effect. Notably, for the corresponding assembled ASSBs, the first charge capacity of the cell with Li//β-Li3AlF6 + γ-Al2O3//LiMn2O4 can reach 108 mAh g−1, which is 33 mAh g−1 higher than that of the cell based on β-Li3AlF6 under the same test conditions. This difference is primarily attributed to the substantially enhanced ionic conductivity of β-Li3AlF6 induced by heterogeneous compositing with nanocrystalline alumina. In addition, Miyazakiz et al. [85] reported that the ionic conductivity of nanocrystalline β-Li3AlF6 at RT can be dramatically increased from 7 × 10−7 to 2 × 10−6 S cm−1 through compositing with LiCl, mainly ascribed to the mixing anion effect and the dispersion effect related to the space charge region. The heterogeneous compositing strategy exhibits an easily operated advantage by coupling uniformly dispersed materials to enhance the ionic conductivity of the prepared halide compounds, which also offers a potential way to optimize the performance of fluorine-based halide electrolytes for realizing their application in ASSBs.
3.5 Regulating the Composition
Regulating the composition of halide compounds enables optimization of metal/vacancy site occupations, which is conducive to achieving a balance between the Li+ carrier concentration and total vacancy content for enhancing the ionic conductivity of halide SSEs. Variation of the halide composition within a certain range can simultaneously induce a gradual transformation of the crystallite structure, which presents an opportunity for obtaining optimally uninterrupted Li+ diffusion channels in halide compounds. Liang et al. [57] discovered a series of LixScCl3+x (x = 2.5, 3, 3.5 and 4) superionic halide electrolytes with RT ionic conductivities over 1 mS cm−1 based on the ccp anion sublattice and increasing the x value in LixScCl3+x gave rise to augmentation of the Li+ carrier, while the opposite trend was observed for the total vacancy content. The optimal balance was obtained for Li3ScCl6 with appropriate Li+ carrier and vacancy concentrations, and consequently, Li3ScCl6 achieved the highest ionic conductivity of 3.02 mS cm−1 at RT among the as-synthesized Li–Sc–Cl halide compounds. Wang et al. [53] investigated the effect of composition variation on the ionic conductivity of Li–Zr–Cl-type halide SSEs and reported that Li2ZrCl6 delivered the highest ionic conductivity of 0.81 mS cm−1 at RT compared to LiZrCl5 (0.15 mS cm−1) and Li3ZrCl7 (0.27 mS cm−1), although these halide compounds could remain phase-pure. Xu et al. [80] synthesized a series of Li3−xYb1+xCl6 (x = 0.017, 0.034, 0.053, 0.071) materials with various degrees of Li deficiency to improve the ionic conductivity of Li–Yb–Cl-type halide electrolytes by increasing the Li vacancies in the lattice structure. Although all prepared samples exhibited an orthogonal crystal structure, Li2.966Yb1.034Cl6 showed the highest ionic conductivity of 0.5 mS cm−1 at RT, benefiting from the optimal balance between the carrier and vacancy concentrations.
Optimizing the composition of halide compounds can be simultaneously employed to regulate their initial crystal structure to enhance the ionic conductivity. Liang et al. [60] reported a series of Li3−3xHo1+xCl6 (− 0.14 < x \(\leqslant\) 0.5) halide superionic conductors with a transition from the trigonal to orthorhombic structure when x was gradually increased. The halide Li2.73Ho1.09Cl6 with the orthogonal structure can exhibit the highest ionic conductivity of 1.3 mS cm−1 at RT, which is four times higher than that of Li3HoCl6 with the trigonal structure, mainly because of the facile diffusion in the z-direction in the orthorhombic structure for Li2.73Ho1.09Cl6. Moreover, the trigonal-to-orthorhombic phase transition has been proven to be universal in Li3−3xM1+xCl6 (− 0.14 < x \(\leqslant\) 0.5, M = Dy, Y, Er, Tm). Specifically, the orthorhombic phase of Li2.73M1.09Cl6 containing Dy, Y, Er, and Tm delivers superior ionic conductivities of 0.9, 0.7, 0.64 and 0.89 mS cm−1 at RT, respectively, which are an order of magnitude higher than the conductivities of the corresponding trigonal phases of Li3MCl6. This trigonal-to-orthogonal structural transition may be a prevailing phenomenon in many other types of Li3−3xM1+xCl6, and further investigation of it is highly desirable to discover new superionic halide electrolytes. Consequently, precise control of the composition of halide compounds can be considered a feasible strategy for improving the ionic conductivity of halide SSEs by achieving the optimal ion diffusion channel in their corresponding microcrystalline structures.
4 Chemical Stability
The chemical stability of halide SSEs is regarded as a crucial performance indicator that could significantly affect their implementation in their whole life, from synthesis, storage, and transportation to application, and is closely related to the operation of the material and the application cost of manufacturing ASSBs. Chemically unstable electrolyte materials suffer severe mechanical and chemical degradation as a result of the destruction of the crystalline structure, which can occur even prior to electrochemical cycling, resulting in unsatisfactory ionic conductivity, high interfacial resistance, and degraded cell performance [24]. Consequently, rational optimization of the chemical stability enables effective regulation of production processes and battery configurations, which in turn offers a favorable guarantee for reducing production costs to maintain performance standards and large-scale commercial applications.
4.1 Air/Humidity Stability
4.1.1 Degradation Mechanisms under Humid Air
An in-depth and comprehensive understanding of the influencing factors of humid air on various halide SSEs and the corresponding degradation mechanisms is highly beneficial for facilitating efficient application of different halide compounds and obtaining feasible strategies against degradation. Halide solid electrolytes exhibit better air stability than sulfide electrolytes and can avoid the generation of toxic H2S, which is favorable for safe operation [21, 28]. Moreover, halide solid electrolytes that can maintain decent stability in dry oxygen environments, such as Li3YCl6 and Li3YBr6 [44], will not undergo degradation in terms of decomposition, phase transition and oxidation reactions. Nevertheless, recent studies have indicated that the majority of halide electrolytes are unstable when exposed to ambient atmosphere and can react with moisture to form different hydrated products, including MCl3·xH2O and LiCl·xH2O [86, 87]. As a consequence, an irreversible degradation reaction occurs, and MCl3·xH2O is transformed into M2O3 and HCl during the thermal dehydration process. In the case of Li3YCl6, the corresponding degradation reactions can be described by Eqs. (1) and (2) [21, 87, 88].
The halide Li3InCl6 exhibits superior tolerance to ambient air compared to other halide compounds, which is mainly attributable to the preferential formation of hydrated intermediates (Li3InCl6·xH2O) that can effectively resist the constant occurrence of harmful hydrolysis reactions (Fig. 10a) [87]. Notably, the conversion from Li3InCl6 to Li3InCl6·xH2O is reversible, which means that the initial structure and ionic conductivity of Li3InCl6 can be recovered after removing the crystal water through a simple dehydration process [52, 81].

Reproduced with permission from Ref. [87]. Copyright 2022, Wiley–VCH. c Hydrolysis reaction energy for binary chlorides M–Cl, ternary lithium chlorides Li–M–Cl, and ternary sodium chlorides Na–M–Cl. Reproduced with permission from Ref. [89]. Copyright 2020, Wiley–VCH. d Illustration of the humidity stabilities of Li3Y1−xInxCl6 and Li3YCl6. e Arrhenius plots of Li3Y0.2In0.8Cl6 before and after air exposure. Reproduced with permission from Ref. [81]. Copyright 2020, American Chemical Society. f Schematic illustration of coating Li3InCl6 with Al2O3 by powder atomic layer deposition. g Digital photos of powder and pellet samples for Li3InCl6 over time in ambient air with (35 ± 5)% relative humidity. Reproduced with permission from Ref. [87]. Copyright 2022, Wiley–VCH
Moisture stability of halide SSEs. a Schematic diagram of the Li3InCl6 degradation mechanism under humid air, and b XRD pattern and digital photo of Li3InCl6 reacted with Al foil for 12 h.
However, Li3InCl6 will inevitably undergo decomposition to form InCl3 and LiCl with long-term exposure, and InCl3 further hydrolyzes to produce In2O3 and HCl through the formation of the unstable intermediate phase In(OH)3; the related reactions are shown in Eqs. (3) and (4) [87, 88]. The formation of the acidic product HCl can corrode aluminum foil (Fig. 10b) [87], which is commonly used as a current collector for battery assembly, and thus possibly cause a safety hazard in application. In addition, Wang et al. [53] demonstrated that Li2ZrCl6 exhibits better stability than Li3InCl6 in an atmosphere with 5% relative humidity. However, the degradation mechanism of Li2ZrCl6 with high humidity tolerance is still unclear and conducting in-depth and comprehensive research in the future is highly desirable.
4.1.2 Strategies to Improve Humid Air Stability
The air/humidity tolerance of halide SSEs is closely associated with the type of central metals in the halide compounds with various valences and ionic radii [89]. In addition, the water absorption rate of the halide electrolytes can be changed by adjusting the contact area of the solid/gas interface and the particle size of halide compounds to effectively regulate the degradation rate of the halide in practical applications.
Zhu et al. [89] investigated the moisture stability of Li- and Na-containing halide compounds A3MCl6 (A = Li/Na, M = cation) by quantifying the hydrolysis reaction energy, and corresponding thermodynamic analyses based on a first-principles computational database were performed, as shown in Fig. 10c. Their results demonstrated that sodium ternary chlorides with lanthanide cations (Tm3+, Er3+, Ho3+, Tb3+, etc.) exhibit better moisture stability, among which the best humidity tolerance is achieved for Na3TmCl6. Lithium ternary chlorides with trivalent metal ions (In3+, Ga3+) and transition metal ions (Cd2+, Zn2+) deliver excellent moisture stability, and halide Li3InCl6 possesses the best moisture resistance, which is also in accordance with the superior humidity tolerance of Li3InCl6 reported experimentally. The results also imply that several of these identified cations with decent moisture stability are viable elements for doping/substitution to enhance the humidity tolerance of halide SSEs.
Li et al. [81] investigated the effect of isovalent In3+ substitution of the central metal in Li3YCl6 on the humidity tolerance of Li3YCl6 and demonstrated that Li3Y1–xInxCl6 (x > 0.5) with a high In3+ content exhibited significantly improved moisture stability. For instance, pristine Li3Y0.2In0.8Cl6 delivered an ionic conductivity of 1.23 mS cm−1 at RT, and the reheated Li3Y0.2In0.8Cl6 after 3%–5% humidity exposure still exhibited a relatively high ionic conductivity of 1.05 mS cm−1 at RT (Fig. 10f), making the corresponding conductivity retention appreciably increase to 85.37% compared to the poor conductivity retention of 0.8% for Li3YCl6. The hydrated intermediate Li3(Y1−xInx)Cl6−xH2O is preferentially formed because of the generation of (Y/In)Cl63− octahedra in the crystallite structure after In-substituted halide compounds are exposed to a humid atmosphere, which is beneficial for the efficient recovery of the initial halide structure after removing crystal water (Fig. 10d); thus, a dramatically improved conductivity retention is achieved. In contrast, the unsubstituted Li3YCl6 is thoroughly hydrolyzed into separated products involving LiCl·H2O and YCl3·6H2O after exposure to moist air under the same conditions, which cannot be converted back by heat treatment (Fig. 10e).
Wang et al. [87] proposed enhancing the moisture stability of Li3InCl6 by coating a layer of Al2O3 on the surface of Li3InCl6 via powder atomic layer deposition (Fig. 10g). They reported that the water absorption rate of Li3InCl6@Al2O3 (5.16 mg h−1) could be decreased to 1/4 of that of the as-synthesized Li3InCl6 (20.83 mg h−1), and the corresponding liquefaction time was significantly improved by seven times. Consequently, coating Li3InCl6 with an Al2O3 protective layer can be regarded as a valuable methodology to hinder hydrolysis degradation of pure Li3InCl6, although the corresponding ionic conductivity slightly decreased after introducing the Al2O3 protective layer. The authors simultaneously reported that the Li3InCl6 pellet samples obtained from cold pressing over 300 MPa could maintain a round shape after exposure to humid air for 8 h, whereas for the Li3InCl6 powder, more than half of the sample turned into a solution after only 1 h of exposure (Fig. 10h) under the same conditions. The relative density of Li3InCl6 is significantly improved from powder to pellet samples, which can greatly reduce the contact area of the solid/gas interface, and thus, the pellet samples exhibited much better moisture stability than the powder samples for Li3InCl6 halide electrolytes.
In summary, the spontaneous degradation of halide SSEs in a humid atmosphere is a crucial challenge hindering large-scale manufacturing and application of halide-based ASSBs. In the future, effective strategies for enhancing the humidity tolerance of halide SSEs to further develop, through optimization of the design, novel moisture-stable halide electrolytes and for exploiting valuable protection approaches are highly desirable.
4.2 Solvent Compatibility
Emerging halide electrolytes are considered promising candidates for facile processing of ASSBs due to the advantages of reliable deformability and high ionic conductivity. Despite this, the solid electrolyte pellets prepared by cold pressing exhibit weak mechanical properties, such as cracks generated during the pressing process [90, 91], that need to be compensated for, which severely limits the large-scale processing of solid electrolytes for the production of commercial ASSBs. Solvent-based processes have received extensive attention for fabricating high-energy–density composite cathodes and ultrathin composite electrolyte membranes with outstanding electrochemical performance [28, 92], which are capable of achieving tight contact between materials and effectively avoiding the occurrence of mechanical failures during the pressing process. Consequently, solvent-based processes can be regarded as a highly reliable strategy for realizing commercial application of halide-based ASSBs.
Generally, maintaining good stability of halide SSEs in solvents is of great importance, and the requirements for an acceptable solvent are that it should not cause chemical or structural detrimental effects or loss of the ionic conductivity of halide SSEs. Unfortunately, the severe reactivity of halide SSEs with polar solvents greatly restricts the widespread application of common solvents [51]. As shown in Fig. 11a, halide SSEs usually exhibit relatively weak interactions with nonpolar or weakly polar solvents. However, because of the strong interaction between halide compounds and polar solvents, halide SSEs can easily react with polar solvents, accompanied by dissolution through the loss of the original microcrystal structures, resulting in reduction of the ionic conductivity to some degree. The possible nucleophilic attacks of solvents on halide solid electrolytes are illustrated in Fig. 11b. Highly polar solvents are considered strong Lewis bases, which contain lone-pair electrons of electronegative elements such as N and O and can react with electrophilic central metals with high valence in halide solid electrolytes according to the hard and soft acids and bases (HSAB) principle [93], giving rise to challenging issues in terms of the poor compatibility of halide compounds with polar solvents.

Reproduced with permission from Ref. [51]. Copyright 2022, American Chemical Society
Solvent compatibility of halide SSEs. a Schematic illustration of the compatibility between halide SSEs and organic solvents with various degrees of polarity. b Possible nucleophilic attacks by different solvents on halide SSEs. c, d Ionic conductivities and dissolved amounts of Zr for Li2ZrCl6 after exposure to various solvents, and corresponding XRD patterns.
Kwak et al. [51] investigated the compatibility of Li2ZrCl6 with different organic solvents by measuring the ionic conductivities and detecting the amount of Zr4+ dissolved in organic solvents. They found that nonpolar dibromoethane (DBM) could only dissolve a very small amount of Li2ZrCl6, corresponding to only 9.9 ppm (1 ppm = 1 μmol mol−1) Zr4+ detected in the mixed suspension with DBM (Fig. 11c), which is also in accordance with the retention of relatively complete XRD peaks and the high ionic conductivity retention achieved after removing the solvent (Fig. 11d). The halide Li2ZrCl6 exhibited severe dissolution in both the strongly polar solvent N-methyl-2-pyrrolidinone and the intermediate-polarity solvent hexyl butyrate. Unexpectedly, even when Li2ZrCl6 was exposed to nonpolar (o-xylene) or weakly polar (toluene) solvents, the main XRD peaks of Li2ZrCl6 disappeared after removing the solvent, accompanied by the appearance of the impurity phase of LiCl (Fig. 11d), indicating that the microcrystal structure of Li2ZrCl6 severely collapsed, leading to a remarkable loss of the ionic conductivity (3 × 10−10 S cm−1 at 30 °C). This work implied that halide electrolytes may have stringent requirements for compatible solvents. To date, reports on acceptable solvents for halide SSEs are rare; considering the relatively high reactivity between common polar solvents and halide compounds, the selection of compatible solvents requires great efforts.
Consequently, to obtain acceptable solvents for realizing application of solvent-based scalable processes for halide SSEs, revealing the degradation mechanisms of halide electrolytes when they are exposed to incompatible solvents and further understanding the stability of halide electrolytes in various types of organic solvents are of great significance.
5 In Situ/Operando Characterization Techniques Applied to Halide-Based ASSBs
The development of high-performance halide-based ASSBs requires an in-depth understanding of the electrochemical reaction processes, degradation mechanisms and dynamic evolution of the electrolyte under realistic operating conditions. This understanding could be achieved through in situ/operando characterization techniques, which offer information on the structural evolution of electrolytes, redox mechanisms and solid-electrolyte interphase (SEI) formation under continuous operation. Compared with traditional ex situ characterization techniques, which are restricted to the understanding of the starting and ending states of materials and batteries and have the characteristics of data uncertainty and hysteresis, in situ/operando characterization techniques are beneficial for gaining deeper insight into the working principle of devices and the underlying reaction mechanisms of materials and are consequently capable of providing a theoretical basis for the design of ASSBs with high energy density and long cycle life.
5.1 In Situ/Operando Characterization of Electrolyte Materials
Recently, computational simulations and experimental results have indicated the existence of metastable state materials during the synthesis of halide electrolytes by the solid-state reaction route, and metastable halides can deliver appreciably enhanced ionic conductivity because of the formation of faster Li+ diffusion pathways in the microcrystalline structure [45, 55, 94]. An in-depth understanding of the synthesis conditions and dynamically stable mechanisms of metastable halides plays an important role in further developing novel halide electrolytes. More importantly, limited by the data hysteresis of ex situ characterization techniques, the underlying degradation mechanisms of halide electrolytes upon humid air exposure are still not completely understood. In situ/operando characterization methods can provide the evolution of the electrolyte microstructure, surface morphology and local chemical state during the dynamic reaction by obtaining continuous monitoring data, which offers an effective strategy to deeply investigate the dynamic structural evolution of halide electrolytes during solid-state synthesis and the degradation mechanisms in continuous operation environments.
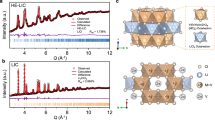
Ito et al. [95] employed in situ synchrotron XRD to investigate the solid-phase reaction of LiCl and YCl3 during the continuous heating process for synthesizing Li3YCl6 and reported that the novel metastable phase β-Li3YCl6 with smaller lattice parameters was formed in the temperature range of 450–600 K, which gradually transformed into α-Li3YCl6 in the process of heating over 600 K (Fig. 12a). The related reactions can be described by Eq. (5) and (6), and the corresponding mass fractions of various phases during the solid-state reaction are shown in Fig. 12b, calculated from the corresponding Rietveld refinement results. Furthermore, the authors found that the synthesis by heating precursor materials to 823 K followed by annealing at 595 K did not yield β-Li3YCl6, suggesting that the phase transition from β- to α-Li3YCl6 was irreversible and that β-Li3YCl6 was a metastable halide electrolyte. β-Li3YCl6 could be assigned to a cell with a 1/3 a-axis and a c-axis similar to that of α-Li3YCl6, leading to electronic repulsion between Y3+ accompanied by a broadened Li+ diffusion channel, and consequently, the ionic conductivity of β-Li3YCl6 with a disordered Y site was appreciably improved by an order of magnitude compared with α-Li3YCl6. Consequently, in situ investigation of solid-state reactions could promote detection of new cation arrangements in the microcrystalline structure of solid electrolytes, which is conducive to the discovery of novel metastable halide electrolytes with high ionic conductivity.

Reproduced with permission from Ref. [95]. Copyright 2021, Wiley–VCH. c Two-dimensional intensity color map of NPD patterns for mechanochemically synthesized α-Li2ZrCl6. d Nyquist plots of α-Li3YCl6 and β-Li3YCl6. Reproduced with permission from Ref. [53]. Copyright 2021, Springer Nature. e Schematic diagram of the operando optical microscopy and Raman spectroscopy device (middle), operando optical microscopy photos obtained with various exposure times (left), and Raman spectra evolution of Li3InCl6 exposed to moist air (right). f, g Illustration of operando SXRD for Li3InCl6, and corresponding patterns of Li3InCl6 during exposure to air with 30% humidity for 120 min. Reproduced with permission from Ref. [88]. Copyright 2020, American Chemical Society
In situ/operando characterization of halide solid electrolytes. a, b In situ synchrotron XRD patterns upon heating the mixture of LiCl and YCl3 to synthesize Li3YCl6, and corresponding mass fractions obtained from Rietveld refinement.
Wang et al. [56] applied in situ neutron powder diffraction (NPD) to study the phase evolution of mechanochemically synthesized α-Li2ZrCl6 in the temperature range from 27 to 427 °C, as shown in Fig. 12c. When the temperature is higher than 275 °C, β-Li2ZrCl6 with a structure similar to that of Li3InCl6 emerges, along with the gradual reduction of the α-Li2ZrCl6 reflection intensities, and β-Li2ZrCl6 is the only phase identified in the NPD pattern at temperatures above 350 °C. The β-Li2ZrCl6 obtained at high temperatures loses the nonperiodic features in terms of the defect structure, strain and amorphous phase, resulting in the ionic conductivity decreasing from a value of 8.08 × 10–4 S cm−1 for the initial α-Li2ZrCl6 to 5.81 × 10–6 S cm−1 (Fig. 12d).
Li et al. [88] revealed the degradation mechanism of halide Li3InCl6 when it was exposed to moist air through various in situ/operando characterization techniques. The optical images obtained from operando optical microscopy suggested that Li3InCl6 particles underwent rapid morphological changes within several minutes of exposure to 30% humidity, which confirmed the hydrophilic characteristic of Li3InCl6 (Fig. 12e). Furthermore, a new peak at 131.5 cm−1 assigned to In2O3 appeared in the operando Raman spectrum, which could be one of the products formed during the degradation of Li3InCl6, and the intensity of the O–H peak significantly increased over time, which provided evidence of hydrated halide formation. Operando synchrotron-based X-ray powder diffraction (SXRD) was employed to detect other hydrolysis products of Li3InCl6, in which a Pilatus pixel area sensitive detector was used to record the scattered X-ray beams (Fig. 12f). A new set of peaks assigned to Li3InCl6·xH2O appeared after exposure to moist air for 10 min, and a mixture of peaks at ~ 19.9° and ~ 23.2° belonging to Li3InCl6·xH2O and LiCl was detected in the SXRD pattern when the exposure time reached 120 min (Fig. 12g), indicating that both were products of the hydrolysis reaction. In addition, in situ X-ray absorption near-edge structure (XANES) results also confirmed the formation of LiCl and Li3InCl6·xH2O when Li3InCl6 was exposed to a humid environment. Based on this, the authors concluded that hydrophilic Li3InCl6 can react with moisture and finally decompose to nonionically conductive In2O3 and LiCl, while part of Li3InCl6 would absorb moisture to form Li3InCl6·xH2O when it was exposed to ambient air.
5.2 In Situ/Operando Characterization of Electrodes and Interfaces
Bulk-type halide-based ASSBs can be directly fabricated through stepwise cold pressing of materials, including solid electrolytes, CAMs and anodes, benefiting from the outstanding mechanical deformability of halide electrolytes, but the related challenging issues of the interfacial stability of the electrolyte and CAMs and the reversible redox reaction in the composite cathode require a thorough understanding [26, 96]. Specifically, the degradation mechanisms of halide electrolytes against lithium anodes during realistic electrochemical reactions require further clarification. Without disassembling batteries and extracting samples for testing, in situ characterization techniques can offer effective information on heterogeneous reactions in high-energy-density cathodes, SEI formation and degradation mechanisms during battery charge and discharge, which in turn can provide crucial strategies for optimization and design of high-performance halide-based ASSBs.
For the first time, Asano et al. [44] performed in situ XRD characterization of a bulk-type cell based on Li3YCl6 to investigate the redox reaction of composite cathodes under realistic operating conditions. A schematic diagram of the in situ XRD measurement geometry and the corresponding cell structure are shown in Fig. 13a. They reported that the (003), (006) and (104) peaks of LiCoO2 shifted to a low angle while the (101) peak slightly shifted to a higher angle during the initial charging cycle (Fig. 13b), indicating that the c-axis of LiCoO2 was elongated and the a-axis was shortened, in accordance with the LiCoO2 evolution behavior detected in situ with liquid electrolytes. However, the in situ XRD peaks of halide Li3YCl6 were not shifted during the charge process, confirming that the charging current is solely produced through Li+ extraction from LiCoO2 cathode materials. Consequently, they concluded that Li3YCl6 with outstanding electrochemical stability can be directly used for ASSBs based on uncoated high-voltage CAMs.

Reproduced with permission from Ref. [44]. Copyright 2018, Wiley–VCH. c, d In situ/operando Raman spectra of the Li3InCl6@LiCoO2 electrode during the charge and discharge processes. e Contour plots of Li3InCl6@LiCoO2 during the charge process. Reproduced with permission from Ref. [97]. Copyright 2020, Elsevier. f Schematic of Li deposition with an argon sputter gun and subsequent in situ XPS measurement. g–i X-ray photoelectron In-3d and Li-1s and Auger In-MNN spectra during/after Li deposition on Li3InCl6. Reproduced with permission from Ref. [98]. Copyright 2021, Wiley–VCH
In situ/operando characterization of electrodes and interfaces for halide-based ASSBs. a Schematic illustration of the in situ XRD measurement geometry and the corresponding cell construction. b In situ XRD patterns of the Li3YCl6-based cell during initial charging.
Wang et al. [97] used in situ/operando Raman spectroscopy to investigate the cathode interfacial stability of ASSBs with Li3InCl6@LiCoO2 composite cathodes prepared by in situ interfacial growth of Li3InCl6 on the LiCoO2 surface. The Raman peaks (191 and 269 cm−1) of Li3InCl6 remained the same at high cutoff voltages above 4 V, and no new peaks were observed, suggesting that Li3InCl6 was stable and that no side reactions occurred during the charge process (Fig. 13c). The intensity of the characteristic peaks (478 and 596 cm−1) of LiCoO2 began to decrease after charging to 4 V (highlighted by a red dashed box) and could be recovered during the discharge process (Fig. 13d), which proves that Li+ can reversibly intercalate into the layered structure of LiCoO2. The contour map of the Li3InCl6@LiCoO2 composite cathodes distinctly shows that the peak intensities at 478 and 596 cm−1 decreased after charging to 4.0 V (Fig. 13e), which is closely associated with the delithiation of LiCoO2 upon charging and LiCoO2 lattice expansion. In addition, the interfacial resistance of the Li3InCl6@LiCoO2 composite cathode was found to be only 6.8 Ω through the use of in situ electrochemical impedance spectroscopy (EIS), which is several orders of magnitude lower than that of mainstream sulfide/oxide-based ASSBs, further demonstrating that the in situ interfacial growth of halide electrolytes on CAMs possesses a significant advantage in eliminating interfacial challenges of halide-based ASSBs.
Riegger et al. [98] employed in situ X-ray photoelectron spectroscopy (XPS) to study the interfacial stability between halide electrolytes (Li3InCl6, Li3YCl6) and lithium metal anodes by sputter-depositing lithium onto halide pellets, as shown in Fig. 13f. The characteristic peak of metallic In at 443.7 eV was detected after ten minutes of lithium deposition and showed a remarkable enhancement after one hour, accompanied by an observably decreased intensity of the Li3InCl6 peak at 446.1 eV (Fig. 13g), indicating that Li3InCl6 could be reduced by lithium metal to form In metal within a short time. Furthermore, a clearly enhanced intensity of the In characteristic peak at 1 076.0 eV was simultaneously observed with increasing contact time after lithium deposition in the In-MNN Auger spectrum (Fig. 13h). In addition, the Li3InCl6/LiCl signal at 56.7 eV significantly decreased in the Li-1s spectrum after Li deposition for one hour, while the Li2O signal at 54.4 eV clearly dominated the Li-1s spectrum (Fig. 13i), indicating the occurrence of continuous interface reactions between Li3InCl6 and the deposited metal lithium. Similar reaction behaviors were also observed between the halide Li3YCl6 and lithium metal anodes, and thus, the authors proposed an idealized net reaction for halide electrolytes and metallic lithium, as shown in Eq. (7).
6 Halide SSEs for All-Solid-State Batteries
Since the discovery of Li3YCl6 and Li3YBr6 with high ionic conductivities of 0.03–1.7 mS cm−1 at RT by Asano’s group in 2018 [44], the exploration of halide compounds as solid electrolytes for advanced ASSBs has received a strong upsurge of interest and has simultaneously achieved impressive progress in recent years. For cathodes using halide SSEs, 4-V-type Ni-rich layered LiMO2 compounds (M = Ni, Co, Mo, and Al mixture) are crucial CAMs for realizing high-energy–density halide-based ASSBs, and recently developed Se- and S-based cathode materials for assembling halide-based ASSBs with higher rate capability have also received extensive attention. For the anodes, the thermodynamic instability of halide electrolytes against lithium metal anodes is caused by the intrinsically poor electrochemical reduction stability of halide compounds, which presents an ongoing challenge, especially for Cl- and Br-based halide electrolytes. The electrochemical performance and cell design of different types of ASSBs using halide SSEs as electrolytes are summarized in Table 2. An in-depth understanding of the electrochemical stability of halide SSEs and the construction of rational interface engineering are of vital significance to effectively control the interface behavior to achieve high-energy and long-life halide-based ASSBs.
6.1 Intrinsic Electrochemical Stability
The intrinsic electrochemical stability of halide SSEs is largely governed by the electrochemical stability window, and a wide electrochemical stability window is also a crucial requirement for realizing halide-based ASSBs with high energy density [21, 102]. The electrochemical stability window originates from the energy separation Eg of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the electrolytes [103]. As a result, high-throughput simulation calculations can be used for effectively analyzing the electrochemical windows of different types of halide electrolytes, which in turn are capable of offering instructive guidance for experimental design.
Wang et al. [102] calculated the thermodynamic intrinsic electrochemical windows of a series of Li–M–X ternary compounds (M = cation; X = F, Cl, Br, I, O, S, N) based on first-principles computations, as shown in Fig. 14a. The thermodynamic electrochemical window is closely related to the species of halide anions in the corresponding halide electrolytes, and the fluorine- and chlorine-based halides exhibit high oxidation stability of over 6 and 4 V vs. Li+/Li, respectively, leading to a wider thermodynamic electrochemical window. In contrast, the sulfide electrolytes show a narrow electrochemical window due to their low oxidation potential of less than 3 V vs. Li+/Li. Similar to the simulation results, related experiments have demonstrated that halide electrolytes have higher oxidation potentials and wider electrochemical windows, such as Li3ErCl6 (0.85–4.26 V) [104] and Li3ScCl6 (0.15–4.08 V) [57], which are significantly superior to those of sulfide and oxide electrolytes, such as Li6PS5Cl (1.71–2.14 V) [105], Li10GeP2S12 (1.72–2.42 V) [106], Li0.33La0.56TiO3 (1.75–3.71 V) [106] and LISICON (1.44–3.39 V) [105]. Qie et al. [35] evaluated the intrinsic electrochemical window of the ternary sodium-based halide electrolytes Na–M–X (M = cation, X = F, Cl, Br, I, O or S), and their theoretical computations showed that nearly all sodium-based halides are capable of exhibiting a 3 V oxidation potential (except for iodides), which is conducive to expanding their electrochemical windows. For instance, Na3YCl6 and Na3YBr6 deliver wide electrochemical windows of 0.51–3.75 and 0.57–3.36 V vs. Na+/Na, respectively, and such superior intrinsic electrochemical stability is more impressive compared with the reported sodium-based oxide and sulfide solid electrolytes involving Na3Zr2Si2PO12 (1.1–2.4 V), Cl-doped t-Na3PS4 (1.2–2.4 V), and Na3AsS4 (1.9–2.1 V) [107, 108]. The high oxidation limits (~ 3.5 V) allow assembly of Na3YCl6 and Na3YBr6 with high-voltage sodium cathode materials, such as the layered oxide material NaCrO2 and polyanionic materials Na2FePO4F and Na3V2(PO4)3 [109,110,111].

Reproduced with permission from Ref. [102]. Copyright 2019, Wiley–VCH. b Calculated thermodynamic equilibrium voltage profile and phase equilibria for Li3ScCl6. c CV curves of Li3ScCl6 at 0.1 mV s−1. d Calculated mutual reaction enthalpy between SSEs and LiCoO2, and corresponding phase equilibria. Reproduced with permission from Ref. [57]. Copyright 2020, American Chemical Society. e Heatmap of the reaction energy between chlorine-based halides and cathode materials. f Calculated band gaps of chlorine-based halides. Reproduced with permission from Ref. [112]. Copyright 2021, American Chemical Society
Thermodynamic intrinsic electrochemical stability of halide SSEs. a Calculated thermodynamic intrinsic electrochemical windows of Li–M–X ternary ionic conductors.
The intrinsically wide electrochemical window of halide SSEs is beneficial for achieving excellent interfacial compatibility with CAMs. Note that an extended electrochemical window can be achieved through the sluggish kinetics or passivation of interphases during experimental operation compared to the theoretically calculated thermodynamic electrochemical stability window. Liang et al. [57] investigated the electrochemical stability window of Li3ScCl6 and the compatibility between Li3ScCl6 and various cathode materials by incorporating experimental measurements with simulation computations. Li3ScCl6 was reported to possess an intrinsic electrochemical window of 0.91–4.26 V vs. Li+/Li based on first-principles calculations, as shown in Fig. 14b. For the Li3ScCl6 SSE, the anodic limit originates from the oxidation of Cl− to produce ScCl3 and Cl2, and the cathodic limit is mainly caused by reduction of Sc3+ to produce LiCl and Sc–Cl compounds, followed by reduction to Sc metal. Two reduction peaks appeared at 0.75 and 0.50 V in the cyclic voltammetry (CV) curves obtained with the Li//Li7P3S11//Li3ScCl6//Li3ScCl6–C cell, and interestingly, there was no obvious oxidation peak up to 5.0 V, which was mainly ascribed to interface formation during CV testing (Fig. 14c). The reaction energy of Li3ScCl6 and LiCoO2 calculated by using the binary model is only 60 meV per atom (Fig. 14d), which is an order of magnitude less than that of sulfide SSEs. The superior compatibility of Li3ScCl6 with LiCoO2 was further demonstrated by XANES and XPS characterization, which was consistent with the calculated results. The high-throughput computational study by Kim et al. [112] also confirmed that most Li3MCl6 materials exhibited a lower reaction energy with different cathode materials based on density functional theory (DFT) calculations, such as LiCoO2 and LiMn2O4, as shown in Fig. 14e, implying that Li3MCl6 with good oxidation stability could serve as appropriate SSEs for high-voltage cathodes. Their calculation results also showed that Li3MCl6 and Li3MBr6 have large band gaps of 6.2 and 5.1 eV on average (Fig. 14f), which is supposed to deliver low electrical conductivity enabling the usage of the solid electrolytes as superior separator materials between the anode and cathode.
The poor electrochemical reduction stability of halide SSEs is not conducive to realizing their utilization with lithium metal anodes. The cathodic limits of halide SSEs have been demonstrated to be mainly governed by the reduction of the central metal cation in halides [112, 113]. Typically, halide compounds with group 3 cations have relatively good reduction stability [102]. For instance, from thermodynamic calculations, Li–M–X ternary halides with Sc3+ and Y3+ cations exhibit reduction potentials of 0.60–0.92 and 0.36–0.62 V vs. Li+/Li, respectively, while In3+, Ga3+, and Zn2+ cations show reduction potentials of 1.98–2.55, 1.85–2.28, and 1.55–1.91 V vs. Li/Li+, respectively [102, 112]. As a result, halide SSEs with group 3 elements are regarded as promising options to provide desirable electrochemical windows due to their lower reduction potential and higher oxidation potential compared with other types of halide SSEs. Nevertheless, no halide SSEs are currently known to be stable with lithium metal, and Li-rich alloys are commonly used as anode materials for assembling halide-based full cells. Consequently, considering the stability in terms of metallic lithium and the electrochemical stability window of halide SSEs, constructing a stable interface between halide SSEs and lithium metal anodes to obtain high-performance halide-based ASSBs is imperative.
6.2 Stability Toward Anodes
Central metal elements with a high-valence state make halide SSEs unstable against lithium metal anodes, inevitably resulting in detrimental side reactions at the interface during the electrochemical cycling process. The interfacial side reactions will further cause continuous augmentation of the interfacial impedance and overpotential, eventually leading to severe impairment of the cell performance [21], which is the prominent issue impeding the application of halide-based ASSBs. Constructing interfacial buffer layers, optimally designing microcrystal structures of halide electrolytes, and developing lithium alloy anodes are considered effective strategies for stabilizing the anode interface of halide-based ASSBs.
6.2.1 Degradation Mechanisms Against Li Anodes
An in-depth understanding of the composition and properties of the interfacial phase as well as the interfacial electronic and ionic conduction behavior can offer instructive guidance for revealing the interaction mechanisms between halide SSEs and metallic lithium.
Fu et al. [114] investigated the interplay between two representative halides Li3YX6 (X = Cl and Br) and Li metal anodes by detecting the molar ratio and properties of the interfacial reaction products. The halide compounds suffered from reductive attack by metallic lithium, causing the generation of an interfacial phase of mixed ionic and electronic conductors involving LiX and Y, of which the reaction product Y could conduct electrons and LiX could transport Li ions. The corresponding interplay between halide compounds and lithium metal is schematically illustrated in Fig. 15a. As a result, both electrons and ions migrate across the reduced interface region to the unreacted materials, uninterruptedly converting more Li3YX6 to Y and LiX byproducts; thus, the formation of the interfacial phase will not stop until either the halide electrolyte or the metallic lithium is completely consumed, and the corresponding redox reactions are shown in Eqs. (8) and (9). Consequently, the formation of a non-self-limiting interfacial phase instead of a passivation layer after the halide comes into contact with metallic lithium is the essence of its incompatibility with the lithium anode. In addition, Li3YBr6 showed a lower reaction rate toward lithium metal than Li3YCl6, which is consistent with the theoretically calculated lower reduction potential of Li3YBr6 compared to Li3YCl6 (0.62 V vs. 0.59 V) [102].

Reproduced with permission from Ref. [98]. Copyright 2021, Wiley–VCH. c, d Time-resolved EIS spectra of Li/Li3YCl6/Li and Li/Li6PS5Cl/Li3YCl6/Li6PS5Cl/Li symmetric cells. e Corresponding cell performance comparison. f, g Continuously growing interface and kinetically stable interface enabled by a thin layer of Li6PS5Cl between Li3YCl6 and lithium metal. Reproduced with permission from Ref. [50]. Copyright 2021, American Association for the Advancement of Science. h LSV curves of Li3YBr6 and Li3YBr5.7F0.3. i Li plating and stripping voltage profiles of the Li3YBr5.7F0.3-based lithium symmetric cell. Reproduced with permission from Ref. [65]. Copyright 2021, Wiley–VCH
Stability of halide SSEs toward lithium anodes. a Schematic illustration of the interplay between lithium and Li3YX6 (X = Cl, Br). b Temporal evolution of the impedance for the Li3InCl6-based lithium symmetric cell.
Riegger et al. [98] also reported that the interface between halides Li3MCl6 (M = In, Y) and lithium metal anodes is thermodynamically unstable. For the Li3InCl6-based lithium symmetric cell, a remarkable increase in the interfacial impedance could be observed (Fig. 15b), exhibiting a high overall resistance of 854 Ω after only 1 h, which confirms the continuous growth of the interfacial phase between the halide and the metallic lithium anode.
Consequently, the thermodynamic incompatibility between halide SSEs and lithium metal anodes can be ascribed to the formation of mixed electronic/ionic conducting interfacial phases including LiX (X = halogen anion) and M0 (M = transition metal element), which give rise to the proceeding interfacial reactions, rather than being self-limiting. Notably, halide SSEs containing different types of metal elements with high-valence states deliver varying degrees of reactivity with lithium metal anodes, which largely depends on the actual reduction potential of the central metals in halide electrolytes.
6.2.2 Interfacial Buffer Layer for Stabilizing Lithium Anodes
An interfacial buffer layer is commonly introduced to the interface between halide SSEs and Li metal anodes to control the interfacial properties. Sulfide electrolyte layers, such as Li6PS5Cl, Li10GeP2S12 and Li7P3S11, are considered to be effective interfacial buffer layers to stabilize lithium metal anodes for realizing normal operation of halide-based ASSBs [33, 53, 64].
Ji et al. [99] employed Li6PS5Cl electrolytes as a protective layer for stabilizing the interface between the halide Li3YCl6 and a lithium metal anode and further revealed the related stabilization mechanism. The Li3YCl6-based lithium symmetric cell with the Li6PS5Cl buffer layer exhibits an overpotential of only 100 mV within 1 000 h, and the assembled LiNi0.8Co0.1Mn0.1O2//Li3YCl6//Li6PS5Cl//Li ASSB shows significantly improved electrochemical cycling performance compared to the full cell without the Li6PS5Cl protective layer. The authors demonstrated that a kinetically stable and ionically conductive self-confined interfacial layer can be formed between the lithium metal anode and Li6PS5Cl, similar to the SEI layer generated in liquid electrolytes, in which the interfacial phase mainly consists of the fast ionic conductor Li3P (~ 10−4 S cm−1) and electronically insulative LiCl and Li2S and could thus effectively suppress the occurrence of continuous interfacial reactions.
Wang et al. [50] found that a semicircle appeared at middle frequencies and enlarged within 24 h in the EIS spectrum of a Li//Li3YCl6//Li symmetric cell (Fig. 15c), indicating continuous interface reactions between Li metal and Li3YCl6. They clarified that Li3YCl6 can be easily reduced by Li metal to generate a highly electronically conductive Li3Y alloy (Fig. 15f), thereby forming a mixed Li+/e− conducting interfacial phase, allowing Li3YCl6 to be continuously reduced. However, the absence of the corresponding semicircle was observed in the EIS spectrum for the Li3YCl6-based lithium symmetric cell with the introduction of the Li6PS5Cl protective layer (Fig. 15d), which is primarily ascribed to the fact that Li6PS5Cl can spontaneously react with Li metal to form a highly Li+-conducting and electronically insulating interfacial phase (Fig. 15g). As a result, the Li6PS5Cl symmetric cell can stably cycle for 500 h with an overpotential of less than 100 mV, while the overpotential of the Li3YCl6 symmetric cell gradually increases to 600 mV under the same conditions (Fig. 15e). In addition, other similar sulfide electrolytes, such as Li10GeP2S12, Li7P3S11, and Li6.7Si0.7Sb0.3S5I, have also been successfully applied to stabilize the anode interface in halide-based ASSBs [47, 64, 115].
6.2.3 Stabilization of Lithium Metal Anodes by an In Situ Formed SEI
Constructing an in situ formed thinner interfacial layer is another effective strategy for stabilizing the interface between halide electrolytes and lithium metal anodes. In principle, the in situ formed interfacial layer should satisfy several requirements, including excellent chemical stability toward halide SSEs and metallic lithium, high ionic conductivity and electronic insulation to suppress the formation of a mixed ionic and electronic conductor (MIEC) interphase and appropriate mechanical strength to avoid the growth of lithium dendrites. The LiF compound can serve as an effective component for a stable SEI to avoid the growth of lithium dendrites due to its high interfacial energy and low electronic conductivity.
Yu et al. [65] successfully synthesized a series of new halide superionic conductors Li3YBr6−xFx (0 \(\leqslant\) x \(\leqslant\) 3) by fluorine doping, expecting to form a F-rich component to stabilize the interface between halide electrolytes and lithium metal. Excessive F doping caused the appearance of an impurity phase, and the obtained Li3YBr5.7F0.3 with an appropriate doping amount could maintain a high ionic conductivity of 1.8 mS cm−1 at RT compared to the value of 2.1 mS cm−1 for Li3YBr6. Linear sweep voltammetry (LSV) analysis of Li3YBr5.7F0.3 showed that the reduction onset potential was significantly reduced to 1 V compared to that of Li3YBr6 (2 V) and that a lower overall current density could be maintained (Fig. 15h). Lithium symmetric cells with Li3YBr5.7F0.3 as the electrolyte can show high stability over 1 000 h at 0.75 mA cm−2 (Fig. 15i), benefiting from the formation of uniformly distributed and highly concentrated LiF interfacial phases at the interface between the halide electrolyte and metallic lithium. The authors found that the in situ formed fluoride-containing interface layer with dense and reticular structures could effectively hinder the growth of lithium dendrites and suppress the occurrence of side reactions at the lithium anode interface, which also exhibited better solid contact area and uniform distribution than the artificial fluorinated interface layer formed by adding additional LiFSI. As a result, markedly enhanced cycling stability and electrochemical performance were achieved for Li3YBr5.7F0.3-based ASSBs directly using lithium metal as the anode and LiCoO2 as the cathode. The synthesis of fluorine-doped novel halide electrolytes is a promising strategy for obtaining an in situ formed SEI, and further investigations should be carried out on designing cell configurations and optimizing electrolyte components to in situ form a stable SEI to stabilize the interface between halide SSEs and lithium metal anodes.
6.2.4 Li Alloys for Stabilizing Lithium Metal Anodes
Li-rich alloy anodes (e.g., In, Si, Sn) have received extensive attention for assembling halide-based ASSBs to address the unstable interface issues associated with bare Li metal materials. The alloy anodes can exhibit chemical stability with halide electrolytes due to their relatively high reduction potential compared to that of Li+/Li and the formation of kinetically stable interphases. Moreover, the alloy anodes could exhibit a lower diffusion barrier than the bare metal lithium, and the enhanced diffusivity is beneficial for rapid diffusion of lithium atoms at the electrode interface, which can effectively reduce the tendency to form lithium dendrites [116]. In addition, the alloy anodes with good mechanical deformation are capable of enhancing the wettability of the halide electrolytes at the interface, which is conducive to maintaining uniform contact between halide electrolytes and alloy anodes. Notably, Li–In alloys are the most popular choice to stabilize metallic lithium anodes for halide-based ASSBs.
Anso et al. [44] assembled halide-based full cells of LiCoO2//Li3YX6 (X = Cl, Br)//Li–In by using a Li–In alloy (the molar ratio of Li:In is 38:62; thicknesses, LLi = 0.2 mm, LIn = 0.4 mm) with a stable voltage at 0.62 V vs. Li+/Li as the negative electrode instead of Li metal to avoid interface side reactions. Galvanostatic cycling of the cell was carried out in the voltage range of 1.9–3.6 V vs. Li+/Li–In, and the full cells showed excellent stability with the Coulombic efficiency remaining above 99% for 100 cycles at 0.1 C. Zhou et al. [58] assembled Li2Sc2/3Cl4-based full cells with high cycling stability by using a Li–In alloy as the negative electrode, obtained by uniformly dispersing ~ 1 mg of Li powder on a thin indium foil (10 mm in diameter, 0.125 mm thick). Analogously, Park et al. [55] prepared a Li–In alloy with a nominal composition of Li0.5In through ball milling of high-purity In and Li and used it as an anode for ASSBs with Li2.60Yb0.60Hf0.40Cl6 halide electrolytes. In addition, other Li-rich alloys have also been demonstrated to stabilize Li metal anodes for halide-based full cells. Park et al. [46] used a mixture of Li11Sn6 and Li3PS4 in a mass ratio of 80:20 as a reference electrode to achieve stable cycling of ASSBs based on the Li2.633Er0.633Zr0.367Cl6 halide electrolyte. However, note that the introduction of a Li–In alloy anode for halide-based ASSBs will reduce the operating voltage, which in turn leads to a certain attenuation of the battery energy density.
6.3 Cathodes Using Halide SSEs
Halide electrolytes exhibit encouraging electrochemical oxidation stability (> 4.2 V vs. Li+/Li), especially chloride-based halide electrolytes, combining the advantages of high ionic conductivity and relatively high oxidation potential [102], which means that halide electrolytes can be directly mixed with CAMs without any protective coatings to prepare cathode composites. Simple grinding enables halide SSE coating of CAMs due to the reliable mechanical deformability of these SSEs, and simultaneously, direct electronic and lithium-ion percolation pathways among CAM particles in cathode composites can be achieved by controlling the volume ratio of CAMs and SSEs (Fig. 16a). Sulfide electrolytes with weak oxidation stability require additional coating layers of high ionic conductivity and low electronic conductivity (such as LiNbO3 and LiTaO3) for CAMs to avoid their high-voltage oxidation, which causes partial electronic conduction through cathode particles and possibly triggers sulfide SSE oxidation (Fig. 16a); thus, halide SSEs exhibit outstanding advantages for the application of composite cathodes. Typically, single-crystalline materials are preferentially chosen for assembling halide-based ASSBs [90], which possess higher mechanical stability, to ensure the mechanical integrity of CAM particles and suppress contact loss in composite cathodes compared with typical polycrystalline materials.

Reproduced with permission from Ref. [26]. Copyright 2021, Wiley‒VCH. e Schematic diagram of the in situ interfacial growth of Li3InCl6 on LiCoO2. Reproduced with permission from Ref. [97]. Copyright 2020, Elsevier. f Illustration of the process of Li3AlF6 surface coating on LiNi0.5Mn1.5O4. Reproduced with permission from Ref. [117]. Copyright 2021, American Chemical Society
a Schematic illustration of ionic and electronic conduction percolation within cathode composites of bare CAMs with halide electrolytes and coated CAMs with sulfide electrolytes. b LSV analysis of Li3InCl4.8F1.2 and Li3InCl6. c Phase equilibria of Li3InCl4.8F1.2 at various potentials versus Li+/Li calculated by first-principles computations. d Charge‒discharge curves of SSE cells with Li3InCl4.8F1.2 and Li3InCl6 cathodes.
To fabricate competitive composite cathodes for halide-based ASSBs, optimizing the composition of the composite cathodes, designing a stable electrolyte microstructure and constructing an electrochemically stable interface between heterogeneous components are of vital importance and beneficial for realizing practical application of halide-based ASSBs with high-voltage, long cycle life and high-specific energy.
6.3.1 High-Voltage Cathodes
High anodic (oxidation) stability is regarded as an essential factor for achieving halide-based ASSBs operating at high voltage. Although halide SSEs exhibit a high theoretically calculated anodic upper limit (> 4 V vs. Li+/Li), as exemplified by recently developed chlorine-based halide SSEs Li3MCl6 (M = Y, In, Er, Sc), combined with good cathode compatibility and high ionic conductivity [102], these SSEs still show restricted electrochemical stability, which is inadequate for ultrahigh-voltage operation. This downside is mainly attributable to the oxidation of Cl− above 4.3V in chlorine-based halides, accompanied by the formation of Li-deficient metal chlorides (YCl3, InCl3, etc.), resulting in blockage of the Li+ conduction pathway in the composite cathodes and further continuous deterioration of the full cell performance.
Zhang et al. [26] synthesized the novel dual-halogen electrolyte Li3InCl4.8F1.2 with a practical anodic limit above 6 V (vs. Li+/Li) and a decent RT ionic conductivity of 0.51 mS cm−1 by employing fluorine to selectively occupy a specific lattice site (Cl-8j) in Li3InCl6. An observably improved anodic potential of Li3InCl4.8F1.2 up to 4.3 V was obtained from the LSV result compared to unsubstituted Li3InCl6 (Fig. 16b). The theoretical anodic potential of Li3InCl4.8F1.2 can reach 4.42 V based on first-principles computations, and the authors found the formation of a fluorinated interphase composed of LiF, LiInF4 and LiInF6 (Fig. 16c). Incorporation with the dual-halogen electrolyte Li3InCl4.8F1.2 and the bare high-voltage LiCoO2 enables stable operation of ASSBs at a high cutoff voltage of 4.8 V vs. Li+/Li (Fig. 16d), which originates from the in situ formation of a cathode-electrolyte interface rich in LiF that can effectively suppress the uninterrupted occurrence of detrimental interfacial reactions at the cathode. Zhou et al. [59] reported the mixed-metal chlorospinel halide Li2In1/3Sc1/3Cl4 with a high ionic conductivity of 2.0 mS cm−1 at RT, and incorporation of as little as 10 wt% Li2In1/3Sc1/3Cl4 with uncoated high-Ni LiNi0.85Co0.1Mn0.05O2 formed a cathode composite that could be successfully applied to assemble ASSBs with a high cutoff potential of 4.8 V versus Li+/Li. Moreover, these high-voltage cells showed a long cycle life of over 3 000 cycles with 80% capacity retention under an operating pressure of 250 MPa, benefiting from the minimal reactivity of Li2In1/3Sc1/3Cl4 with CAMs as well as the extremely low electronic conductivity of 4.7 × 10−10 S cm−1 of Li2In1/3Sc1/3Cl4. Consequently, further optimizing the structure design of halide electrolytes can be considered an effective strategy for facilitating the development and practical application of halide-based ASSBs at ultrahigh voltages.
6.3.2 Interfacial Contact
CAMs are known to undergo an inevitable volume change as a function of the lithium content during cycling, along with the internal stress generated by chemomechanical expansion [118], which results in continuous contact loss between the halide SSEs and CAM particles that deteriorates the cycling stability of ASSBs [119]. In addition, obtaining composite cathodes with close interfacial contact and excellent ion transport requires a higher content of halide electrolytes through mechanical mixing methods, which causes significant dilution of the energy density for ASSBs. In situ construction of high ionic conducting and electronic insulating halide coatings on the surface of CAM particles is beneficial for optimizing the surface chemistry at the cathode interface, which in turn effectively mitigates the contact loss and mechanical failure of halide-based ASSBs during electrochemical cycling.
Wang et al. [97] successfully achieved strong interfacial contact between halide electrolytes and CAMs by synthesizing the continuous film-coated cathode Li3InCl6@LiCoO2 through in situ interfacial growth of Li3InCl6 on the surface of CAMs (Fig. 16e). As little as 15 wt% Li3InCl6 with a high ionic conductivity of 1.5 mS cm−1 at RT was uniformly dispersed in the composite cathode, achieving intimate solid‒solid interfacial contact, thus leading to an ultrasmall interfacial resistance between Li3InCl6 and LiCoO2 (5.3 Ω cm−2) and ensuring an electrode-based gravimetric energy density of ASSBs up to 512 Wh kg−1. Strong interfacial interactions were observed between Li3InCl6 and LiCoO2 by using XPS characterization and DFT calculations, which effectively facilitated uniform interfacial growth of Li3InCl6, thus obtaining a high initial discharge capacity up to 131.7 mAh g−1 and an initial Coulombic efficiency of 92.7% at 0.1 C for Li3InCl6-based ASSBs. Kobayashi et al. [117] reported that β-Li3AlF6 with a lower migration barrier (E = 0.25 eV) can be successfully in situ coated onto LiNi0.5Mn1.5O4 and LiCoO2 cathodes through a simple sol–gel calcination process (Fig. 16f) and found that the assembled liquid full cells based on β-Li3AlF6-coated cathodes exhibited observably improved cycling performance by suppressing the side reactions of the cathode-electrolyte interface (CEI), which implies the potential application of in situ coating of fluorine-based halides on the surface of CAMs to realize close interfacial contact in composite cathodes for ASSBs. In summary, in situ construction of halide coatings on cathode surfaces offers a versatile methodology to overcome longstanding interfacial contact challenges between halide SSEs and CAMs, which is beneficial for the development of ASSBs with high energy density and high stability.
6.3.3 Cathode Composition
The proportions, morphology, and microstructure of the cathode composition dramatically affect the chemomechanical properties of the composite cathode during electrochemical cycling. In addition, in-depth optimization of the preparation process of the composite cathode is of vital importance to achieving long cycle-life as well as high-specific-energy halide-based ASSBs [21, 96]. Simple grinding of halide electrolytes and CAMs followed by a powder cold-pressing operation can obtain composite cathodes for ASSBs, mainly attributed to the superior plasticity of halide SSEs, which is favorable for obtaining relatively close contact of particles in composite cathodes. A rational composition ratio of the composite cathode is considered to be one of the most crucial factors for achieving a continuous ionic and electronic cross-linked diffusion network in the cathode. A high proportion of CAMs could form a decent electronic percolation pathway, accompanied by a significantly improved energy density of the full cell, whereas high-content CAMs can aggregate to form low ionic transport, which causes poor ionic conductivity as a limiting factor.
Single-crystal CAMs exhibit better thermal, high-voltage and mechanical tolerance than polycrystalline materials, which can efficiently ensure the mechanical integrity of the CAMs and suppress contact loss in composite cathodes [120, 121]. In contrast, polycrystalline materials experience structural collapse during electrode pressing and anisotropic volumetric strain during electrochemical cycling [122], giving rise to intergranular cracks that prominently weaken ion transport kinetics and increase the interfacial impedance.
Han et al. [90] investigated the effect of uncoated single-crystalline and polycrystalline LiNi0.88Co0.11Al0.01O2 as cathodes on the performance of ASSBs based on the halide Li3YCl6 and Li6PS5Cl0.5Br0.5 and compared the advantages and disadvantages of the single-crystalline and polycrystalline CAMs as well as the halide and sulfide, as shown in Fig. 17a. They reported that the different composite cathodes with polycrystalline materials all exhibited distinct internal cracks even after the first charge of the corresponding ASSBs, which are primarily attributed to the lattice shrinkage caused by the detrimental H2–H3 phase transition at \(\geqslant\) 4.1 V (vs. Li+/Li). Although the ionic conductivity of the halide Li3YCl6 (0.4 mS cm−1 at 30 °C) is an order of magnitude lower than that of the sulfide Li6PS5Cl0.5Br0.5 (4.8 mS cm−1 at 30 °C), the initial Coulombic efficiency of Li3YCl6-based ASSBs (87.7%) is remarkably higher than that of Li6PS5Cl0.5Br0.5-based ASSBs (77.5%) when employing the same single-crystal cathode at 0.1 C (Fig. 17b), which is attributable to the occurrence of severe interfacial reactions with the sulfide electrolyte with poor electrochemical oxidation stability above 3 V (vs. Li+/Li). The coverage by and heterogeneous contact with Li3YCl6 of CAMs could be effectively improved by introducing a larger amount of Li3YCl6 (from 29.1 to 40.7 wt%) into the composite cathode, which could avoid loosening and loss of ionic contact under volumetric strain during cycling; thus, the capacity retention could be distinctly enhanced from 67.2% to 89.3% at the 200th cycle by using excessive Li3YCl6 in the composite cathode (Fig. 17c).

Reproduced with permission from Ref. [90]. Copyright 2021, Wiley–VCH. d First charge–discharge profiles for Li2.6Er0.6Zr0.4Cl6-based ASSBs with various amounts of VGCFs. e Their corresponding cycling performance. Reproduced with permission from Ref. [104]. Copyright 2022, American Chemical Society. f Illustration of optimizing composite cathode components to obtain high-performance ASSBs. g First cycle profiles of Li3InCl6-based electrodes with different amounts of CAMs. h First cycle profiles of the electrodes with various halide SSEs. Reproduced with permission from Ref. [100]. Copyright 2022, Cell Press
a Pros and cons of Ni-rich layered oxides (single-crystalline vs. polycrystalline) and SSEs (halides vs. sulfides). b Initial charge‒discharge voltage profiles for electrodes with different combinations of LiNi0.88Co0.11Al0.01O2 (NCA) and SSEs. c Cycling performances for the single-crystalline NCA/Li3YCl6 electrodes with different amounts of Li3YCl6.
The introduction of carbon-based conductive agents can provide effectively enhanced electronic transport pathways in the composite cathode, which is beneficial for obtaining lower charge transfer resistance and fast reaction kinetics for ensuring the stability of ASSBs during high-rate cycling. The addition of carbon conductive agents could accelerate the decomposition of sulfide electrolytes due to their lower onset oxidation potential [123]. In contrast, halide electrolytes have higher electrochemical oxidation stability (> 4 V vs. Li+/Li), which means that the introduction of a moderate amount of conductive carbon can serve as a potential option to enhance the electrochemical performance of halide-based ASSBs. Compared with conductive carbon black, which is susceptible to severe agglomeration, vapor-grown carbon fibers (VGCFs) can achieve uniform dispersion and simultaneously offer continuous electronic pathways in the composite cathode, benefiting from the multiple points of contact with the CAMs.
Shao et al. [104] investigated the effect of introducing VGCFs into a composite cathode with LiCoO2 on the electrochemical performance of Li2.6Er0.6Zr0.4Cl6-based ASSBs. They reported that the ASSBs without VGCFs and with 5 wt% VGCFs showed similar high initial Coulombic efficiencies of 97.1% and 97.4%, respectively (Fig. 17d), indicating that the introduction of VGCFs could not induce decomposition of the halide Li2.6Er0.6Zr0.4Cl6. Moreover, the addition of 5 wt% VGCFs was beneficial for achieving significantly improved initial discharge capacity from 125.2 to 147.5 mAh g−1 and capacity retention from 72% to 80% at 0.1 C after 100 cycles (Fig. 17e), which is due to the effectively enhanced electron percolation pathways in the composite cathode.
Gao et al. [100] systematically investigated the effects of the carbon nanofiber (CNF) proportion (1–10 wt%) and CAM loading (65–75 wt%) in composite cathodes and different halide SSEs (Li3InCl6, Li3YCl6 and Li3AlCl6) on the electrochemical performance of ASSBs with LiNi0.83Mn0.06Co0.11O2 (Fig. 17f). They found that ASSBs with a suitable CNF content of 5 wt% in the composite cathode exhibited the highest capacity, which was primarily ascribed to the lower CNF percentages not being able to provide sufficient electronically conducting pathways to ensure the activity of CAMs and the higher CNF percentages disrupting ionic pathways in the composite cathode. Furthermore, modest augmentation of the CAM loading from 65 to 70 wt% reduced the capacity to some extent (Fig. 17g), mainly attributed to the decreased SSE quantity resulting in a lower capacity utilization of the CAMs. In addition, the ASSBs with Li3InCl6 as an ionic conductor in the composite cathode achieved the highest capacity (Fig. 17h) since Li3InCl6 possesses the best ionic conductivity among the different halide SSEs, which can tolerate a high loading of CAMs for their high utilization.
6.4 Industrial Perspective for Halide-Based ASSBs
6.4.1 Fabrication of Sheet-Type Electrodes
While most laboratory-scale ASSBs based on halide SSEs were tested by using binder-free pellets (containing CAMs, conductive carbon, and SSEs), the pelletized electrodes are not conducive to implementation of scalable processing [31, 124], which in turn significantly restricts the practical application of halide-based ASSBs. Wet-slurry fabrication is regarded as a well-established scalable strategy to manufacture sheet-type electrodes by using a small amount of polymeric binders to achieve intimate contact between uniformly dispersed particles [125,126,127]. Different from the slurry process for electrodes of traditional batteries with liquid electrolytes, the slurry process for ASSB electrodes comprises homogenization of the four components of CAMs, conductive carbon, polymer binders and additional SSEs. Unfortunately, the violent reaction between halide electrolytes and conventional polar solvents severely limits the application of common organic solvents during the slurry fabrication process [51]. Consequently, nonpolar or low-polarity solvents, such as anisole, toluene, p-xylene and heptane, are usually selected as processing solvents for fabricating halide-based slurries [128, 129], which in turn limits the choice of acceptable polymeric binders [28]. In addition, the introduction of ionic insulating polymer binders could block ionic transport across the bulk of SSEs, consequently resulting in impaired electrochemical performance of ASSBs [130, 131]. These remaining challenges provide instructive guidance for the research direction of fabricating continuous sheet-type electrodes for halide-based ASSBs.
Wang et al. [128] fabricated slurry-cast composite cathodes with Li3InCl6 and LiNi0.8Co0.1Mn0.1O2 using toluene with a weakly polar solvent and investigated different kinds of polymer binders, including polymethyl methacrylate (PMMA), ethyl cellulose (EC), styrene-butadiene rubber (SBR), and nitrile rubber (NBR), as well as the effect of their contents on the electrochemical performance of Li3InCl6-based ASSBs. The content of the polymer binder in the composite cathode exerts a crucial influence on the performance of ASSBs (Fig. 18a–c), and a 2 wt% binder content is the most suitable ratio, which enables close contact between CAMs and SSEs as well as decent lithium-ion transport in the composite cathode. All these polymer binders can be used for fabricating composite cathodes with low interfacial resistance to achieve excellent electrochemical performance of Li3InCl6-based ASSBs.

Reproduced with permission from Ref. [128]. Copyright 2021, Frontiers Media S.A. d Schematic illustration of a solvent-free process for an all-solid-state pouch cell with dry-fabricated sheet electrodes. e, f Charge–discharge curves of the Li3InCl6@LiCoO2/Li3InCl6 + Li6PS5Cl/graphite@Li6PS5Cl cell, and corresponding cycling stability. A photograph of the dry-fabricated electrode is shown in the inset of (e). Reproduced with permission from Ref. [132]. Copyright 2021, American Chemical Society
Electrochemical performance of halide-based ASSBs with flexible electrodes. a–c Initial charge–discharge profiles of composite cathodes with various amounts of binder: PMMA, EC and NBR for Li3InCl6-based ASSBs.
A solvent-free process is another promising strategy to fabricate sheet-type electrodes for halide-based ASSBs, which can avoid the possible chemical reactions between solvents and halide SSEs and simultaneously take advantage of effectively simplifying the technological process as well as reducing environmental pollution. Wang et al. [132] reported a solvent-free approach to fabricate freestanding sheet-type electrodes by mixing Li3InCl6@LiCoO2 and fibrous polytetrafluoroethylene (PTFE), followed by roll-to-roll pressing to obtain the desired thickness, as shown in Fig. 18d. The representative halide electrolyte (Li3InCl6) and sulfide electrolyte (Li6PS5Cl) were fabricated into ultrathin membranes with thicknesses of 15–20 μm and a dimension of 8 cm × 6 cm. The assembled all-solid-state pouch cell with a configuration of Li3InCl6@LiCoO2//Li3InCl6 + Li6PS5Cl//graphite@Li6PS5Cl delivered a high capacity of 121.2 mAh g−1 and an initial Coulombic efficiency of 71.8% at 0.1 C (Fig. 18e) and retained a capacity of 81.3 mA g−1 after 50 cycles (Fig. 18f). The solvent-free approach presents several advantages in terms of scalable production, environmental friendliness and cost effectiveness and could thus be a feasible technology to ensure factory-scale manufacturing to effectively facilitate commercialization of halide-based ASSBs.
6.4.2 Cost
The cost of inorganic SSEs is known to be a decisive factor in allowing their efficient large-scale production for commercial application of ASSBs. Based on laboratory-scale prices (Table 3), the bulk prices of the raw materials required to synthesize different types of solid electrolytes (Table 4) were estimated using the general correlation between the unit price and purchase quantity of the chemicals shown in Eq. (10) or Eq. (11) (logarithmic form) [133].
where P and Q represent the unit price and the purchase quantity of chemical reagents, respectively, and a and b are constants for a given raw material.
For the synthesis of sulfide electrolytes, the precursor Li2S with a high bulk price of 653.87 USD kg−1 is nearly irreplaceable for preparing all types of sulfide SSEs (Table 4), which is also the highest consumption cost for synthesizing the various sulfide SSEs. Moreover, Li2S compounds are unstable when exposed to humid air and need to be preserved in an inert atmosphere, thus resulting in additional costs for fabricating sulfide SSEs. In contrast, for the synthesis of halide electrolytes, the bulk price of the precursor LiCl is only 5.88 USD kg−1, which is much lower than that of the raw material Li2S mentioned above (Table 4), and LiCl could simultaneously present decent stability in an ambient environment. From this perspective, the synthesis of halide SSEs seems to hold great promise for cost effectiveness. Unfortunately, this unique superiority is no longer maintained when considering the cost of non-Li-containing precursors for preparing halide SSEs. That is, the central metal elements of most halides are rare-earth metals, which are found in low abundance in the Earth’s crust, as shown in Fig. 19a [53]. For example, the abundances of Y and In are 33 and 25 ppm, respectively, and such values are far below those of nonlithium cations in sulfide electrolytes, such as the abundance of P, which is 1 050 ppm. As a result, the cost of chloride precursors for synthesizing halide SSEs is relatively high, as shown in Fig. 19b [53], which is not conducive to the large-scale preparation of halide electrolytes.

Reproduced with permission from Ref. [53]. Copyright 2021, Nature Publishing Group. c Costs of precursors for Li6PS5Cl and different chlorine-based halide SSEs
Raw material costs of the state-of-the-art chloride solid electrolytes. a Estimated unit prices of different chlorides for synthesizing halide SSEs. b Abundance of the central metal elements of halide SSEs in the Earth’s crust.
Recent investigations have found that Zr is a promising central metal element to obtain the cost-effective Li2ZrCl6 halide electrolyte [53, 56]. Zr presents a relatively high abundance of 165 ppm in the Earth’s crust (Fig. 19b), which enables the bulk price of the precursor ZrCl4 to be several orders of magnitude lower than that of other chloride precursors (Fig. 19a). More importantly, the substitution of Zr for the central metal element in halides can significantly enhance the ionic conductivity at RT, such as that of Li2.5Y0.5Zr0.5Cl6 (1.4 mS cm−1) and Li2.6In0.6Zr0.4Cl6 (1.25 mS cm−1) [46, 48], which provides an effective strategy for obtaining performance-enhanced and cost-effective halide superionic conductors. The prices of precursor materials required to synthesize halide and sulfide SSEs are shown in Fig. 19c. Both Li2ZrCl6 and Zr-substituted halides are less expensive than the sulfide Li6PS5Cl, especially Li2ZrCl6, which has a bulk price of only 10.76 USD kg−1, 26 times lower than that of Li6PS5Cl. In summary, although expensive non-Li-containing raw materials remarkably increase the production cost of rare-earth-based halide solid electrolytes, novel low-cost Li2ZrCl6 and Zr-substituted superionic halide conductors can be regarded as promising candidates for realizing large-scale manufacturing of halide SSEs.
7 Conclusion and Perspective
In summary, emerging halide SSEs with high ionic conductivity, exceptional mechanical deformability and outstanding oxidation stability represent a promising family of materials for realizing the application of high-performance ASSBs. In this review, we comprehensively summarize the remarkable progress in halide solid electrolytes in ASSBs for energy storage.
Herein, the major conclusions can be drawn based on the in-depth understanding and comprehensive review of the recent advances in the newly emerging halide electrolytes as follows. (1) Compared with the conventional solid-state reaction synthesis with high energy and longtime consumption as well as harsh equipment requirements, the recently developed universal ammonium-assisted and water-mediated synthesis routes are more attractive due to the appreciable advantages of the time-saving processes, nanoscale uniformity and easily scalable production. (2) Engineering strategies in terms of optimization of synthesis parameters, partial substitution of constituent elements and tuning of cation site disorder offer valuable opportunities for acquiring halide SSEs with high ionic conductivity. In particular, heterovalent substitution of rare-earth-based halide electrolytes with Zr4+ could achieve high ionic conductivities over 1 mS cm−1 while retaining exceptional oxidation stability and significant advantages in cost. (3) Poor chemical stability is considered the crucial factor impeding the synthesis and application of halide electrolytes, which is manifested in the irreversible degradation of most halides when they are exposed to humid air, accompanied by the formation of corrosive hydrochloric acid, and the strong interaction between the halide compounds and polar solvents inevitably causes depletion of the halide microcrystal structures and loss of the ionic conductivity. (4) The intrinsically poor electrochemical reduction stability of halide electrolytes with high-valence-state metal elements is responsible for the severe interfacial reactions with the lithium metal anodes in ASSBs. Subtle design strategies, including construction of an in situ LiF-rich interfacial layer and interfacial buffer layers, are effective protocols for suppressing the occurrence of interfacial side reactions. (5) Superior interfacial compatibility can be achieved between uncoated high-voltage oxide cathode materials and halide electrolytes with high oxidation potential, and simultaneously, single-crystal cathode materials are preferentially selected to assemble halide-based ASSBs to guarantee the mechanical integrity of the composite cathode and avoid interfacial contact loss during cycling.
Although promising results have been achieved for halide electrolytes in terms of universal wet-chemistry synthesis, comprehensive performance improvement and attempted utilization in ASSBs, there are still challenges to be addressed and advanced properties to be developed in the future. The attractive research directions and prospects are outlined in detail as follows.
-
(1)
Enhancement of chemical and electrochemical stability
Although halide Li3InCl6 presents superior tolerance toward ambient atmosphere due to the formation of hydrated intermediates, in which crystal water can be readily removed by a simple dehydration process, the majority of halide solid electrolytes exposed to humid air undergo irreversible degradation reactions, leading to a decrease in the ionic conductivity and the formation of corrosive acid. The strong interaction between conventional polar solvents and halide electrolytes inevitably causes severe destruction of the crystal phase structure and loss of the ionic conductivity, which greatly restricts the application of halides in solvent-based processes. An in-depth understanding of the harmful degradation mechanisms of halides in humid air and polar solvents can offer fundamental theoretical guidance for the design of novel halide electrolytes with high humidity tolerance and for the development of reliable solvents with good compatibility.
In addition, the thermodynamic instability of halide electrolytes with high-valence cations against lithium metal anodes is a prominent issue impeding extensive application of halide-based ASSBs. The protocol of introducing a sulfide interfacial buffer layer to stabilize the lithium metal anode possibly causes the instability issue of halides with sulfides at elevated temperatures and the issue of reduced energy density. The anode interface of halide-based ASSBs can be stabilized by optimally designing the microcrystal structure of halide electrolytes and employing lithium alloy anodes. Simultaneously, the construction of in situ formed LiF-rich thin interfaces to stabilize lithium anodes and the application of the lithium-free composite anode of Ag–C offer a prospective direction for addressing the instability of lithium anodes in halide-based ASSBs.
-
(2)
Improving the ionic conductivity
State-of-the-art halide solid electrolytes with trivalent metal elements such as Li3MX6 (M = Sc, Y, Ho, In; X = Cl, Br) and the corresponding Zr4+-substituted halides Li3−xM1−xZrxCl6 show ionic conductivities on the order of mS cm−1 at RT to date, which still has a significant gap compared to sulfide electrolytes. Moreover, fluorine-based halide SSEs with exceptional oxidation stability are considered promising electrolytes enabling access to cathodes with a high voltage of > 5 V, but they suffer from severe limitations due to their unsatisfactory ionic conductivity of approximately 10−5 S cm−1 at RT. Similarly, the currently discovered Na-based halides, such as Na3YCl6, Na3ErCl6 and Na3−xM1−xZrxCl6 (M = Y, Er), only exhibit a maximum ionic conductivity of 0.04 mS cm−1, implying that there is great potential for further increasing the ionic conductivity. A comprehensive understanding of the underlying local structural evolution and the ionic transport mechanism of different halide electrolytes is of great significance to effectively guiding engineering strategies in terms of optimizing synthesis parameters, tuning cation site disorder and regulating the lithium vacancy concentration to enhance the ionic conductivity.
-
(3)
Application of advanced in situ/operando characterization as well as theoretical computation and simulation
In situ/operando characterization techniques can provide the evolution of the properties of halide electrolytes, including the microstructure, surface morphology, local composition and chemical state, during the dynamic reaction, which offers an effective strategy to thoroughly understand the SEI formation, degradation mechanisms and redox mechanisms under realistic operating conditions in halide-based ASSBs. Implementation of advanced in situ/operando characterization techniques involving in situ NPD, in situ XANES, in situ Raman/Fourier transform infrared spectroscopy, operando SXRD, etc. capable of monitoring the metastable phase and valence evolution and revealing the degradation mechanisms of halide electrolytes during the dynamic reaction, as well as identifying the SEI components and tracing the interfacial processes under working conditions, is highly desired.
In addition, establishing design principles based on high-throughput computation through aliovalent element substitution and tailoring of anion/cation sublattices is favorable for providing feasible protocols to synthesize novel halide electrolytes with excellent (electro)chemical stability and ionic conductivity. A comprehensive thermodynamic analysis of the origin of the electrochemical and moisture instability for different types of halides can also offer theoretical guidance for revealing the degradation mechanisms of halide electrolytes. Furthermore, high-throughput simulation is beneficial for discovering novel halide electrolytes with high ionic conductivities and good thermophysical properties, which is also highly desirable for identifying attractive halide candidates to accommodate high-energy–density ASSBs with high-voltage cathodes by screening a wide range of halide systems.
-
(4)
Fabrication of thin electrolytes and electrodes
Bulk-type ASSBs fabricated from cold-pressed halide and composite cathode powders require compensating for the weak mechanical properties in terms of the formation of microcracks during the pressing process, and the poor interfacial contact requires application of high additional pressure, which are the prominent challenges to large-scale manufacture of electrolytes and electrodes for achieving commercial application of halide-based ASSBs. Tape casting is considered to be a practical technique from the perspective of mass fabrication of both sheet-type electrodes and thin electrolyte membranes and for simultaneously realizing compact-interfacial-contact and high-energy–density ASSBs. To date, investigations on such topics are scarce. Considering the high reactivity of halide electrolytes toward conventional polar solvents, the selection of proper solvents and binders as well as compositional control are research directions that need to be intensively studied in the future. In addition, the solvent-free process protocol to fabricate ultrathin inorganic or inorganic/polymer composite electrolyte membranes also provides a promising strategy for large-scale manufacturing of halide-based ASSBs.
References
Goodenough, J.B.: Electrochemical energy storage in a sustainable modern society. Energy Environ. Sci. 7, 14–18 (2014). https://doi.org/10.1039/c3ee42613k
Sun, C.W., Liu, J., Gong, Y.D., et al.: Recent advances in all-solid-state rechargeable lithium batteries. Nano Energy 33, 363–386 (2017). https://doi.org/10.1016/j.nanoen.2017.01.028
Trahey, L., Brushett, F.R., Balsara, N.P., et al.: Energy storage emerging: a perspective from the Joint Center for Energy Storage Research. PNAS 117, 12550–12557 (2020). https://doi.org/10.1073/pnas.1821672117
Goodenough, J.B., Park, K.S.: The Li-ion rechargeable battery: a perspective. J. Am. Chem. Soc. 135, 1167–1176 (2013). https://doi.org/10.1021/ja3091438
Armand, M., Tarascon, J.M.: Building better batteries. Nature 451, 652–657 (2008). https://doi.org/10.1038/451652a
Xie, J., Lu, Y.C.: A retrospective on lithium-ion batteries. Nat. Commun. 11, 2499 (2020). https://doi.org/10.1038/s41467-020-16259-9
Kwade, A., Haselrieder, W., Leithoff, R., et al.: Current status and challenges for automotive battery production technologies. Nat. Energy 3, 290–300 (2018). https://doi.org/10.1038/s41560-018-0130-3
Kang, X.: Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 104, 4303–4418 (2004). https://doi.org/10.1021/cr030203g
Tarascon, J.M., Armand, M.: Issues and challenges facing rechargeable lithium batteries. In: Dusastre, V. (ed.) Materials for Sustainable Energy, pp. 171–179. World Scientific, London (2011). https://doi.org/10.1142/9789814317665_0024
Ding, Y.L., Cano, Z.P., Yu, A.P., et al.: Automotive Li-ion batteries: current status and future perspectives. Electrochem. Energy Rev. 2, 1–28 (2019). https://doi.org/10.1007/s41918-018-0022-z
Manthiram, A., Yu, X.W., Wang, S.F.: Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2, 16103 (2017). https://doi.org/10.1038/natrevmats.2016.103
Bachman, J.C., Muy, S., Grimaud, A., et al.: Inorganic solid-state electrolytes for lithium batteries: mechanisms and properties governing ion conduction. Chem. Rev. 116, 140–162 (2016). https://doi.org/10.1021/acs.chemrev.5b00563
Zhao, Q., Stalin, S., Zhao, C.Z., et al.: Designing solid-state electrolytes for safe, energy-dense batteries. Nat. Rev. Mater. 5, 229–252 (2020). https://doi.org/10.1038/s41578-019-0165-5
Mauger, A., Julien, A., Paolella, A., et al.: Building better batteries in the solid state: a review. Materials 12, 3892 (2019). https://doi.org/10.3390/ma12233892
Randau, S., Weber, D.A., Kötz, O., et al.: Benchmarking the performance of all-solid-state lithium batteries. Nat. Energy 5, 259–270 (2020). https://doi.org/10.1038/s41560-020-0565-1
Kamaya, N., Homma, K., Yamakawa, Y., et al.: A lithium superionic conductor. Nat. Mater. 10, 682–686 (2011). https://doi.org/10.1038/nmat3066
Sun, C.W., Alonso, J.A., Bian, J.J.: Recent advances in perovskite-type oxides for energy conversion and storage applications. Adv. Energy Mater. 11, 2000459 (2021). https://doi.org/10.1002/aenm.202000459
Cui, G.L.: Reasonable design of high-energy-density solid-state lithium-metal batteries. Matter 2, 805–815 (2020). https://doi.org/10.1016/j.matt.2020.02.003
Kato, Y., Hori, S., Saito, T., et al.: High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy 1, 16030 (2016). https://doi.org/10.1038/nenergy.2016.30
Abakumov, A.M., Fedotov, S.S., Antipov, E.V., et al.: Solid state chemistry for developing better metal-ion batteries. Nat. Commun. 11, 4976 (2020). https://doi.org/10.1038/s41467-020-18736-7
Li, X.N., Liang, J.W., Yang, X.F., et al.: Progress and perspectives on halide lithium conductors for all-solid-state lithium batteries. Energy Environ. Sci. 13, 1429–1461 (2020). https://doi.org/10.1039/c9ee03828k
Yang, K., Chen, L.K., Ma, J.B., et al.: Progress and perspective of Li1+xAlxTi2−x(PO4)3 ceramic electrolyte in lithium batteries. InfoMat 3, 1195–1217 (2021). https://doi.org/10.1002/inf2.12222
van den Broek, J., Afyon, S., Rupp, J.L.M.: Interface-engineered all-solid-state Li-ion batteries based on garnet-type fast Li+ conductors. Adv. Energy Mater. 6, 1600736 (2016). https://doi.org/10.1002/aenm.201600736
Chen, R.S., Li, Q.H., Yu, X.Q., et al.: Approaching practically accessible solid-state batteries: stability issues related to solid electrolytes and interfaces. Chem. Rev. 120, 6820–6877 (2019). https://doi.org/10.1021/acs.chemrev.9b00268
Yu, S., Schmidt, R.D., Garcia-Mendez, R., et al.: Elastic properties of the solid electrolyte Li7La3Zr2O12 (LLZO). Chem. Mater. 28, 197–206 (2016). https://doi.org/10.1021/acs.chemmater.5b03854
Zhang, S.M., Zhao, F.P., Wang, S., et al.: Advanced high-voltage all-solid-state Li-ion batteries enabled by a dual-halogen solid electrolyte. Adv. Energy Mater. 11, 2100836 (2021). https://doi.org/10.1002/aenm.202100836
Zhang, Q., Cao, D.X., Ma, Y., et al.: Sulfide-based solid-state electrolytes: synthesis, stability, and potential for all-solid-state batteries. Adv. Mater. 31, 1901131 (2019). https://doi.org/10.1002/adma.201901131
Nikodimos, Y., Huang, C.J., Taklu, B.W., et al.: Chemical stability of sulfide solid-state electrolytes: stability toward humid air and compatibility with solvents and binders. Energy Environ. Sci. 15, 991–1033 (2022). https://doi.org/10.1039/d1ee03032a
Yu, C., Zhao, F.P., Luo, J., et al.: Recent development of lithium argyrodite solid-state electrolytes for solid-state batteries: synthesis structure stability and dynamics. Nano Energy 83, 105858 (2021). https://doi.org/10.1016/j.nanoen.2021.105858
Zhao, W.J., Yi, J., He, P., et al.: Solid-state electrolytes for lithium-ion batteries: fundamentals, challenges and perspectives. Electrochem. Energy Rev. 2, 574–605 (2019). https://doi.org/10.1007/s41918-019-00048-0
Wang, C.H., Liang, J.W., Zhao, Y., et al.: All-solid-state lithium batteries enabled by sulfide electrolytes: from fundamental research to practical engineering design. Energy Environ. Sci. 14, 2577–2619 (2021). https://doi.org/10.1039/d1ee00551k
Choi, S., Jeon, M., Kim, B.K., et al.: Electrochemical behaviors of Li-argyrodite-based all-solid-state batteries under deep-freezing conditions. Chem. Commun. 54, 14116–14119 (2018). https://doi.org/10.1039/c8cc08030e
Li, X.N., Liang, J.W., Chen, N., et al.: Water-mediated synthesis of a superionic halide solid electrolyte. Angew. Chem. 131, 16579–16584 (2019). https://doi.org/10.1002/ange.201909805
Liu, B., Wang, D., Avdeev, M., et al.: High-throughput computational screening of Li-containing fluorides for battery cathode coatings. ACS Sustainable Chem. Eng. 8, 948–957 (2020). https://doi.org/10.1021/acssuschemeng.9b05557
Qie, Y., Wang, S., Fu, S.J., et al.: Yttrium-sodium halides as promising solid-state electrolytes with high ionic conductivity and stability for Na-ion batteries. J. Phys. Chem. Lett. 11, 3376–3383 (2020). https://doi.org/10.1021/acs.jpclett.0c00010
Ginnings, D.C., Phipps, T.E.: Temperature-conductance curves of solid salts. III. Halides of lithium. J. Am. Chem. Soc. 52, 1340–1345 (1930). https://doi.org/10.1021/ja01367a006
Liang, C.C., Epstein, J., Boyle, G.H.: A high-voltage solid-state battery system. II. Fabrication of thin-film cells. J. Electrochem. Soc. 116, 1452–1454 (1969). https://doi.org/10.1149/1.2411560
Kanno, R., Takeda, Y., Takada, K., et al.: Ionic conductivity and phase transition of the spinel system Li2−2xM1+xCl4 (M = Mg, Mn, Cd). J. Electrochem. Soc. 131, 469–474 (1984). https://doi.org/10.1149/1.2115611
Kanno, R., Takeda, Y., Yamamoto, O.: Structure, ionic conductivity and phase transformation of double chloride spinels. Solid State Ion. 28, 1276–1281 (1988). https://doi.org/10.1016/0167-2738(88)90370-0
Lutz, H.D., Schmidt, W., Haeuseler, H.: Chloride spinels: a new group of solid lithium electrolytes. J. Phys. Chem. Solids 42, 287–289 (1981). https://doi.org/10.1016/0022-3697(81)90142-6
Wickleder, M.S., Meyer, G.: Ternre halogenide vom typ A3MX6. III. Synthese, strukturen und ionenleitfhigkeit der halogenide Na3MX6 (X = Cl, Br). Z. Anorg. Allg. Chem. 621, 457–463 (1995). https://doi.org/10.1002/zaac.19956210321
Bohnsack, A., Balzer, G., Güdel, H., et al.: Ternre halogenide vom typ A3MX6. VII [1]. Die bromide Li3MBr6 (M = Sm, Lu, Y): synthese kristallstruktur ionenbeweglichkeit. Z. Anorg. Allg. Chem. 623, 1352–1356 (1997). https://doi.org/10.1002/zaac.19976230905
Tomita, Y., Matsushita, H., Kobayashi, K., et al.: Substitution effect of ionic conductivity in lithium ion conductor, Li3InBr6−x C1x. Solid State Ion. 179, 867–870 (2008). https://doi.org/10.1016/j.ssi.2008.02.012
Asano, T., Sakai, A., Ouchi, S., et al.: Solid halide electrolytes with high lithium-ion conductivity for application in 4 V class bulk-type all-solid-state batteries. Adv. Mater. 30, 1803075 (2018). https://doi.org/10.1002/adma.201803075
Liu, B., Wang, D., Avdeev, M., et al.: High-throughput computational screening of Li-containing fluorides for battery cathode coatings. ACS Sustainable Chem. Eng. 8, 948–957 (2019). https://doi.org/10.1021/acssuschemeng.9b05557
Park, K.H., Kaup, K., Assoud, A., et al.: High-voltage superionic halide solid electrolytes for all-solid-state Li-ion batteries. ACS Energy Lett. 5, 533–539 (2020). https://doi.org/10.1021/acsenergylett.9b02599
Kim, S.Y., Kaup, K., Park, K.H., et al.: Lithium ytterbium-based halide solid electrolytes for high voltage all-solid-state batteries. ACS Mater. Lett. 3, 930–938 (2021). https://doi.org/10.1021/acsmaterialslett.1c00142
Helm, B., Schlem, R., Wankmiller, B., et al.: Exploring aliovalent substitutions in the lithium halide superionic conductor Li3−xIn1−xZrxCl6 (0\(\leqslant\)x\(\leqslant\)0.5). Chem. Mater. 33, 4773–4782 (2021). https://doi.org/10.1021/acs.chemmater.1c01348
Shi, X., Zeng, Z., Zhang, H., et al.: Gram-scale synthesis of nanosized Li.HoBr6. solid electrolyte for all-solid-state Li–Se battery. Small Methods 5, 2101002 (2021). https://doi.org/10.1002/smtd.202101002
Wang, C.H., Liang, J.W., Luo, J., et al.: A universal wet-chemistry synthesis of solid-state halide electrolytes for all-solid-state lithium-metal batteries. Sci. Adv. 7, eabh1896 (2021). https://doi.org/10.1126/sciadv.abh1896
Kwak, H., Wang, S., Park, J., et al.: Emerging halide superionic conductors for all-solid-state batteries: design, synthesis, and practical applications. ACS Energy Lett. 7, 1776–1805 (2022). https://doi.org/10.1021/acsenergylett.2c00438
Li, X.N., Liang, J.W., Luo, J., et al.: Air-stable Li3InCl6 electrolyte with high voltage compatibility for all-solid-state batteries. Energy Environ. Sci. 12, 2665–2671 (2019). https://doi.org/10.1039/c9ee02311a
Wang, K., Ren, Q.Y., Gu, Z.Q., et al.: A cost-effective and humidity-tolerant chloride solid electrolyte for lithium batteries. Nat. Commun. 12, 4410 (2021). https://doi.org/10.1038/s41467-021-24697-2
Muy, S., Voss, J., Schlem, R., et al.: High-throughput screening of solid-state Li-ion conductors using lattice-dynamics descriptors. iScience 16, 270–282 (2019). https://doi.org/10.1016/j.isci.2019.05.036
Park, J., Han, D., Kwak, H., et al.: Heat treatment protocol for modulating ionic conductivity via structural evolution of Li3−xYb1−xMxCl6 (M = Hf4+, Zr4+) new halide superionic conductors for all-solid-state batteries. Chem. Eng. J. 425, 130630 (2021). https://doi.org/10.1016/j.cej.2021.130630
Kwak, H., Han, D., Lyoo, J., et al.: New cost-effective halide solid electrolytes for all-solid-state batteries: mechanochemically prepared Fe3+-substituted Li2ZrCl6. Adv. Energy Mater. 11, 2170045 (2021). https://doi.org/10.1002/aenm.202170045
Liang, J.W., Li, X.N., Wang, S., et al.: Site-occupation-tuned superionic LixScCl3+x halide solid electrolytes for all-solid-state batteries. J. Am. Chem. Soc. 142, 7012–7022 (2020). https://doi.org/10.1021/jacs.0c00134
Zhou, L.D., Kwok, C.Y., Shyamsunder, A., et al.: A new halospinel superionic conductor for high-voltage all solid state lithium batteries. Energy Environ. Sci. 13, 2056–2063 (2020). https://doi.org/10.1039/d0ee01017k
Zhou, L.D., Zuo, T.T., Kwok, C.Y., et al.: High areal capacity, long cycle life 4 V ceramic all-solid-state Li-ion batteries enabled by chloride solid electrolytes. Nat. Energy 7, 83–93 (2022). https://doi.org/10.1038/s41560-021-00952-0
Liang, J.W., Van der Maas, E., Luo, J., et al.: A series of ternary metal chloride superionic conductors for high-performance all-solid-state lithium batteries. Adv. Energy Mater. 12, 2103921 (2022). https://doi.org/10.1002/aenm.202103921
Flores González, N., Minafra, N., Dewald, G., et al.: Mechanochemical synthesis and structure of lithium tetrahaloaluminates, LiAlX4 (X = Cl, Br, I): a family of Li-ion conducting ternary halides. ACS Mater. Lett. 3, 652–657 (2021). https://doi.org/10.1021/acsmaterialslett.1c00055
Kwak, H., Han, D., Son, J.P., et al.: Li+ conduction in aliovalent-substituted monoclinic Li2ZrCl6 for all-solid-state batteries: Li2+xZr1−xMxCl6 (M = In, Sc). Chem. Eng. J. 437, 135413 (2022). https://doi.org/10.1016/j.cej.2022.135413
Liu, Z.T., Ma, S., Liu, J., et al.: High ionic conductivity achieved in Li3Y(Br3Cl3) mixed halide solid electrolyte via promoted diffusion pathways and enhanced grain boundary. ACS Energy Lett. 6, 298–304 (2020). https://doi.org/10.1021/acsenergylett.0c01690
Shi, X.M., Zeng, Z.C., Sun, M.Z., et al.: Fast Li-ion conductor of Li3HoBr6 for stable all-solid-state lithium–sulfur battery. Nano Lett. 21, 9325–9331 (2021). https://doi.org/10.1021/acs.nanolett.1c03573
Yu, T.W., Liang, J.W., Luo, L., et al.: Superionic fluorinated halide solid electrolytes for highly stable Li–metal in all-solid-state Li batteries. Adv. Energy Mater. 11, 2101915 (2021). https://doi.org/10.1002/aenm.202101915
Wu, E.A., Banerjee, S., Tang, H., et al.: A stable cathode-solid electrolyte composite for high-voltage, long-cycle-life solid-state sodium-ion batteries. Nat. Commun. 12, 1256 (2021). https://doi.org/10.1038/s41467-021-21488-7
Kwak, H., Lyoo, J., Park, J., et al.: Na2ZrCl6 enabling highly stable 3 V all-solid-state Na-ion batteries. Energy Storage Mater. 37, 47–54 (2021). https://doi.org/10.1016/j.ensm.2021.01.026
Schlem, R., Banik, A., Eckardt, M., et al.: Na3−xEr1−xZrxCl6: a halide-based fast sodium-ion conductor with vacancy-driven ionic transport. ACS Appl. Energy Mater. 3, 10164–10173 (2020). https://doi.org/10.1021/acsaem.0c01870
Do, J.L., Friščić, T.: Mechanochemistry: a force of synthesis. ACS Cent. Sci. 3, 13–19 (2017). https://doi.org/10.1021/acscentsci.6b00277
Schlem, R., Burmeister, C.F., Michalowski, P., et al.: Energy storage materials for solid-state batteries: design by mechanochemistry. Adv. Energy Mater. 11, 2101022 (2021). https://doi.org/10.1002/aenm.202101022
Sebti, E., Evans, H.A., Chen, H., et al.: Stacking faults assist lithium-ion conduction in a halide-based superionic conductor. J. Am. Chem. Soc. 144, 5795–5811 (2022). https://doi.org/10.1021/jacs.1c11335
Schlem, R., Muy, S., Prinz, N., et al.: Mechanochemical synthesis: a tool to tune cation site disorder and ionic transport properties of Li3MCl6 (M = Y, Er) superionic conductors. Adv. Energy Mater. 10, 1903719 (2020). https://doi.org/10.1002/aenm.201903719
Yu, C., Li, Y., Adair, K.R., et al.: Tuning ionic conductivity and electrode compatibility of Li3YBr6 for high-performance all solid-state Li batteries. Nano Energy 77, 105097 (2020). https://doi.org/10.1016/j.nanoen.2020.105097
Schlem, R., Banik, A., Ohno, S., et al.: Insights into the lithium sub-structure of superionic conductors Li3YCl6 and Li3YBr6. Chem. Mater. 33, 327–337 (2021). https://doi.org/10.1021/acs.chemmater.0c04352
Zhao, B., Lu, Y.F., Yuan, B.H., et al.: Preparation of free-standing Li3InCl6 solid electrolytes film with infiltration-method enable roll-to-roll manufacture. Mater. Lett. 310, 131463 (2022). https://doi.org/10.1016/j.matlet.2021.131463
Oi, T., Miyauchi, K.: Amorphous thin film ionic conductors of mLiF.nAlF3. Mater. Res. Bull. 16, 1281–1289 (1981). https://doi.org/10.1016/0025-5408(81)90099-4
Oi, T.: Ionic conductivity of amorphous mLiFnMF3 thin films (M = Al, Cr, Sc or Al+Sc). Mater. Res. Bull. 19, 1343–1348 (1984). https://doi.org/10.1016/0025-5408(84)90198-3
Jin, X., Sendek, A.D., Cubuk, E.D., et al.: Atomic layer deposition of stable LiAlF4 lithium ion conductive interfacial layer for stable cathode cycling. ACS Nano 11, 7019–7027 (2017). https://doi.org/10.1021/acsnano.7b02561
Wan, T.H., Ciucci, F.: Ab initio study of the defect chemistry and substitutional strategies for highly conductive Li3YX6 (X = F, Cl, Br, and I) electrolyte for the application of solid-state batteries. ACS Appl. Energy Mater. 4, 7930–7941 (2021). https://doi.org/10.1021/acsaem.1c01262
Xu, G.F., Luo, L., Liang, J.W., et al.: Origin of high electrochemical stability of multi-metal chloride solid electrolytes for high energy all-solid-state lithium-ion batteries. Nano Energy 92, 106674 (2022). https://doi.org/10.1016/j.nanoen.2021.106674
Li, X., Liang, J., Adair, K.R., et al.: Origin of superionic Li3Y1–xInxCl6 halide solid electrolytes with high humidity tolerance. Nano Lett. 20, 4384–4392 (2020). https://doi.org/10.1021/acs.nanolett.0c01156
Saito, Y., Maier, J.: Ionic conductivity enhancement of the fluoride conductor CaF2 by grain boundary activation using Lewis acids. J. Electrochem. Soc. 142, 3078–3083 (1995). https://doi.org/10.1149/1.2048691
Rongeat, C., Reddy, M.A., Witter, R., et al.: Nanostructured fluorite-type fluorides as electrolytes for fluoride ion batteries. J. Phys. Chem. C. 117, 4943–4950 (2013). https://doi.org/10.1021/jp3117825
Feinauer, M., Euchner, H., Fichtner, M., et al.: Unlocking the potential of fluoride-based solid electrolytes for solid-state lithium batteries. ACS Appl. Energy Mater. 2, 7196–7203 (2019). https://doi.org/10.1021/acsaem.9b01166
Miyazaki, R., Maekawa, H.: Li+-ion conduction of Li3AlF6 mechanically milled with LiCl. ECS Electrochem. Lett. 1, A87–A89 (2012). https://doi.org/10.1149/2.012206eel
Sacci, R.L., Bennett, T.H., Drews, A.R., et al.: Phase evolution during lithium–indium halide superionic conductor dehydration. J. Mater. Chem. A 9, 990–996 (2021). https://doi.org/10.1039/d0ta10012a
Wang, S.H., Xu, X., Cui, C., et al.: Air sensitivity and degradation evolution of halide solid state electrolytes upon exposure. Adv. Funct. Mater. 32, 2108805 (2022). https://doi.org/10.1002/adfm.202108805
Li, W.H., Liang, J., Li, M., et al.: Unraveling the origin of moisture stability of halide solid-state electrolytes by in situ and operando synchrotron X-ray analytical techniques. Chem. Mater. 32, 7019–7027 (2020). https://doi.org/10.1021/acs.chemmater.0c02419
Zhu, Y.Z., Mo, Y.F.: Materials design principles for air-stable lithium/sodium solid electrolytes. Angew. Chem. Int. Ed. 59, 17472–17476 (2020). https://doi.org/10.1002/anie.202007621
Han, Y., Jung, S.H., Kwak, H., et al.: Single- or poly-crystalline Ni-rich layered cathode, sulfide or halide solid electrolyte: which will be the winners for all-solid-state batteries? Adv. Energy Mater. 11, 2100126 (2021). https://doi.org/10.1002/aenm.202100126
Liang, Y.H., Liu, H., Wang, G.X., et al.: Challenges, interface engineering, and processing strategies toward practical sulfide-based all-solid-state lithium batteries. InfoMat 4, e12292 (2022). https://doi.org/10.1002/inf2.12292
Park, K.H., Bai, Q., Kim, D.H., et al.: Design strategies, practical considerations, and new solution processes of sulfide solid electrolytes for all-solid-state batteries. Adv. Energy Mater. 8, 1800035 (2018). https://doi.org/10.1002/aenm.201800035
Sahu, G., Lin, Z., Li, J.C., et al.: Air-stable, high-conduction solid electrolytes of arsenic-substituted Li4SnS4. Energy Environ. Sci. 7, 1053–1058 (2014). https://doi.org/10.1039/c3ee43357a
Gombotz, M., Wilkening, H.M.R.: Fast Li ion dynamics in the mechanosynthesized nanostructured form of the solid electrolyte Li3YBr6. ACS Sustainable Chem. Eng. 9, 743–755 (2021). https://doi.org/10.1021/acssuschemeng.0c06694
Ito, H., Shitara, K., Wang, Y.M., et al.: Kinetically stabilized cation arrangement in Li3YCl6 superionic conductor during solid-state reaction. Adv. Sci. 8, 2101413 (2021). https://doi.org/10.1002/advs.202101413
Zahiri, B., Patra, A., Kiggins, C., et al.: Revealing the role of the cathode–electrolyte interface on solid-state batteries. Nat. Mater. 20, 1392–1400 (2021). https://doi.org/10.1038/s41563-021-01016-0
Wang, C.H., Liang, J.W., Jiang, M., et al.: Interface-assisted in situ growth of halide electrolytes eliminating interfacial challenges of all-inorganic solid-state batteries. Nano Energy 76, 105015 (2020). https://doi.org/10.1016/j.nanoen.2020.105015
Riegger, L.M., Schlem, R., Sann, J., et al.: Lithium–metal anode instability of the superionic halide solid electrolytes and the implications for solid-state batteries. Angew. Chem. Int. Ed. 60, 6718–6723 (2021). https://doi.org/10.1002/anie.202015238
Ji, W.X., Zheng, D., Zhang, X.X., et al.: A kinetically stable anode interface for Li3YCl6-based all-solid-state lithium batteries. J. Mater. Chem. A 9, 15012–15018 (2021). https://doi.org/10.1039/d1ta03042f
Gao, X.W., Liu, B.Y., Hu, B.K., et al.: Solid-state lithium battery cathodes operating at low pressures. Joule 6, 636–646 (2022). https://doi.org/10.1016/j.joule.2022.02.008
Li, X.N., Liang, J.W., Kim, J.T., et al.: Highly stable halide-electrolyte-based all-solid-state Li–Se batteries. Adv. Mater. 34, 2200856 (2022). https://doi.org/10.1002/adma.202200856
Wang, S., Bai, Q., Nolan, A.M., et al.: Lithium chlorides and bromides as promising solid-state chemistries for fast ion conductors with good electrochemical stability. Angew. Chem. Int. Ed. 58, 8039–8043 (2019). https://doi.org/10.1002/anie.201901938
Goodenough, J.B., Kim, Y.: Challenges for rechargeable Li batteries. Chem. Mater. 22, 587–603 (2010). https://doi.org/10.1021/cm901452z
Shao, Q.N., Yan, C.H., Gao, M.X., et al.: New insights into the effects of Zr substitution and carbon additive on Li3–xEr1–xZrxCl6 halide solid electrolytes. ACS Appl. Mater. Interfaces 14, 8095–8105 (2022). https://doi.org/10.1021/acsami.1c25087
Zhu, Y.Z., He, X.F., Mo, Y.F.: Origin of outstanding stability in the lithium solid electrolyte materials: insights from thermodynamic analyses based on first-principles calculations. ACS Appl. Mater. Interfaces 7, 23685–23693 (2015). https://doi.org/10.1021/acsami.5b07517
Nolan, A.M., Zhu, Y.Z., He, X.F., et al.: Computation-accelerated design of materials and interfaces for all-solid-state lithium-ion batteries. Joule 2, 2016–2046 (2018). https://doi.org/10.1016/j.joule.2018.08.017
Tang, H.M., Deng, Z., Lin, Z.N., et al.: Probing solid–solid interfacial reactions in all-solid-state sodium-ion batteries with first-principles calculations. Chem. Mater. 30, 163–173 (2018). https://doi.org/10.1021/acs.chemmater.7b04096
Chu, I.H., Kompella, C.S., Nguyen, H., et al.: Room-temperature all-solid-state rechargeable sodium-ion batteries with a Cl-doped Na3PS4 superionic conductor. Sci. Rep. 6, 33733 (2016). https://doi.org/10.1038/srep33733
Komaba, S., Nakayama, T., Ogata, A., et al.: Electrochemically reversible sodium intercalation of layered NaNi0.5Mn0.5O2 and NaCrO2. ECS Trans. 16, 43–55 (2009). https://doi.org/10.1149/1.3112727
Ellis, B.L., Makahnouk, W.R.M., Rowan-Weetaluktuk, W.N., et al.: Crystal structure and electrochemical properties of A2MPO4F fluorophosphates (A = Na, Li; M = Fe, Mn Co, Ni). Chem. Mater. 22, 1059–1070 (2010). https://doi.org/10.1002/chin.201031010
Jian, Z.L., Han, W.Z., Lu, X., et al.: Superior electrochemical performance and storage mechanism of Na3V2(PO4)3 cathode for room-temperature sodium-ion batteries. Adv. Energy Mater. 3, 156–160 (2013). https://doi.org/10.1002/aenm.201200558
Kim, K., Park, D., Jung, H.G., et al.: Material design strategy for halide solid electrolytes Li3MX6 (X = Cl, Br, and I) for all-solid-state high-voltage Li-ion batteries. Chem. Mater. 33, 3669–3677 (2021). https://doi.org/10.1021/acs.chemmater.1c00555
Richards, W.D., Miara, L.J., Wang, Y., et al.: Interface stability in solid-state batteries. Chem. Mater. 28, 266–273 (2016). https://doi.org/10.1021/acs.chemmater.5b04082
Fu, Y.Y., Ma, C.: Interplay between Li3YX6 (X = Cl or Br) solid electrolytes and the Li metal anode. Sci. China Mater. 64, 1378–1385 (2021). https://doi.org/10.1007/s40843-020-1580-3
Luo, X.M., Wu, X.Z., Xiang, J.Y., et al.: Heterovalent cation substitution to enhance the ionic conductivity of halide electrolytes. ACS Appl. Mater. Interfaces 13, 47610–47618 (2021). https://doi.org/10.1021/acsami.1c13295
Zhang, Y.Z., Sun, C.W.: Composite lithium protective layer formed in situ for stable lithium metal batteries. ACS Appl. Mater. Interfaces 13, 12099–12105 (2021). https://doi.org/10.1021/acsami.1c00745
Kobayashi, H., Yuan, G.H., Gambe, Y., et al.: Effective Li3AlF6 surface coating for high-voltage lithium-ion battery operation. ACS Appl. Energy Mater. 4, 9866–9870 (2021). https://doi.org/10.1021/acsaem.1c01885
Koerver, R., Zhang, W.B., de Biasi, L., et al.: Chemo-mechanical expansion of lithium electrode materials: on the route to mechanically optimized all-solid-state batteries. Energy Environ. Sci. 11, 2142–2158 (2018). https://doi.org/10.1039/c8ee00907d
Pang, Y.P., Pan, J.Y., Yang, J.H., et al.: Electrolyte/electrode interfaces in all-solid-state lithium batteries: a review. Electrochem. Energy Rev. 4, 169–193 (2021). https://doi.org/10.1007/s41918-020-00092-1
Fan, X.M., Hu, G.R., Zhang, B., et al.: Crack-free single-crystalline Ni-rich layered NCM cathode enable superior cycling performance of lithium-ion batteries. Nano Energy 70, 104450 (2020). https://doi.org/10.1016/j.nanoen.2020.104450
Trevisanello, E., Ruess, R., Conforto, G., et al.: Polycrystalline and single crystalline NCM cathode materials: quantifying particle cracking, active surface area, and lithium diffusion. Adv. Energy Mater. 11, 2003400 (2021). https://doi.org/10.1002/aenm.202003400
Cha, H., Kim, J., Lee, H., et al.: Boosting reaction homogeneity in high-energy lithium-ion battery cathode materials. Adv. Mater. 32, 2003040 (2020). https://doi.org/10.1002/adma.202003040
Zhang, W.B., Leichtweiß, T., Culver, S.P., et al.: The detrimental effects of carbon additives in Li10GeP2S12-based solid-state batteries. ACS Appl. Mater. Interfaces 9, 35888–35896 (2017). https://doi.org/10.1021/acsami.7b11530
Nam, Y.J., Oh, D.Y., Jung, S.H., et al.: Toward practical all-solid-state lithium-ion batteries with high energy density and safety: comparative study for electrodes fabricated by dry- and slurry-mixing processes. J. Power Sources 375, 93–101 (2018). https://doi.org/10.1016/j.jpowsour.2017.11.031
Lee, J., Lee, T., Char, K., et al.: Issues and advances in scaling up sulfide-based all-solid-state batteries. Acc. Chem. Res. 54, 3390–3402 (2021). https://doi.org/10.1021/acs.accounts.1c00333
Wu, J.Y., Yuan, L.X., Zhang, W.X., et al.: Reducing the thickness of solid-state electrolyte membranes for high-energy lithium batteries. Energy Environ. Sci. 14, 12–36 (2021). https://doi.org/10.1039/d0ee02241a
Yang, X.F., Adair, K.R., Gao, X.J., et al.: Recent advances and perspectives on thin electrolytes for high-energy-density solid-state lithium batteries. Energy Environ. Sci. 14, 643–671 (2021). https://doi.org/10.1039/d0ee02714f
Wang, K., Ye, Q., Zhang, J., et al.: Halide electrolyte Li3InCl6-based all-solid-state lithium batteries with slurry-coated LiNi0.8Co0.1Mn0.1O2 composite cathode effect of binders. Front Mater. 8, 727617 (2021). https://doi.org/10.3389/fmats.2021.727617
Tan, D.H.S., Banerjee, A., Deng, Z., et al.: Enabling thin and flexible solid-state composite electrolytes by the scalable solution process. ACS Appl. Energy Mater. 2, 6542–6550 (2019). https://doi.org/10.1021/acsaem.9b01111
Lee, K., Kim, S., Park, J., et al.: Selection of binder and solvent for solution-processed all-solid-state battery. J. Electrochem. Soc. 164, A2075–A2081 (2017). https://doi.org/10.1149/2.1341709jes
Oh, D.Y., Nam, Y.J., Park, K.H., et al.: Slurry-fabricable Li +-conductive polymeric binders for practical all-solid-state lithium-ion batteries enabled by solvate ionic liquids. Adv. Energy Mater. 9, 1802927 (2019). https://doi.org/10.1002/aenm.201802927
Wang, C.H., Yu, R.Z., Duan, H., et al.: Solvent-free approach for interweaving freestanding and ultrathin inorganic solid electrolyte membranes. ACS Energy Lett. 7, 410–416 (2021). https://doi.org/10.1021/acsenergylett.1c02261
Hart, P.W., Sommerfeld, J.T.: Cost estimation of specialty chemicals from laboratory-scale prices. Cost Eng. 39, 31–35 (1997)
Acknowledgements
The authors acknowledge the financial support of the Beijing National Laboratory for Condensed Matter Physics, 21C Innovation Laboratory, Contemporary Amperex Technology Ltd. through project No. 21C-OP-202212; the Foundation of Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University; the Foundation of State Key Laboratory of High-Efficiency Utilization of Coal and Green Chemical Engineering (Grant No. 2022-K15), China University of Mining & Technology (Beijing); and the National Natural Science Foundation of China (Nos. 51672029 and 51372271).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tuo, K., Sun, C. & Liu, S. Recent Progress in and Perspectives on Emerging Halide Superionic Conductors for All-Solid-State Batteries. Electrochem. Energy Rev. 6, 17 (2023). https://doi.org/10.1007/s41918-023-00179-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41918-023-00179-5