
Abstract
Fuel cell reactors can be tailored to simultaneously cogenerate value-added chemicals and electrical energy while releasing negligible CO2 emissions or other pollution; moreover, some of these reactors can even “breathe in” poisonous gas as feedstock. Such clean cogeneration favorably offsets the fast depletion of fossil fuel resources and eases growing environmental concerns. These unique reactors inherit advantages from fuel cells: a high energy conversion efficiency and high selectivity. Compared with similar energy conversion devices with sandwich structures, fuel cell reactors have successfully “hit three birds with one stone” by generating power, producing chemicals, and maintaining eco-friendliness. In this review, we provide a systematic summary on the state of the art regarding fuel cell reactors and key components, as well as the typical cogeneration reactions accomplished in these reactors. Most strategies fall short in reaching a win–win situation that meets production demand while concurrently addressing environmental issues. The use of fuel cells (FCs) as reactors to simultaneously produce value-added chemicals and electrical power without environmental pollution has emerged as a promising direction. The FC reactor has been well recognized due to its “one stone hitting three birds” merit, namely, efficient chemical production, electrical power generation, and environmental friendliness. Fuel cell reactors for cogeneration provide multidisciplinary perspectives on clean chemical production, effective energy utilization, and even pollutant treatment, with far-reaching implications for the wider scientific community and society. The scope of this review focuses on unique reactors that can convert low-value reactants and/or industrial wastes to value-added chemicals while simultaneously cogenerating electrical power in an environmentally friendly manner.
Graphical Abstract
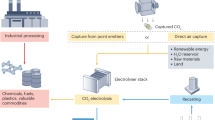
A schematic diagram for the concept of fuel cell reactors for cogeneration of electrical energy and value-added chemicals

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Electricity and value-added chemicals are among the indispensable necessities of modern human society [1,2,3,4,5,6]. However, energy generation and chemical production have led to the fast depletion of fossil fuel resources [7,8,9] and related environmental issues [10, 11]. Therefore, the global search for a more efficient and environmentally friendly approach to clean power generation and the production of downstream value-added chemicals has drawn extensive attention in the past few decades because of increasing demand. To date, most strategies have fallen short in addressing these issues to reach a win–win solution that simultaneously meets production needs and environmental concerns [12,13,14,15,16,17,18,19].
Inspired by this, the ideal method should be tailored to simultaneously cogenerate value-added chemicals with high conversion efficiency and high-density power in the same reactor without any external electrical energy or emission of pollutants. Considering their combined function as both chemical reactors and electric generators, fuel cells, which emerged in 1838 [20, 21], have been adopted as a base to realize the concept of “cogeneration” [22,23,24]. In fuel cell reactors, value-added chemicals and electrical energy can be simultaneously harvested with negligible emissions of CO2 or other pollution; furthermore, this kind of reactor can even “breathe in” poisonous gas as a feedstock. Therefore, these unique reactors can result in great contributions to a low-carbon economy [25, 26], which coincides with the concept of “carbon neutrality” [27,28,29]. Multinational groups of scientists have collaborated on clean energy that surpasses carbon neutrality by absorbing CO2 from the atmosphere. These unique reactors would provide the potential to release less CO2 and/or to capture CO2, which could be one strategy to achieve carbon neutrality commitments.
As a kind of electrical power-generating device, fuel cells can be understood as electrochemical reactors that convert chemical energy from fuels and oxygen (or oxygen in the air) into electrical energy through electrochemical reactions with a maximum theoretical energy efficiency of 83%; this is in contrast to the only 58% of internal combustion engines due to the omission of the Carnot cycle in fuel cells [30, 31]. As chemical synthesizers, fuel cells can be classified as “electrosynthesis” devices [32,33,34], i.e., electrochemical cells for the production of chemical compounds. By controlling the cell potential, catalyst components, surface structure, and reagent concentration, electrosynthesis has considerable advantages over ordinary redox reactions, such as a regulatable reaction rate and selectivity, reduced waste heat, and higher selectivity and yield [35]. Electrosynthesis has been a zealously researched science topic [36,37,38] and is also economically beneficial in industrial applications, e.g., wastewater treatment [39, 40]. In fuel cell reactors, both oxidation reactions (at the anode) and reduction reactions (at the cathode) can be utilized, often invoking radical intermediates. The intermediates can be produced in initial reactions at the electrode surface, which then diffuse into the solution and participate in subsequent reactions [34, 35], thereby providing the possibility to broaden the range beyond only direct redox reactions at the electrode.
All these superior attributes of fuel cell reactors guarantee energy efficiency when used as electricity generation systems, and the flexible selection of oxidation and reduction reactions at the separated anode and cathode also makes it possible to produce a multitude of value-added chemicals. Furthermore, the use of membranes in a sandwich structure to separate the anodic and cathodic parts ensures the purity of the product [23, 41], as shown in Fig. 1.

Schematic diagram showing the concept of fuel cell reactors that cogenerate electrical energy and value-added chemicals
2 Scope of the Review
This review paper starts with an overall view of all fuel cell reactors for cogeneration, covering the in-depth and distinct aspects of the reactors from their fundamentals to their evaluation criteria. Then, the main categories of fuel cell reactors, including polymer electrolyte membrane fuel cell (PEMFC) reactors and solid oxide fuel cell (SOFC) reactors, will be introduced. Furthermore, other electrochemical reactors based on batteries, which are not actual fuel cells but have borrowed the concept of cogeneration in their discharge processes, are also included in this review. Next, the structural design and electrode material selection of various fuel cell reactors will be comprehensively described and discussed in terms of their universality and individuality. Finally, state-of-the-art reactor technologies will be systematically summarized, and the review paper will end with our vision of future developmental perspectives in this field.
Certain aspects of this review have been covered in other reviews in the past decades: fuel cells for chemicals and energy cogeneration [42], solid electrolyte membrane reactors–current experience and future outlook [23, 24], methane conversion to C2 hydrocarbons in solid electrolyte membrane reactors [43, 44], research progress in ethane dehydrogenation to cogenerate power and value-added chemicals in solid oxide fuel cells [45], fuel cells for carbon capture applications [46], alkaline anion exchange membrane fuel cells for cogeneration of electricity and valuable chemicals [22], progress in solid oxide fuel cells with nickel-based anodes operating on methane and related fuels [47], and insights into the interfacial effects in heterogeneous metal nanocatalysts toward selective hydrogenation [48]. However, the above review papers either focused on one unique type of fuel cell reactor/reaction with a limited literature summary or reactors without cogeneration, which coproduce value-added chemicals and electrical energy, sorted out separately. Therefore, it is imperative to publish a review paper covering all the types of fuel cell reactors that simultaneously cogenerate value-added chemicals and electrical energy in an environmentally friendly manner. A comprehensive summary combined with a thorough analysis of the latest research breakthroughs and advances, aiming for a solid introduction to the universality and individuality of these unique reactors, is urgently needed.
3 Fundamentals of Cogeneration
The “electrogenerative process”, i.e., “cogeneration”, was defined in 1963 by Langer and Landi [49] as an electrochemical reaction with negative free reaction energy \(\left( {\Delta_{{\text{R}}} G^{\Theta } } \right)\) for the overall process that simultaneously produces desired chemicals and electrical energy.
Under the hypothesis that all the pure reactants are converted into pure products, with each species in its standard state, the change in free reaction energy \(\left( {\Delta_{{\text{R}}} G^{\Theta } } \right)\) can be easily calculated according to the stoichiometric factor νi of each reactant i according to the following equation when the standard free energy of formation \(\left( {\Delta_{{\text{F}}} G^{\Theta } } \right)\) [50, 51] of reactant i is available:
The standard equilibrium voltage U0 (V) can also be obtained as follows:
where n is the number of electrons and F is the Faraday constant (96 485 C mol–1).
According to the thermodynamic understanding of electrochemical cogeneration, reactions should be chosen according to the following criteria.
-
1.
The overall reactions must follow \({\Delta_{{\text{R}}} G^{\Theta}}\) < 0, i.e., must be thermodynamically favorable.
-
2.
The chemicals reacting at the anode must be oxidizable, e.g., must have olefinic double or triple bonds; accordingly, the chemicals at the cathode must be reducible, e.g., must have oxidizing groups or elements with high valence.
-
3.
The reactants should be easily soluble in the electrolyte or easily adsorbable at the catalytic sites on the corresponding electrode for the electrode reaction to take place.
Several fuel cell reactors have been developed to shorten complicated chemical formation processes to single-step processes [52, 53]. Compared to conventional catalytic routes, those in fuel cell reactors possess the following advantages.
-
1.
Because the reactants are fed separately into the system, i.e., the oxidation and reduction reactions are never competing at the same sites and the risk of explosion can be reduced.
-
2.
The size of the reactors and the corrosion that occurs in them can be reduced; moreover, by taking advantage of the electrochemical reactions, the required operating temperature can also be much lower.
-
3.
By altering either the electrode potential or external load, the type of product and the corresponding chemical selectivity in the process can be varied; in addition, the variety of particle sizes and the surface status of the catalysts in the electrodes play important roles in selectivity and efficiency.
-
4.
Since the reactants are recyclable, their use is economical, thereby realizing fuel cell reactors that can meet all the requirements while providing an additional benefit in the form of power generation.
4 Evaluation Criteria of Cogeneration Systems
According to the concept of cogeneration, the two main functions of such systems are the harvesting of value-added chemicals and the production of electrical energy, so the corresponding evaluation standards for chemical synthesis reactors and energy-producing systems (fuel cells) [54, 55] can be borrowed, i.e., conversion and selectivity, as well as power density and energy density.
4.1 Conversion and Selectivity
In chemical reaction engineering, conversion and its related terms, i.e., yield and selectivity, are very important for effective holistic reactors. These terms are expressed as ratios of how much of a reactant has reacted (X—conversion, 0 < X < 1), how much of the desired product has been formed (Y—yield, 0 < Y < 1), and how much of the desired product has been formed in proportion to the total amount of reactant(s) consumed (S—selectivity) [56,57,58].
According to the chemical reaction taking place, the stoichiometric coefficients satisfy the following relation:
with the following assumptions.
This definition can also be applied for multiple parallel reactions, either per reaction or simply for the limiting reaction. A batch reaction assumes that all reactants are added at the beginning; a semibatch reaction assumes that some reactants are added at the beginning, while the rest are fed during the batch process; and a continuous reaction assumes that reactants are fed and products leave the reactor continuously in a steady-state process.
Conversion can be defined for both (semi)batch and continuous reactors as the overall conversion and instantaneous conversion. When fuel cells are used as reactors, the internal reactions are continuous [23, 43]. For continuous processes, both the overall and instantaneous conversions are the same. Furthermore, for multiple reactants, the conversion can be defined as overall or per reactant:
In general, the yield is defined as the amount of product(s) produced per amount of product(s) that could be produced:
The overall selectivity is defined as the amount of the reactant required to form a product per total amount of that reactant consumed:
For continuous reactors, the three concepts can be combined:
For example, according to the reactions in Sect. 6.2.2.3, ethylene can be produced from ethane in a fuel cell reactor [59, 60] with the simultaneous cogeneration of electrical energy; the conversion, yield, and selectivity can be calculated as follows:
In addition, some more complicated reactions, such as methane reforming inside SOFC reactors [61, 62], which is described in Sect. 6.2.2.1 can be calculated as follows:
To calculate the conversion, selectivity, and yield, the species and production concentrations need to be defined both qualitatively and quantitatively. Most of these parameters can be determined by directly detecting the outcomes with nuclear magnetic resonance (NMR) spectroscopy [63, 64] and/or chromatographic techniques [65,66,67], such as high-performance liquid chromatography (HPLC), gas chromatography (GC), or even gas chromatography-mass spectrometry (GC–MS).
Another scheme is to “mark” the product with unique molecules as probes, which provides more possibilities for designing the monitoring part of fuel cell reactors. Sombatmankhong et al. reported a strategy to understand the formation, decomposition, and removal of hydrogen peroxide in the cathodic chamber by sensing a fluorescence signal in it using confocal microscopy [68]. Dichlorofluorescein (DCFH) can be used as a highly sensitive fluorescent probe that can be detected by confocal laser scanning microscopy. The quantitative hydrogen peroxide production, effect of operating potential on current efficiency, and electrochemical cogeneration performance have been characterized and investigated with this detection scheme.
4.2 Energy Density and Power Density
In a given region of space or in a system, the energy density is the amount of energy stored per unit volume. In most situations, only the extractable or useful energy is measured, i.e., the inaccessible energy (e.g., rest-mass energy) is ignored [69, 70].
The power density, i.e., the rate of energy flux per unit of area/mass, is a key determinant of the nature and dynamics of energy systems and is an important but largely overlooked parameter [71, 72], because any understanding of complex energy systems must rely on quantitative measurements of many fundamental variables. In energy transformers, such as batteries, fuel cells, motors, and other similar systems (e.g., power supply units), the power density refers to a volume. Therefore, this parameter is also called the volume power density and is expressed as W m–3 [73, 74]. Sometimes the mass power density is an important consideration where weight is constrained, such as in electrical vehicles [71, 75].
Different from pure fuel cell systems, in which the energy density is mainly determined by the fuel type and operating conditions [76,77,78], in our cogeneration systems, “fuels” (or the oxidizing agents fed to the cathodes) have two roles: as feedstocks for realizing value-added chemicals and for producing electrical energy [43]. Therefore, controlling both the input type and operating conditions should be considered first to produce chemicals with higher value and purity, and the energy density or power density should be a secondary evaluation standard after conversion and selectivity [79,80,81].
5 System Approaches for Cogeneration
5.1 Solid Oxide Fuel Cell (SOFC) Reactors
As electrochemical energy conversion devices, SOFCs offer extraordinary promise for delivering significant electrical efficiency and important environmental benefits. In terms of fuel flexibility, as well as efficient (with fuel regeneration of higher than 70%) and clean electric power generation [82, 83], SOFCs produce valuable electricity via reactions between a fuel and pure oxygen (or oxygen in the air); this is accomplished by the diffusion of oxide ions through an ion-conducting solid electrolyte layer from the cathode to the anode or by protons produced from the fuel that diffuse in the opposite direction [84, 85]. SOFCs are composed of a dense electrolyte layer sandwiched between two porous electrodes, i.e., the cathode and anode, as shown in Fig. 2, using an oxide ion (Fig. 2a) and/or a proton (Fig. 2b) conductor as the electrolyte. The external circuit is completed when the electrons generated from fuel oxidation at the anode are accepted for oxygen reduction at the cathode [86, 87].

Schematic diagram of a solid oxide fuel cell (SOFC) showing a an oxide ion-conducting electrolyte and b a proton-conducting electrolyte during operation. A key advantage of SOFCs is their fuel flexibility (i.e., they can also utilize hydrocarbons as a fuel source)
Figure 2 schematically shows the configuration of an SOFC. In this system, the solid electrolyte can be an oxygen ion (O2–) conductor, proton (H+) conductor, or another type of conductor. The materials for oxygen ion conductors, proton conductors and another type of conductors are summarized in Tables 1, 2, and 3, respectively. A typical cell consists of a dense solid electrolyte membrane and two porous electrodes. To realize whole redox reactions in the cell, the cathode in the figure is exposed to an oxygen-containing gas or other kinds of oxidizing agent in a gaseous state; the other electrode, i.e., the anode, is exposed to the reacting mixture, which is mostly composed of a reducing agent in a gaseous state that acts as the fuel.
For simplicity, most of the overall chemical reactions can be written stoichiometrically as follows:
or
As shown in Fig. 3, taking an SOFC with an oxygen ion (O2–) conductor as an example, the two electrodes can be connected to a voltmeter (Case a), to an external resistive load (Case b), or to an external power source (Case c) [23]. Due to the different chemical potentials of oxygen at the two sides (the anode and cathode) in the cell, the driving force for oxygen transport across the electrolyte may operate the cell in one of the following modes [23], which are marked as a, b, and c in Fig. 3.
-
(a)
In the open-circuit configuration, which can also be applied in oxygen activity sensors, the spontaneous movement of oxygen to the low-oxygen side is counterbalanced by the large resistance of the voltmeter; hence, there is no net current through the electrolyte. The difference in chemical potential is converted into the open-circuit electromotive force (emf) of the cell.
-
(b)
In the closed-circuit configuration, oxygen ions travel from the cathode to the anode to react with the “fuel” (reducing agent), which is oxidized to H2O and/or CO2 or partially oxidized to value-added products, according to Reaction (14). Simultaneously, the electric circuit is closed, and if the goal is to produce electricity only, chemical energy can be directly converted into electrical energy.
-
(c)
If, on the other hand, the primary goal is not electricity production but the electrochemical production of (partially) oxidized products, the external power source can be used to impose a current, which is equivalent to an oxygen flux, through the cell in the desired direction. This operation mode is called electrochemical oxygen “pumping”. The term “pumping” can be used to describe an externally driven flux regardless of the flow direction, especially when oxygen is forced to flow opposite to the thermodynamically expected direction. This case represents an electrical energy-consuming mode.

Copyright © 2013, the American Chemical Society
Three modes of operation of solid electrolyte cells: a as oxygen activity sensors, b as fuel cells, and c as electrolyzers or electropromotion reactors. Reprinted with permission from Ref. [88].
It has been stated for decades that one important goal for energy technologies and chemical reactor development is efficiency and environmental benefits. At the forefront of these kinds of technologies are SOFCs, which have been most intensively studied for the direct conversion from chemical energy in a fuel (hydrocarbon, hydrogen, ammonia, hydrothion, etc.) into electricity with high thermodynamic efficiency and minimal environmental pollution [89, 90].
To simultaneously harvest value-added chemicals and electrical energy based on the strength of the superiorities mentioned above, many efforts have been made to selectively oxidize hydrocarbons to valuable compounds with the simultaneous cogeneration of power in SOFC reactors [91, 92]. To estimate the thermodynamic tendency of the reaction, the open-circuit voltages (OCVs) for a single cell based on the selective oxidation reaction can be calculated as shown below.
Taking electrochemical oxidative ethane dehydrogenation as an example [59]:
where E0 is the thermodynamic reversible potential, as determined from the Gibbs free energy \({\Delta }G^{0} \left( T \right)\); this value can be obtained from heat capacity software, such as HSC Chemistry [93]. It has been found that under closed-circuit conditions, the conversion, i.e., selective oxidization of the fuel, is strongly dependent on the current [94], which is adjustable by changing the loading (resistance) in the external circuit so that the conversion increases with increasing current.
In reactors based on SOFCs, most cogeneration reactions employ the anodic side as a zone for value-added chemical production, i.e., selective oxidation of the “fuels”. Selective oxidation mainly uses the O2– ions transferred from the cathodic side (Reaction (14)) [95, 96] or dehydrogenizes the fuel to eliminate some H atoms and generate unsaturated bonds (Reaction (15)) [97, 98]; notably, O2–-conducting and H+-conducting electrolyte membranes are used, respectively, for such reactors. However, due to the poor controllability of the oxidation degree, the latter system has much higher selectivity, which will be discussed in detail in Sect. 6.1.
“Selective oxidation” can be understood not only as “selective” groups in the fuel but also as “selective” unique compositions of the mixed gaseous fluid oxidized in the cogeneration process. The latter definition is beneficial for adjusting the components according to a theoretical ideal, which is significant for application and achieving economic value. One possible strategy for a cogeneration system adopts a novel CH4–CO2 dry reforming [99] process before selectively oxidizing the in situ produced H2 in high-performance proton-conducting SOFCs to coproduce CO-concentrated syngas and electricity; this can be achieved by optimally using the heat generated in the selective H2 oxidation step.
An additional layered structure reported by Luo et al. [61] can be incorporated to form an anode support in a layered SOFC with a Ni0.8Co0.2-La0.2Ce0.8O1.9 (NiCo-LDC) composite for a dry reforming process. Figure 4 is a schematic illustration of this novel layered SOFC design. The cross-sectional scanning electron microscopy (SEM) images of the layered proton-conducting SOFCs (an H-SOFC or PC-SOFC) show how the designed construction was achieved. A feed stream composed of CH4 and CO2 was fully reformed through the high-performance NiCo-LDC catalyst layer, yielding syngas (CO and H2 with a ratio of 1:1) in anode reactions in which part of the H2 in the syngas was consumed by selective oxidation on the anodic catalyst Ni-BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb). In this selective oxidation step, not only were electrical power and CO-enriched syngas generated but the total amount of heat required for internal dry reforming was provided. According to the thermodynamic data below, thermal independence could be obtained when H2 utilization was higher than 52% (corresponding to a total fuel utilization of 26%). Thus, the energy input for conventional methane reforming [100, 101] can be significantly counterbalanced.

Reproduced with permission from Ref. [61]. Copyright © 2016, the Royal Society of Chemistry
a Schematic showing the configuration of the novel layered H-SOFC. Cross-sectional SEM images of the layered H-SOFC: b microstructure of the NiO-BZCYYb/BZCYYb/NBCaC-BZCYYb triple layer, c magnified image of the NBCaC-BZCYYb cathode, d microstructure of the NiCoO-LDC/NiO-BZCYYb interphase; and e EDX elemental mappings of (d).
Similar to the above approach for selectively oxidizing the sweep gas immediately after the reactions in the reforming layer, another alternative structure can be designed in the opposite order as the route of successive reactions, i.e., to reform the off-gas from a fuel cell reactor, in which partial oxidation occurs simultaneously with the harvesting of electrical energy from the difference in Gibbs free energy of the feed gas and the selectively oxidized products. Shao et al. [79] integrated a downstream catalyst in a single-chamber SOFC to maximize its cogeneration function. A bilayer electrolyte fuel cell reactor operated in an atmosphere of CH4:O2 = 2:1 achieved an OCV of 1.07 V and a maximum peak power density of approximately 1 500 mW cm–2 at 700 °C while also achieving the partial oxidation of methane to CO, CO2, H2, and H2O. Then, the effluent gas passed through a GdNi/Al2O3 catalyst at 850 °C, and synthesis gas (syngas) was obtained. In the whole reaction from methane to syngas, the conversion was higher than 95%, with a greater than 98% CO and H2 selectivity and a H2/CO ratio of approximately two. The authors also found that neither the syngas formation rate nor the H2/CO molar ratio was affected by the polarization current density. This design successfully split Reaction (21) into two steps as follows (Fig. 5):

Copyright © 2011, Wiley
Fuel cell reactor system used for the cogeneration of synthesis gas and electric power from methane. The fuel cell and the GdNi-Al2O3 catalyst were located in a single quartz tube reactor (Φ = 14 mm and L = 300 mm). The feed gas was composed of methane and oxygen at a molar ratio of 2:1, which was introduced from one side of the reactor, first passed through the fuel cell, and then the catalyst layer. Both the fuel cell and catalyst were heated in electric furnaces. Reprinted with permission from Ref. [79].
Step one:
Step two:
Selectivity is guaranteed by reduction of the “overoxidized” CO2 and H2O in Step one back to CO and H2 in Step two, and part of the combustion energy in Reaction (21) is converted to electrical energy in Step one.
To explain and guide the design of reactors based on SOFCs, some modeling results have been published [102,103,104]. Barnett et al. [102] evaluated strategies for scaling electrochemical partial oxidation (EPOX) processes from the laboratory scale to the practical application scale using computational models. As shown in Fig. 6, the reaction in their research comprised the partial oxidation of hydrocarbon fuel streams to simultaneously produce syngas (H2 and CO) and electrical energy. Because a practical limitation in this reaction was that carbon tended to deposit on Ni-based anode catalysts, the authors explored the use of barrier layers to overcome carbon deposition. Syngas and electricity could be delivered through a designed tubular cell from methane as the primary fuel. The effects of the inlet velocity, barrier layer length, and cell potential were also investigated. The anodic catalysts are summarized in Table 4.

Copyright © 2008, Elsevier
a Illustration of an anode-supported tubular solid oxide fuel cell reactor with a porous barrier layer in the entrance region. b Solution profiles for the nominal tube geometry and operating conditions. The middle panel shows the gas-phase composition of the fuel stream along the channel length. The lower panel shows the local current density, temperatures of the fuel and air streams, and the MEA structure as functions of axial position. The upper panels show the gas-phase composition in the pore spaces and the adsorbed-species coverages on Ni surfaces within the Ni-YSZ anode at three axial positions along the tube. The bottom of these graphs is at the channel interface, and the top is at the dense electrolyte interface. Reprinted with permission from Ref. [102].
Similar research has been published by Ni et al. [103] (Fig. 7). The authors employed a 2D model developed for a tubular direct carbon solid oxide fuel cell (DC-SOFC) reactor for the cogeneration of CO and electricity. Factors including the distance between the carbon chamber and anode electrode (Dce), the operating temperature, and the cell length were all considered. A comparison between electrolyte-supported and anode-supported DC-SOFCs was also made to understand the effects of the support type on the CO generation characteristics and electrical power output.

Copyright © 2016, Elsevier
Schematic of an a electrolyte-supported DC-SOFC and b anode-supported DC-SOFC. Reprinted with permission from Ref. [103].
5.2 Polymer Electrolyte Membrane Fuel Cell (PEMFC) Reactors
PEMFCs have been developed as electrical power sources for portable and stationary power applications, even for vehicles [105]. Most PEMFCs work at temperatures between 20 and 100 °C and pressures between 1 and 5 bar (1 bar = 100 kPa) [106]; thus, not only is a highly proton-conductive polymer membrane, e.g., Nafion™, needed but also a supporting electrolyte or additional product-separating unit is required. As shown in Fig. 8, in the PEMFC anode, hydrogen, methanol, or other fuels can be oxidized, and protons can be produced (Reaction (29) or (30)) and transferred through the Nafion membrane to the cathode, at which pure oxygen or oxygen in the air is employed as the oxidizing agent of the overall fuel cell reaction to generate H2O with protons (Reaction (31)).
or

Schematic of a typical fuel cell reactor based on a polymer electrolyte membrane fuel cell (PEMFC) reactor
The proton-producing reaction at the anode can be understood as a “dehydrogenation reaction”, which can also be used for (partial) oxidation. Borrowing the concept of PEMFCs, the types of reagents for the cathode can be extended from oxygen to other substances with oxidizing ability. As mentioned above, most reactions with an inorganic or unsaturated organic material as an oxidizing agent can occur at the cathode in a PEMFC [68, 106,107,108,109,110,111] (Fig. 9), making use of the “hydrogenation reaction” at the cathode. Therefore, in some reactors, the zones for value-added chemical generation are the cathodic sides, which use H+ from the anode or produce OH– for the anode [22, 112]:

Copyright © 2012, Elsevier
Schematic representation of the gas diffusion electrodes under investigation. Reprinted with permission from Ref. [111].
The standard free enthalpies \(\left( {\Delta_{{\text{R}}} G^{\Theta } } \right)\) [113] of the overall fuel cell reactions can be calculated to be negative, from which the equilibrium voltages U0 can also be obtained. In this way, the cogeneration of desired products and electrical energy in the same fuel cell reactor can be successfully approached.
Similar to PEMFC reactors, another option for cell design is a free electrolyte liquid phase with an additional proton-permeable membrane placed in the electrolyte between the anode and cathode to prevent the crossover of reagents or products [113, 114].
In principle, as shown in Fig. 10, alkaline anion exchange membrane fuel cell (AAEMFC) reactors have similar structures to PEMFC reactors; the main difference is the use of an alkaline anion exchange membrane (AAEM) rather than a polymer electrolyte membrane (PEM) [22]. The corresponding catalysts are summarized in Table 4.

Schematic of a typical fuel cell reactor based on AAEMFCs
Similar to Mode (b) in Fig. 3 of Sect. 5.1 (closed-circuit operation mode in a fuel cell), selective oxidation on the anodic side in combined reactors based on PEMFC or AAEMFC reactors employs dehydrogenation using protons transferred through the membrane to the cathode or OH– produced on the cathodic side [106, 108, 109, 115, 116]:
or
One special case in this type is a biofuel cell reactor, in which microbes (including enzymes) are employed as the catalyst [117,118,119,120]. In these reactors, waste can be easily used as fuel [121], realizing simultaneous waste disposal, as shown in Fig. 11.

Copyright © 2012, Science
Conversion of wastes into bioelectricity and chemicals by using microbial electrochemical technologies. Reprinted with permission from Ref. [121].
5.3 Other Types of Fuel Cell reactors
In addition to the predominant candidates, namely, PEMFCs and SOFCs, alkaline fuel cells (AFCs), phosphoric acid fuel cells (PAFCs), molten salt fuel cells (MSFCs), flow fuel cells (FFCs), and photocatalytic fuel cells (PFCs) have been employed as a basis for cogeneration reactors. Similarly, most reactors based on AFCs, PAFCs, MSFCs, and PFCs use H+ transferred through the electrolyte for reactions in the anode because this approach is more flexible than that of FFCs.
5.3.1 Alkaline Fuel Cell (AFC) Reactors
Initially, AFCs were designed with two electrodes separated by a porous matrix saturated in an aqueous alkaline solution, e.g., potassium hydroxide (KOH) [122, 123]. Because the aqueous alkaline solution can become “poisoned” by the conversion from KOH to K2CO3 due to CO2 generated from fuel oxidation at the anode, more flexible choices have been provided by adopting different combinations of the electrolyte, catholyte, and anolyte. This kind of fuel cell has also been used as a basic cogeneration reactor model.
Zhou and Wu et al. [112] (Fig. 12) reported a reactor based on an alkaline ethanol fuel cell (AEFC) reactor for harvesting electricity and simultaneously reducing Cr2O72– in wastewater, i.e., the contaminant (Cr(VI)) also acts as the oxidant:

Copyright © 2013, the American Chemical Society
Schematic diagram of the fuel cell reactor based on the AEFC. Reprinted with permission from Ref. [112].
An agar-gel electrolyte containing saturated KNO3 was chosen as the catholyte (1 M H2SO4) and anolyte (0.5 M KOH) (1 M = 1 mol L−1). The electrolytes and the cathodic catalysts are summarized in Tables 3 and 5, which are in Sects. 6.1.3 and 6.3.2, respectively.
Similarly, Wen et al. [124] assembled an alkaline-acid Zn-H2O fuel cell for the simultaneous generation of electricity. The OCV was approximately 1.25 V, and H2 was produced with almost 100% Faradic efficiency (FE). The energy was harvested from both electrochemical Zn oxidation and electrochemical neutralization, as demonstrated in Fig. 13 and the equations below.

Copyright © 2018, Wiley
Schematic illustration of the as‐proposed Zn-H2O fuel cell, with a Zn plate in 4.0 M NaOH and Pt/CNTs loaded on a carbon‐cloth cathode in 2.0 M H2SO4, separated by a bipolar membrane. Reprinted with permission from Ref. [124].
Denayer et al. [125] (Fig. 14) employed a tank in series reactor (TSR) model for an AFC that cogenerated H2O2 and electricity in potentiostatic mode. The authors revealed the influence of mass transfer on the distribution of the current density and concentration.

Copyright © 2012, Elsevier
TSR model of an AFC. Reprinted with permission from Ref. [125].
5.3.2 Phosphoric Acid Fuel Cell (PAFC) Reactors
PAFCs employ liquid phosphoric acid as the electrolyte and were the first kind of fuel cell to be successfully commercialized [122, 123]. PAFCs are also the generation preceding PEMFCs; liquid phosphoric acid was replaced with a solid PEM to perform the same function of conducting protons.
Otsuka et al. [126] (Fig. 15) adopted a cell with the configuration [alkene, H2O|(Pd + vapor-grown carbon fiber (VGCF)) anode|H3PO4/silica wool|VGCF-cathode|O2, NO] to realize NO2 reduction and electricity generation. The alkene oxidation rate was accelerated by more than ten times with the addition of NO into the O2 stream to the cathode. The cathode overpotential in this reactor was very small due to the very fast rate of reduction of NO2 to NO and H2O.

Reproduced with permission from Ref. [126]. Copyright © 2003, The Electrochemical Society
Reaction mechanism for the electrochemical oxidation of propylene in the a [C3H6-O2] and b [C3H6-(O2 + NO)] fuel cell reactor systems.
Similarly, Pescarmona et al. [127] constructed a H2-NO PAFC reactor for the cogeneration of hydroxylamine (NH2OH) and electricity.
5.3.3 Molten Salt Fuel Cell (MSFC) Reactors
MSFCs use an electrolyte composed of a molten salt mixture suspended in a porous matrix [128, 129], mostly with operating temperatures at 650 °C or above [130, 131], especially for molten-carbonate fuel cells (MCFCs).
Datta et al. [132] (Fig. 16) used MSFC reactors in a supported molten salt electrocatalysis technique for the partial oxidation of ethylene with homogeneous (liquid phase) catalysts. In this system, to allow operating temperatures of up to 165 °C, the authors chose Nafion membranes impregnated with different electrolytic materials (molten salt containing dispersed or dissolved transition metal complex catalysts occupying a fraction of the pore space) as electrolytes. The reactions and mechanism of the reactor are provided in Sect. 6.2.2.4.

Reproduced with permission from Ref. [132]. Copyright © 1996, The Electrochemical Society
Schematic diagram of a cogeneration reactor based on a supported molten salt electrocatalytic (SMSEC) fuel cell.
Freni et al. [133] (Fig. 17) presented an MCFC reactor system operated with and without a cogeneration bottoming cycle. The thermodynamic yield for direct internal methane catalytic partial oxidation (CPOX) could be accomplished with a theoretical energy efficiency ranging from 100% to 54.9%. This system appeared to be very flexible for application in plants to produce syngas for CH2OH or NH2 synthesis with electricity as a byproduct.

Copyright © 1999, Elsevier
Schematic description of a CPOX-MCFC system. Reprinted with permission from Ref. [133].
5.3.4 Flow Fuel Cell (FFC) Reactors
FFCs use features of laminar flow to form stable interfaces between the anolyte and catholyte, removing the need for expensive semipermeable membranes [134,135,136]. FFCs, especially laminar flow fuel cells (LFFCs), can also be used as cogeneration reactors due to the following advantages.
-
1.
Stable interfaces between the catholyte and anolyte [136].
-
2.
Reduced cost.
- 3.
- 4.
- 5.
-
6.
Independently controllable pH for both the anolyte and catholyte [141].
Wouters et al. [142] (Fig. 18) used a microfluidic fuel cell-type reactor for nitrobenzene reduction and electricity production.

Copyright © 2016, Elsevier
Exploded view of the cogeneration colaminar flow cell (CLFC). (1) POM piece with milled flow channels. (2) Electrocatalyst-coated graphite electrodes were placed in a Delrin® polyoxymethylene (POM) piece and glued to electrical wires with conductive epoxy. (3) Topas® cyclic olefin copolymer (COC) plate and rubber seal covering the channel. (4) Polymethylmethacrylate (PMMA) piece with holes that are used as nanoports for tubing. (5) Aluminum plates that hold the construction together. Reprinted with permission from Ref. [142].
Anode: methanol oxidation
The overall cell reaction is as follows:
Godoi et al. [143] employed a flow battery, which was designed with a hollow fiber of carbon nanotubes (cathode), Zn wire (anode), and 1‐ethyl‐3‐methylimidazolium tetrafluoroborate (electrolyte), to produce CH4 from CO2 under ambient conditions while also promptly outputting the gaseous product through the hollow fiber; this system exhibits a FE of up to 94%. The mechanistic scheme is shown in Fig. 19.

Copyright © 2021, Springer
Proposed mechanistic scheme for the reactions on the a Zn anode and b–d CHF cathode. The inset in (a) shows the numbered [EMIM] + . Reprinted with permission from Ref. [143].
5.3.5 Multiple-Membrane Fuel Cell (MMFC) Reactors
Xia et al. [144] (Fig. 20) reported a different kind of fuel cell reactor with a multiple-membrane structure (three layers): the cationic and anionic products were formed and then combined after diffusion.

Copyright © 2019, Science
Schematic illustration of the two different H2O2 synthesis methods using H2 and O2. a Synthesis of H2O2 using diluted H2 and O2 at high pressure. Methanol, which is used to improve the solubility of the reacting gases in the medium, must then be removed downstream. Other studies that avoid alcohols have been performed in acidic solutions of either HCl or H2SO4, with NaBr or NaCl as promotors. b Electrosynthesis of H2O2 using pure H2 and O2 streams separately introduced to the anode and cathode, respectively. The solid electrolyte (SE) consisted of either functionalized styrene–divinylbenzene copolymer microspheres or inorganic CsxH3–xPW12O40. Electrochemically generated cations (H+) and anions (HO2–), driven by the electric field, cross in the porous SE layer and recombine to form H2O2. DI water flows through the porous SE layer then dissolves H2O2 without any impurities. Reprinted with permission from Ref. [144].
2e– O2 reduction reaction (2e–-ORR):
H2 oxidation reaction (HOR):
H2O2 formation:
5.4 Photoelectrochemical Cell (PEC) Reactors
Photoelectrochemical cells (PECs) electrooxidize water to hydrogen and oxygen by irradiating the anode with electromagnetic radiation. This approach has been referred to as artificial photosynthesis and has been suggested as a way of storing solar energy in the form of hydrogen for use as a fuel [145, 146].
In other words, the cogeneration of chemical and electrical energy, even nonspontaneous reactions, can proceed in this kind of cell by assimilating optical energy.
Li and Wang et al. [147] reported a reactor (Fig. 21) using one photochemical-chemical loop linked with redox couples (Fe2+/Fe3+ and I–/I3–) for H2S chemical absorption redox reactions and photoelectrochemical H2 production as a proof of concept. H2S was directionally oxidized into α-S rather than polysulfide, which was guaranteed by the “oxidizing agent”, namely Fe3+ or I3–, and could be restored to its reduced state to close the loop. Therefore, in this reactor, the hazardous H2S waste could be converted to value-added α-S and electrical energy.

Copyright © 2014, Wiley
a Conventional photoelectrochemical approach and b proposed photoelectrochemical-chemical loop for H2S splitting on an n-type photoelectrode linked with redox couples. Reprinted with permission from Ref. [147].
As a flexible template, Li and Wang et al. [148] further improved the reactors in Fig. 21: the authors used I–/I3– instead as the redox couple in the photoanode loop for the oxidation of H2S to S; in the cathode, the reduction of O2 was accomplished with anthrahydroquinone (H2AQ) to produce hydrogen peroxide (H2O2) and anthraquinone (AQ), which was then electroreduced to H2AQ. The photooxidation of H2S to S and the indirect photoreduction of O2 to H2O2 were integrated into one photochemical-chemical cycle, as shown in Fig. 22.

Reproduced with permission from Ref. [148]. Copyright © 2014, The Royal Society of Chemistry
Schematic illustration of the selective production of H2O2 and S from O2 and H2S on the n-type electrode.
Zheng et al. [149] (Fig. 23) designed and implemented a new type of unassisted PEC system, essentially a light-driven fuel cell reactor, that could produce chemicals (with H2O2 as the main product) at both the BiVO4 photoanode and C cathode. Their work was the first to demonstrate two-sided H2O2 generation with a maximum power density of 0.194 mW cm–2. Both the water oxidation reaction (WOR, Reaction (49)) at the anode and the oxygen reduction reaction (ORR, Reaction (50)) are two-electron pathways.

Copyright 2018, Wiley
a Schematic illustration of the design of a light-driven fuel cell reactor with spontaneous H2O2 generation. The light bulb illustrates the simultaneously produced electricity that flows through the external circuit. b Band diagram of the system. The conduction band (CB) and valence band (VB) edge positions of BiVO4 straddle the redox potentials of O2/H2O2 and H2O/H2O2, suggesting the possibility of unassisted H2O2 production. The theoretical Voc of the light-driven fuel cell reactor is 0.693 V, as estimated from the CB of BiVO4 and O2/H2O2 redox potential. Reprinted with permission from Ref. [149].
The competing reactions (fourelectron pathway and oneelectron pathway) can be repressed by the selection of electrode materials.
For some systems, the reactor design above cannot meet the requirements of mass transfer and light absorption; thus, effort has been put into improving the photoelectrode structure. Jia et al. [150] (Fig. 24) reported a rotating disk photocatalytic fuel cell (RDPFC) reactor using a highly efficient aqueous-film rotating disk; the thin aqueous film on the upper part was continuously refreshed via mass transfer for the simultaneous production of electricity and hydrogen with the concomitant degradation of a high concentration of dye in wastewater.

Copyright © 2014, Elsevier
a Schematic diagram of the aqueous-film RDPFC reactor. b Direct photoexcitation pathway of TiO2 under UV light irradiation. Configuration of the anode compartments in c the aqueous-film RDPFC and d the conventional immersion reactor. The cell is filled with the sample solution. The figure is not to scale. Reprinted with permission from Ref. [150].
5.5 New Types of Cogeneration Reactors
The concept of “cogeneration”, i.e., the simultaneous generation of electrical energy and production of value-added chemicals, can be expanded into reactors based on energy-converting devices other than fuel cells. For example, considering only batteries, the discharge reaction of the whole battery can be understood as a fuel cell reaction that converts chemical energy to electrical energy through electrochemical reactions at both the anode and cathode. Here, we take a nitrogen fixation reaction in a battery as an example.
Zhang et al. [27] (Fig. 25) experimentally proved a proposed reversible Reaction (54) and reported the successful implementation of this reaction based on a reversible N2 cycle in a rechargeable lithium-nitrogen (Li-N2) battery. A Li anode, ether-based electrolyte, and carbon cloth cathode composed the assembled N2-fixation battery system, which is not only a strategy for next-generation electrochemical energy storage systems (Reactions (52) to (53)) but also provides promising candidates for N2 fixation.

Copyright © 2017, Elsevier
Based on a rechargeable lithium-nitrogen battery, an advanced strategy for reversible nitrogen fixation and energy conversion has been successfully implemented at room temperature and atmospheric pressure. It shows a promising nitrogen fixation FE and superior cyclability. Reprinted with permission from Ref. [27].
Furthermore, to the best of our knowledge, NH3 can be produced from Li3N by reacting with water:
The production of NH3 can be verified by the hydrolysis reaction upon combination with Nessler’s reagent to form NH2Hg2OI, which produces a yellow complex:
Considering only discharge reactions, Li can be oxidized as a fuel with the simultaneous reduction of N2 and production of NH3 in the same electrochemical reactor. Because Li is recyclable, ammonia synthesis can be carried out on the cathodic side in this reactor. Nitrogen fixation is very important for industrial engineering and agriculture, and this reactor design may provide a solution.
Similarly, Zhi et al. [151] (Fig. 26) and Ma et al. [152] reported an Al-N2 battery.

Reproduced with permission from Ref. [151]. Copyright © 2020, the Royal Society of Chemistry
Schematic diagram showing the achievement of both energy storage and N2 fixation using the Al-N2 battery system.
More similar cases have been revealed for other batteries involving low-priced reagents at the cathode and the reversible and/or irreversible generation of value-added intermediate products during discharge.
Wang et al. [153] reported a novel fuel-gas CO-generating Li-CO2 battery with a porous fractal Zn cathode capable of tuning CO formation within a wide discharge current range and obtaining a maximum FE of 67% (Fig. 27). In this battery, the proposed CO production mechanism is as follows:

Reproduced with permission from Ref. [153]. Copyright © 2018, the Royal Society of Chemistry
Li-CO2 battery with a porous fractal Zn (PF-Zn) cathode producing CO and electrical energy. a Schematic diagram showing Li-CO2 batteries generating fuel-gas CO. b Galvanostatic discharge potentials at several discharge currents from 0.010 to 0.225 mA. c Galvanostatic discharge potential during long-term discharge at 0.1 mA. Open-circuit potential before 0 min. d Max Faradaic efficiency (FE) of CO at several currents during 10 h of discharge. e FE of CO during long-term discharge at 0.1 mA.
In most Li-CO2, Na-CO2, and metal-CO2/O2 batteries, solid discharge products, such as Li2C2O4 [154] and peroxodicarbonate [154, 155], or solid-state carbon [156] can be obtained, as suggested by the following reaction:
The obtained carbon can exhibit different morphologies, e.g., a network of fibers [157]; if the shape and ingredients are controllable and the product has a high value, the discharge process can successfully realize cogeneration.
With additional reports on Metal-CO2 battery cogeneration reactions [158,159,160,161,162,163], Xie et al. [162] presented metal-CO2 batteries as the crossroad for realizing CO2 cycling, in which simultaneous energy transformations and chemical changes exist in the same system (Fig. 28). This could be understood as an extension of cogeneration reactions.

Reproduced with permission from Ref. [162]. Copyright © 2020, Wiley
Metal-CO2 batteries as the crossroad between the Li/Na‐CO2 system and Zn/Al‐CO2 system to better support sustainable human life.
6 Material Design Strategies for Cogeneration Systems
6.1 Electrolyte Membranes for Fuel Cell Reactors
6.1.1 O2−-conducting Electrolyte Membranes
As mentioned above, in some cogeneration reactors, especially those based on SOFCs, O2−-conducting electrolyte membranes are employed as separators to split the anodic and cathodic reactions and to transfer the O2− generated in the cathode to the anode. These ions act as the oxidizing agent for selective oxidation for achieving the simultaneous cogeneration of electrical energy and value-added chemicals.
Yang et al. [94] (Fig. 29) reported the use of an SOFC as a reactor with Bi4Cu0.2V1.8O11−δ as the O2−-conducting electrolyte membrane for the selective electrochemical oxidation of propane with the cogeneration of electric power and acrylic acid; notably, the selectivity reached 73%.

Reproduced with permission from Ref. [94]. Copyright © 2009, the Royal Society of Chemistry
Mechanism of the selective oxidation of propane in the Bi4Cu0.2V1.8O11−δ (BICUVOX.10) SOFC reactor.
However, due to the poor controllability of selective oxidation from deep oxidation, the selectivity of reactors with O2−-conducting electrolyte membranes is not satisfactory.
6.1.2 H+-Conducting Electrolyte Membranes
Due to their benefits for the simultaneous cogeneration of chemicals and electricity with low or even no CO2 emissions [60, 97, 194], PC-SOFCs have been developed to convert fuel to desired (value-added) chemicals and generate cleaner energy [113]. Additionally, compared with SOFC reactors with O2−-conducting electrolyte membranes, PC-SOFC reactors can better control the degree of oxidation.
As shown in Fig. 30a, due to the presence of oxygen in the reaction, it is not easy to control the degree of deep oxidation from ethane to CO2 and thereby achieve high ethylene selectivity [195, 196], i.e., a large number of byproducts, such as CO and CO2, are produced, which decreases the selectivity of ethylene and the total value of the products [197]. This problem can be overcome in PC-SOFCs because the dehydrogenation reactions are all realized by protons conducted through the electrolyte from the anode to the cathode.

Reproduced with permission from The Royal Society of Chemistry. Reproduced with permission from Ref. [97]. Copyright © 2011, the PCCP Owner Societies
Schematic working principles of hydrocarbon SOFCs with a an oxide ion electrolyte and anode for hydrocarbon deep oxidation and b a proton-conducting electrolyte and dehydrogenation anode.
Proton-conducting electrolytes normally have higher ionic conductivity than oxide ion-conducting electrolytes; thus, PC-SOFCs, which can be operated at intermediate or low temperatures, have attracted increasing attention in recent years [198,199,200,201]. As shown in Fig. 30b, PC-SOFCs are employed as membrane reactors when the anode catalyzes the selective dehydrogenation of a dry hydrocarbon fuel to provide protons. More cogeneration reactors based on SOFCs with proton-conducting electrolyte membranes are summarized in Table 2.
Similarly, in most PEMFCs, Nafion membranes [202, 203] are used as H+-conducting electrolyte membranes (shown in Fig. 31). Nafion, a sulfonated tetrafluoroethylene-based fluoropolymer-copolymer, was discovered by Walther Grot of DuPont in the late 1960s [204, 205]. The unique ionic properties of Nafion are a result of the incorporation of perfluorovinyl ether groups terminated with sulfonate groups onto a tetrafluoroethylene (Teflon) backbone. Nafion has gained a considerable amount of attention as a proton-conducting membrane for PEMFCs [206, 207] because of its excellent thermal and mechanical stability [208, 209]. When PEMFCs are adopted for cogeneration reactors, the selection of proton-conducting electrolyte membranes is optimized; thus, these membranes have been applied in many practical devices, as listed in Table 2.

Copyright © 2010, Elsevier
An idealized Nafion pore. Reprinted with permission from Ref. [210].
6.1.3 Mixed-Ion/Other Ion-Conducting Electrolyte Membranes
In addition to the above cogeneration reactors that use O2−- or H+-conducting electrolyte membranes with O2− or H+ as the reactants or products of electrode reactions, another option in reactor design is to assume that electrolyte membranes are only used as separators between the anodic and cathodic sides to make a closed circle. In this case, mixed-ion or other ion-conducting electrolyte membranes could be an effective design choice.
As mentioned in Sect. 5.3.1, the AEFC reactor assembled by Zhou and Wu et al. [112] used an agar-gel electrolyte containing saturated KNO3 as the “electrolyte membrane”, with Cr(VI) in H2SO4 (aq.) as the catholyte and C2H5OH in KOH (aq.) as the anolyte. The agar-gel electrolyte served only to split the two reactions and to constitute a closed circle, rather than perform mass transport for the reactions, as clearly demonstrated in Fig. 32.

Copyright © 2013, American Chemical Society
Schematic diagram of AEFC. Reprinted with permission from Ref. [112].
In the example above, the authors classified their device as alkaline based on the anolyte rather than the electrolyte membrane. The most important difference between this structure and those in Sects. 6.1.1 and 6.1.2 is that the electrode reactions happen independently, with only electron shifts in the external circuit and no ion transfer through the electrolyte membranes, similar to Zn-H2O fuel cell reactors [125]. The advantage of this kind of reactor is the flexibility of reaction selection on the two sides, and the disadvantage is the consumption of catholyte and anolyte in these reactions, which makes a circulation mechanism necessary for continuous operation.
6.2 Catalyst Materials for Value-Added Chemical Production at the Anode in Fuel Cell Reactors
Anode catalysts in fuel cell reactors, which produce target chemicals on the anodic side, should provide sites for selective oxidation to take place. In reactors for targeted product synthesis, according to the definition of “selective oxidation”, i.e., oxygenation and dehydrogenation, the corresponding catalysts can be classified into two categories, which are described below. For each category, typical examples and catalyst designs are presented.
6.2.1 Anode Catalysts for Oxygenation in Fuel Cell Reactors
Oxygenation reactions in organic chemistry can be defined as reactions increasing the oxidation number of C atoms or other atoms. This kind of chemical change should be accomplished on the anodic side of fuel cell reactors. Properly speaking, the dehydrogenation reaction should be a subset of the oxygenation reaction, but in considering holistic fuel cell reactors, we classify the catalysts for dehydrogenation separately in Sect. 6.2.2; such catalysts accomplish the target reactions by exporting dehydrogenated H atoms through proton-conducting electrolyte membranes.
In this part, several typical oxygenation reactions that take place at the three-phase boundary (shown in Fig. 33) will be introduced. These reactions employ O2− ions imported from the cathode as the oxidizing agent, similar to anodic reactions in SOFCs with O2−-conducting electrolyte membranes; this is shown in the following figure. As mentioned in Sect. 6.1.1 The selectivity is insufficient due to the poor controllability of selective oxidation from deep oxidation.

Copyright © 2002, Elsevier
Schematic illustration of the three-phase boundary at the anode. Reprinted with permission from Ref. [190].
6.2.1.1 Steam-Carbon Fuel Cell for the Cogeneration of H2
The steam-carbon fuel cell concept has been proposed and investigated [95, 227, 228] by adopting and modifying the fluidized bed direct carbon fuel cell (FB-DCFC) concept [229, 230]. A bed of solid carbon particles in the anode compartment contacts the electrode surface, and the cathode compartment is exposed to steam [231].
The overall cell configuration (Fig. 34) is as follows:

Reproduced with permission from Ref. [95]. Copyright © 2011, The Electrochemical Society
Schematic describing the operating principle of steam-carbon fuel cell reactors.
The OCV can be calculated with the Nernst equation:
The OCV can be eliminated by Reaction (66), and the cell potential can guarantee that the reactor is operated as a fuel cell, which enables the cogeneration of electrical power and hydrogen without the need for externally applied energy.
6.2.1.2 Oxidation of NH3 to NO
A few papers have reported types of fuel cells that use NH3 as a kind of fuel to simultaneously produce electrical energy and nitric oxide. For example, Vayenas et al. obtained nitric oxide with a yield higher than 60% along with simultaneous electrical energy production in an SOFC reactor: NH3, NO, N2, Pt/ZrO2(8% Y2O3)/Pt, air [92, 185, 232]; notably, selectivity for NO was obtained between 900 and 1 200 K. Above 1 200 K, direct NH3 decomposition to N2 and H2 became very fast. The selectivity was limited by the flux through the electrolyte, and thinner electrolyte walls must be used to maintain high selectivity at higher NH3 molar flow rates.
6.2.1.3 Producing HCN from CH3OH and NH3
Hydrogen cyanide, which is important for the production of a variety of chemicals, is produced on a large scale worldwide. There are three main commercial hydrogen cyanide production routes, namely, the Andrussow ammoxidation process [233, 234] (Reaction (68)), the Blausäure-Methan-Ammoniak ammonolysis process [235] (Reaction (69)), and the Nitto ammoxidation process [236] (Reaction (70)):
However, all three classical reactions require high-temperature and expensive noble metal catalysts, such as Pt and Ru.
Atkinson et al. accomplished this process by using an SOFC reactor and simultaneously obtained electrochemical energy [166]. The authors used a cermet anode composed of Ni and Ce0.9Gd0.1O1.95 (CGO10), a CGO10 electrolyte, and a La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF)/CGO10 composite cathode at an intermediate temperature (500–650 °C). The electrode reactions are as follows.
HCN in the anode off-gas reached a maximum yield of 40%, and the selectivity of HCN reached 47.5%. CO2, N2, H2, and CH4 were obtained as byproducts.
6.2.2 Anode Catalysts for Dehydrogenation in Fuel Cell Reactors
For dehydrogenation, anode catalysts in fuel cell reactors possess the vital function of pulling off unique H atoms from the reactants; these H atoms are transferred in the form of protons through the proton-conducting electrolyte membrane to participate in cathodic reactions. Most of these kinds of reactions occur at the anodes of PC-SOFC reactors. Representative reactions are classified and summarized below, and details for each type of reaction can be found in Table 6.
6.2.2.1 Oxidation of CH4 to CO2 and H2 or to CO and H2 (Synthesis Gas (Syngas))
Since no oxygen ions can be moved to the anode to directly oxidize carbon species, the direct utilization of hydrocarbon fuels in PC-SOFCs as the only source of electrochemical energy remains challenging. However, this challenge can be overcome through an internal reforming process by reaction with either steam or CO2 [61, 257,258,259]:
In particular, the latter reaction, which is the production of syngas from CH4 and CO2, is defined as “dry reforming” [260,261,262]. An efficient approach based on the SOFC system for this energy conversion process is to include a microreformer [62] in the anode chamber [263, 264] for the internal reforming of biogas (IRB), which can effectively decrease the thermal gradients in the anode.
The high-temperature operation of SOFCs is beneficial to the endothermic reaction step and kinetics of all reactions. IRB can reduce both the system cost and complexity and promote intrinsic thermal coupling between endothermic (IRB, Reaction (75)) and exothermic (electrooxidation, Reactions (76) and (77)) reactions.
From the above reactions, it is clear that heat balance can be easily achieved with the utilization of more than 52% H2.
Luo et al. [81] employed the characteristics of a PC-SOFC and a double perovskite oxide material (PrBaMn2O5+δ (PBM) [265], with excellent coke/sulfur resistance and crystal-structure stability in a reducing atmosphere) to accomplish in situ methane reforming, effective CO2 utilization, and the cogeneration of electrical power through a NiCo/PBM bifunctional nanoarchitecture on BZCYYb in a porous nickel cermet anode using readily available H2S-containing hydrocarbon fuels [252, 266].
Alternatively, the strategy of adding a reforming layer can be used in other fuel cell reactor structure designs [62]. Shao et al. [216] designed a La2NiO4 catalyst layer on the conventional anode of a PC-SOFC for CO2 dry reforming, in which CO2 is reformed with CH4 to yield CO and H2 before entering the anode of the PC-SOFC. The authors employed a Ruddlesden-Popper structure as a catalyst layer on a conventional NiO + BaZr0.4Ce0.4Y0.2O3−δ (NiO + BZCY4) anode to accomplish the in situ CO2 dry reforming of methane with a corresponding CO selectivity of up to 93.3% (Table 6).
Another option is to selectively oxidize H2 in the syngas to coproduce electricity and CO-enriched syngas with few CO2 byproducts. Luo et al. [61] achieved the NiCo-LDC internal dry reforming of methane using a PC-SOFC with a layered structure. It has proven that multiple-twinned bimetallic nanoparticles (Fig. 35) have superior activity toward in situ dry reforming. From the results revealed in Fig. 35, compared to conventional designs, this layer-structured SOFC demonstrated high internal reforming performance (CO2 conversion reached 91.5% at 700 °C) and drastically improved CO2 resistance. With a CH4–CO2 feed stream of 1 A cm−2, the structure achieved up to 100 h of galvanostatic stability. Moreover, by effectively and exclusively converting H2 by electrochemical oxidation, CO-concentrated syngas with no CO2 could be harvested in the anode effluent with a maximum power density of more than 910 mW cm−2 at 700 °C and a polarization resistance as low as 0.121 Ω cm2.

Reproduced with permission from Ref. [61]. Copyright © 2016, the Royal Society of Chemistry
TEM analyses of NiCo-LDC catalysts: a BF micrograph; b–d HRTEM images of NiCo bimetallic nanoparticles at the corresponding sites labeled in (a), with the dotted lines indicating the twin boundaries; and e schematic of a multiple-twinned nanoparticle. H2 selective oxidation was evaluated at 700 °C in layered SOFCs fed with syngas containing various levels of H2. f H2 selectivity and exhaust gas composition as a function of H2 percentage in the syngas and g corresponding voltage responses with different feeds at a constant current load of 2 A cm−2.
From the examples above, we can categorize strategies for dry reforming into two types: one strategy is to accomplish the process in an anodic reaction, and the other is to add a reforming layer before the anodic reaction to optimize the composition for the subsequent electrochemical conversion. The corresponding catalyst designs for the PC-SOFC anodic reaction and the front reforming step are clear.
6.2.2.2 Olefine Acids Produced from Alkanes
As mentioned in Sect. 6.1.1 and demonstrated in Fig. 29, Yang et al. [94] reported a highly efficient catalyst, namely, MoV0.3Te0.17Nb0.12O, as the anodic catalyst in an SOFC reactor for the production of acrylic acid from propane with a selectivity of up to approximately 73% along with the simultaneous production of electricity at 400–450 °C [94]. Selective oxidation from propane to acrylic acid can be realized by transferring O2− through the electrolyte membrane from the cathode:
6.2.2.3 Olefins Produced from Alkanes
For industrial purposes, the predominant method for the production of ethylene is steam cracking [267,268,269]. Because the reaction is reversible, highly endothermic, and severely limited by thermodynamic equilibrium, high-temperature operating conditions are critical but consume energy.
As mentioned in Sect. 6.1.2, due to the high selectivity, i.e., low CO/CO2 emission, of fuel cell reactors with proton-conducting electrolytes [62, 270,271,272], increasingly good results have been reported, especially in alkane elimination reactions. In this kind of cogeneration system, the anode serves as a selective electrocatalytic dehydrogenation reaction zone, which is a key challenge for direct hydrocarbon PC-SOFC reactors [60, 194].
Fu et al. [97] reported the use of a new, inexpensive, high-performance Cu-Cr2O3 nanocomposite catalyst derived from CuCrO2 nanoparticles in a dehydrogenation anode for use in hydrocarbon PC-SOFC reactors (Fig. 36). The efficacy of the new catalyst was demonstrated through the efficient and selective conversion of ethane to ethylene, with an ethylene yield ranging from approximately 9% to 39% and a selectivity ranging from 99% to 90% at operating temperatures ranging from 650 to 750 °C, without any CO2 emission. The simultaneously cogenerated electrical energy possessed a maximum power density of 170 mW cm−2. The mechanism for ethane electrocatalytic dehydrogenation in the SOFC reactor was also proposed.

Reproduced with permission from The Royal Society of Chemistry. Reproduced with permission from Ref. [97]. Copyright © 2011, the PCCP Owner Societies
Proposed mechanism for electrocatalytic ethane dehydrogenation to ethylene over a Cu-Cr2O3 composite anode in a PC-SOFC reactor.
A new kind of anode material composed of double-layered perovskite ((Pr0.4Sr0.6)3(Fe0.85Mo0.15)2O7 (DLP-PSFM)) with a uniformly dispersed in situ exsolution of Co-Fe alloy nanoparticles was synthesized by Luo et al. [59] (Fig. 37) by annealing cubic perovskite (Pr0.4Sr0.6Co0.2Fe0.7Mo0.1O3−δ) in a reducing atmosphere. A maximal output power density of 496.2 mW cm−2 in H2 and 348.84 mW cm−2 in C2H6 at 750 °C was achieved. Moreover, due to the considerably efficient catalysis on the in situ Co-Fe alloy nanoparticles that are homogeneously distributed on the DLP-PSFM backbone, a high ethylene yield was achieved, increasing from 13.2% at 650 °C to 41.5% at 750 °C with a remarkable ethylene selectivity of over 91% and no CO2 emission. Lu et al. [253] also used proton-conducting fuel cells and (Pr0.3Sr0.7)(0.9)Ni0.1Ti0.9O3 as the anodic material to selectively convert propane to propylene and ethylene, respectively, as shown in Fig. 38.

Copyright © 2016, the American Chemical Society
BaCe0.7Zr0.1Y0.2O3−δ electrolyte-supported PC-EFC single cell fabricated with the new DLP-PSFM anode material. Reprinted with permission from [59].

Copyright 2010, Elsevier
Cogeneration of electricity and olefin via proton-conducting fuel cells using (Pr0.3Sr0.7)(0.9)Ni0.1Ti0.9O3 catalyst layers. Reprinted with permission from Ref. [253].
6.2.2.4 Aldehyde Production from Alcohol
Datta et al. used an MSFC with a homogeneous liquid phase to realize the partial oxidation of alcohol to aldehyde and simultaneously coproduce electric power.
Overall for the anode:
Cathode:
Experiments demonstrated the efficiency of the Wacker catalyst (PdC12/CuCl2) (Fig. 39) dispersed in molten salt for the production of acetaldehyde from ethanol at relatively mild temperatures, in which the overall reaction mechanism was assumed to be analogous to that of the Wacker process for ethylene [273].

Reproduced with permission from Ref. [131]. Copyright © 1996, The Electrochemical Society
Proposed mechanism for acetaldehyde synthesis by oxidizing the dehydrogenation of ethanol on a Wacker catalyst.
6.2.2.5 Oxidation of CH4 to CH3OH
The conversion of methane and steam to methanol can be classified as an expanded form of dehydrogenation. Hibino et al. [219] reported this process in a fuel cell reactor where a mixture of methane and H2O vapor was supplied to the anode and air was supplied to the cathode.
At the anode, there were two competing reactions:
and
Therefore, when decreasing the oxidizing ability of active oxygen species for methane, the selective oxidation of methane to methanol (Reaction (85)) was achieved by maintaining a high current efficiency.
Furthermore, fundamental limitations [274] (Fig. 40), materials and device targets [275] have also been studied.

Reproduced with permission from Ref. [274]. Copyright © 2018, the PCCP Owner Societies
Schematic figure of an electrochemical fuel cell converting CH4 and O2 into CH3OH and H2O.
6.3 Catalyst Materials for the Production of Value-Added Chemicals at the Cathode in Fuel Cell Reactors
Similar to fuel cells, i.e., the predecessors of fuel cell reactors, cathodes can be categorized as reaction zones for reduction. In most fuel cells, oxygen or air is fed to the cathodes, at which O2 is converted to O2− (O2−-conducting electrolyte membrane fuel cells) or H+ is transformed to H2O (PC-FCs). When considering the utilization of this part as a value-added chemical production site, the types of reactions can theoretically be chosen from any reaction that has a reduction process, reaction conditions compatible with the whole system, and an overall negative Gibbs free energy. However, focusing on chemical-producing reactors, most reported studies of cathode applications have focused on imported protons (H+).
6.3.1 Cathode Catalysts for Consuming H+ in Fuel Cell Reactors
6.3.1.1 Hydrogenation Reactions for Unsaturated Organics
Schmidt et al. reported that several unsaturated organic alcohols and acids can be fed as oxidants to the cathode with hydrogen as fuel in a PEMFC [106], in which U0 (mentioned in Sect. 3) is in the range of 0.4–0.6 V, i.e., the electrogenerative hydrogenation process takes place at a positive potential relative to the anodic hydrogen electrode [277]. Additionally, the electrogenerative hydrogenation process can be carried out at lower H2 pressures and temperatures compared to conventional catalytic hydrogenation with molecular hydrogen.
In representative reactors based on PEMFCs that use hydrogenation reactions for the cogeneration of electricity, humidified hydrogen is fed to the anode, where protons can be produced. In the presence of H2O, H3O+ can be conducted from the anodic side to the cathodic side through the PEM. Then, the H3O+ react with the unsaturated organics at the cathode. The electrode reactions are as follows.
where R1 and R2 are equal to –H, –CH3, –(CH2)n–CH3, –CH2–OH, –CHO, –COOH, etc.
Schmidt et al. [106] reported a fuel cell reactor employing Nafion® 117 as the PEM and 20% Pt/C on Vulcan XC-72 as the cathodic catalyst for hydrogenation reactions with several unsaturated organic alcohols (allyl alcohol, propargyl alcohol, 2-butyne-1,4-diol, and 2-butene-1,4-diol) and acids (maleic acid, acrylic acid, crotonic acid, and acetylenedicarboxylic acid); the results are listed in Table 7. The authors explained the adsorption of unsaturated alcohols/acids and hydrogenation of double or triple bonds as follows:
Similar reactions can be found in the cathodes of PEMFC reactors for the cogeneration of cyclohexylamine (CHA) by the selective reduction of nitrobenzene (NB) [110],
and the hydrogenation of allyl alcohol to 1-propanol [115],
6.3.1.2 Reduction of Nitric Oxide
The main industrial process for the production of hydroxylamine, a commodity chemical mainly used in caprolactam synthesis, is nitric oxide (NO) hydrogenation in 20% sulfuric acid with a Pt catalyst in a catalytic-slurry reactor [278, 279]:
Analogous production can also be achieved in a PEMFC reactor with more moderate reaction conditions by employing the anode to realize the classical hydrogen oxidation reaction:
At the cathode, the electrochemical reduction products of NO in an acidic medium are as follows [114, 280]:
and
The selectivity for a particular product is sensitive to the electrode potential; for example, nitrous oxide (N2O) is favored at high potentials (E > 0.5 V vs. NHE) while more hydrogenated products predominate at lower potentials [114, 280]. Thus, the selectivity/current efficiency for electrocatalytic reactions to produce hydroxylamine strongly depends on the electrode composition, i.e., the electrocatalyst.
These steps can also be accomplished in a H2-NO PAFC [127] with the mechanism as shown in Fig. 41.

Reproduced with permission from Ref. [127]. Copyright © 2016, the Royal Society of Chemistry
Proposed mechanism for the reduction of NO to NH2OH over isolated Fe sites.
A modified TSR modeled after a H2-NO fuel cell reactor operated in potentiostatic mode with a cocurrent flow direction was presented by Denayer et al. [107] (Fig. 42). The components, the gas channel energy balances in the liquid catholyte, the catalyst layers, and the charge balances at the electrolyte/electrode interfaces were all accounted for by the developed model. The simulation results revealed that the current density, temperature, and concentration distribution were influenced by the mass transfer and catalyst selectivity.

Copyright © 2012, Elsevier
Cocurrent flow pattern for the TSR model of a planar H2-NO fuel cell reactor. Reprinted with permission from Ref. [107].
As introduced in Sect. 5.3.2, Otsuka et al. [126] studied cells operated at 373 K with the configuration [alkene, H2O(Pd + VGCF) anode|H3PO4/silica-woo|׀VGCF-cathode|O2, NO]. The alkene oxidation rate, i.e., the current density, was increased by more than ten times by the addition of NO into the O2 stream at the cathode. In this system, NO acted as an electrochemical reduction catalyst for the conversion of O2 to H2O, according to the following chemical reaction formulas:
6.3.1.3 Hydrogen Peroxide Production
Hydrogen peroxide (H2O2) is widely used in the pulp and paper industry, medicine, water treatment, and detergents. The industrial chemical production method is based on the cyclic reduction of oxygen with hydrogen and uses anthraquinone as the catalyst [281, 282]; an alternative method has been designed based on the electrochemical reduction of oxygen (in the air) at carbonaceous cathodes through a two-electron reaction route [283, 284]. However, neither of the above strategies is safe, clean, and energy economical. Therefore, schemes to cogenerate H2O2 in the cathode of a PEMFC have been investigated.
Agladze et al. [108] accomplished this reaction in a PEMFC reactor with methanol as the fuel. At the cathode, oxygen was reduced to hydrogen peroxide according to the following formula when producing electrical energy:
One possible extension of this reaction is to utilize the in situ produced H2O2 as an oxidizing agent. Yang et al. [109] reported this kind of PEMFC reactor for the cogeneration of electrical energy and phenol, which is a valuable chemical for petrochemicals and is industrially produced via the so-called “cumene process” [285]. The authors used H2 as the fuel and source of protons, which participated as the reactant for the generation of H2O2 and the subsequent phenol, as shown in Fig. 43.

Copyright © 2005, Elsevier
Reaction scheme for phenol synthesis using in situ-generated H2O2 in a PEMFC reactor. Reprinted with permission from Ref. [109].
6.3.1.4 Oxidation with Byproducts at the Cathode
The partial oxidation of methane can be performed at the cathode in a fuel cell reactor. Hinino et al. [217, 218] (Fig. 44) reported a one-pass process for the production of methanol from methane at an intermediate temperature and low pressure in a micro- or nanosized electrocatalyst system. The oxidant for the methane oxidation step was O*, which was a byproduct of the ORR at the cathode.

Copyright © 2010, Elsevier
Schematic illustrations of a macrosized and b micro- or nanosized electrochemical cells. Reprinted with permission from Ref. [217].
The mechanisms for Reactions (112) and (113) on Pd-Au-Cu/C catalysts can be explained according to the Fenton mechanism [217, 218]:
Analogously, under these circumstances, Fenton chemistry predicts the hydroxylation of benzene to phenol [220]:
Furthermore, the oxidation of cyclohexane (CyH) to CyOH or CyO over SmCl3/graphite cathodes can be expected [225, 226].
6.3.2 Cathode Catalysts for Other Reactions in Fuel Cell Reactors
As discussed in Sects. 5.3.1 and 6.1.3, cathodic reactions in cogeneration reactors can be applied to reduce ions from higher to lower valences [112, 124]. Because H+ imported from the anode is not consumed and O2− is not produced at the anode, different analytes and catholytes are employed to participate in the reactions on the electrodes, and a mixed conducting electrolyte membrane closes the circle.
As classified above, in most fuel cell reactors that simultaneously produce electrical energy, value-added chemicals can be harvested at the anode or cathode with the import or export of H+ or O2−. The electrolyte membrane can play an important role in both mass transfer and closing the loop. However, there are also some exceptions, in which no mass transfer occurs through the electrolyte membrane. Some of the typical cogeneration reactions are summarized in Fig. 45.

Summary of the typical cogeneration reactions in Sect. 6: a value-added chemicals produced at the anode and b value-added chemicals produced at the cathode
7 Conclusions and Perspectives
This review presents several typical works focusing on fuel cell reactors in which value-added chemicals and electrical energy are simultaneously produced, killing two birds with only one stone. In this kind of reactor, thermodynamically favorable reactions represent the “stone”: at least one of the electrode reactions is employed for chemical synthesis, and most of the free energy difference between the reactant and product is converted into the form of clean energy. Cogeneration reactions can be classified according to their baseline (i.e., the type of fuel cell used to approach cogeneration), the type of conducting electrolyte membrane, or the electrode reactions adopted for chemical production. However, from the perspective of value-added chemical production, most “synthesis reactions” involve O2− consumption (or H+ production) at the anode or H+ use at the cathode. Due to the poor controllability of the oxidation degree, lower selectivity can be obtained during selective oxidation with O2− due to deep oxidation also occurring. In addition to selectivity, conversion and yield need to be investigated in regard to various chemical synthesis reactors, while the energy density and power density are also integral for the other functions of a fuel cell reactor.
Based on the principles above, several typical cogeneration reactions and corresponding key assembly units (e.g., electrolyte membranes and catalysts) have been demonstrated and categorized. However, for fuel cell reactors with higher selectivity for the production of a unique chemical and better energy conversion efficiency, more effort should be put into the following directions.
-
1.
The material optimization of key components is still imperative for improving product selectivity, energy conversion efficiency, and system stability. Fuel cells can be a fundamental reference, but unlike fuel cells, the target products of the catalysts in cogeneration reactors are value-added chemicals rather than complete oxidation products, e.g., CO2 and H2O; thus, they should be the focus of the electrocatalysis reactions. Additionally, the elucidation of mechanisms would be helpful for the performance optimization of next-generation materials.
-
2.
Product detection remains difficult, especially when gaseous products and liquid products coexist with supporting electrolytic ions. In low-temperature (room temperature) systems, the gaseous products and/or liquid products can dissolve in the electrolyte and/or solvent, which then needs to be mostly neutralized and/or separated before being injected into an IC, HPLC, or other instrument or used in any practical application. In most systems, more than one product can be obtained, and target product separation could be important for value-added chemical production. As an essential part of the whole system design, universally applicable and/or in situ product detection strategies still have considerable room for improvement.
-
3.
Due to the flexibility of reaction selection and the potential for power production, the application of cogeneration reactions is appealing. In addition to the harvesting of electrical energy and value-added chemical production, more environmental management strategies, such as the biosafe disposal of organic matter or toxic ions in industrial wastewater, could be explored.
References
Reshad, A.S., Tiwari, P., Goud, V.V.: Thermo-chemical conversion of waste rubber seed shell to produce fuel and value-added chemicals. J. Energy Inst. 91, 940–950 (2018). https://doi.org/10.1016/j.joei.2017.09.002
Saptal, V.B., Bhanage, B.M.: N-heterocyclic olefins as robust organocatalyst for the chemical conversion of carbon dioxide to value-added chemicals. Chemsuschem 9, 1980–1985 (2016). https://doi.org/10.1002/cssc.201600467
Sayama, K.: Production of high-value-added chemicals on oxide semiconductor photoanodes under visible light for solar chemical-conversion processes. ACS Energy Lett. 3, 1093–1101 (2018). https://doi.org/10.1021/acsenergylett.8b00318
Rodriguez, G., Molina, R.: The competence model and the human potential development for the electricity production. In: 2014 International Conference on Mechatronics, Electronics and Automotive Engineering. Cuernavaca, Mexico. IEEE, pp. 201–205 (2014). https://doi.org/10.1109/ICMEAE.2014.44
Zeliger, H.I.: Toxicology of global warming. In: Zeliger, H.I. (ed.) Human Toxicology of Chemical Mixtures: Toxic Consequences Beyond the Impact of One-Component Product and Environmental Exposures, 2nd edn., pp. 507–519. William Andrew Applied Science Publishers, Norwich (2011). https://doi.org/10.1016/B978-1-4377-3463-8.00037-0
Béchaux, C., Dorne, J.L.C.M.: Mechanistic approaches for human and ecological risk assessment of chemical mixtures. Toxicol. Lett. 229, S18 (2014). https://doi.org/10.1016/j.toxlet.2014.06.098
Whiting, K., Carmona, L.G., Sousa, T.: A review of the use of exergy to evaluate the sustainability of fossil fuels and non-fuel mineral depletion. Renew. Sustain. Energy Rev. 76, 202–211 (2017). https://doi.org/10.1016/j.rser.2017.03.059
Capellán-Pérez, I., Mediavilla, M., de Castro, C., et al.: Fossil fuel depletion and socio-economic scenarios: an integrated approach. Energy 77, 641–666 (2014). https://doi.org/10.1016/j.energy.2014.09.063
Höök, M., Tang, X.: Depletion of fossil fuels and anthropogenic climate change—a review. Energy Policy 52, 797–809 (2013). https://doi.org/10.1016/j.enpol.2012.10.046
Place, S.E., Mitloehner, F.M.: Invited review: Contemporary environmental issues: a review of the dairy industry’s role in climate change and air quality and the potential of mitigation through improved production efficiency. J. Dairy Sci. 93, 3407–3416 (2010). https://doi.org/10.3168/jds.2009-2719
Dai, K.S., Bergot, A., Liang, C., et al.: Environmental issues associated with wind energy: a review. Renew. Energy 75, 911–921 (2015). https://doi.org/10.1016/j.renene.2014.10.074
Huber, G.W., Iborra, S., Corma, A.: Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering. Chem. Rev. 106, 4044–4098 (2006). https://doi.org/10.1021/cr068360d
Trost, B.M., Crawley, M.L.: Asymmetric transition-metal-catalyzed allylic alkylations: applications in total synthesis. Chem. Rev. 103, 2921–2944 (2003). https://doi.org/10.1021/cr020027w
DiSalvo, F.J.: Thermoelectric cooling and power generation. Science 285, 703–706 (1999). https://doi.org/10.1126/science.285.5428.703
Prival, M.J., Dellarco, V.L.: Evolution of social concerns and environmental policies for chemical mutagens. J. Label. Compd. Radiopharm. 14, 46–50 (1989). https://doi.org/10.1002/em.2850140611
Veziroglu, A., Macario, R.: Fuel cell vehicles: state of the art with economic and environmental concerns. Int. J. Hydrog. Energy 36, 25–43 (2011). https://doi.org/10.1016/j.ijhydene.2010.08.145
Temple, G.: Static and dynamic electricity. Nature 146, 446 (1940). https://doi.org/10.1038/146446a0
MacDonald, A.E., Clack, C.T.M., Alexander, A., et al.: Future cost-competitive electricity systems and their impact on US CO2 emissions. Nat. Clim. Change 6, 526–531 (2016). https://doi.org/10.1038/nclimate2921
Khare, A.R., Kumar, B.Y.: Multiagent structures in hybrid renewable power system: a review. J. Renew. Sustain. Energy 7, 063101 (2015). https://doi.org/10.1063/1.4934668
Steele, B.C., Heinzel, A.: Materials for fuel-cell technologies. Nature 414, 345–352 (2001). https://doi.org/10.1038/35104620
Winter, M., Brodd, R.J.: What are batteries, fuel cells, and supercapacitors? Chem. Rev. 105, 1021 (2005). https://doi.org/10.1021/cr040110e
Pan, Z.F., Chen, R., An, L., et al.: Alkaline anion exchange membrane fuel cells for cogeneration of electricity and valuable chemicals. J. Power Sources 365, 430–445 (2017). https://doi.org/10.1016/j.jpowsour.2017.09.013
Garagounis, I., Kyriakou, V., Anagnostou, C., et al.: Solid electrolytes: applications in heterogeneous catalysis and chemical cogeneration. Ind. Eng. Chem. Res. 50, 431–472 (2011). https://doi.org/10.1021/ie1001058
Ferreira, V.J., Figueiredo, J.L., Faria, J.L.: Fuel cells: cogeneration of C2 hydrocarbons or simultaneous production/separation of H2 and C2 hydrocarbons. In: Ferreira, G. (ed.) Alternative Energies. Advanced Structured Materials, vol. 34, pp. 221–239. Springer, Heidelberg (2013). https://doi.org/10.1007/978-3-642-40680-5_10
Jiang, B., Sun, Z.Q., Liu, M.Q.: China’s energy development strategy under the low-carbon economy. Energy 35, 4257–4264 (2010). https://doi.org/10.1016/j.energy.2009.12.040
Chen, Q.X., Kang, C.Q., Xia, Q., et al.: Power generation expansion planning model towards low-carbon economy and its application in China. IEEE Trans. Power Syst. 25, 1117–1125 (2010). https://doi.org/10.1109/TPWRS.2009.2036925
Ma, J. Bao, D., Shi, M., et al.: Reversible nitrogen fixation based on a rechargeable lithium-nitrogen battery for energy storage. Chem 2, 525–532 (2017). https://doi.org/10.1016/j.chempr.2017.03.016
Yao, T.T., Chen, Z.Y.: The explanation of international standard about carbon neutral. Electron. Qual. 59–61 (2011)
Zeng, S.J., Cen, N.S.: “碳中和”与北京绿色奥运 (Carbon neutrality and Beijing green olympics). Soc. Sci. Beijing 2, 4–8 (2008). https://doi.org/10.13262/j.bjsshkxy.bjshkx.2008.02.013
Liebhafsky, H.A.: The fuel cell and the Carnot cycle. J. Electrochem. Soc. 106, 1068–1071 (1959). https://doi.org/10.1149/1.2427213
Lutz, A.E., Larson, R.S., Keller, J.O.: Thermodynamic comparison of fuel cells to the Carnot cycle. Int. J. Hydrog. Energy 27, 1103–1111 (2002). https://doi.org/10.1016/S0360-3199(02)00016-2
Turygin, V.V., Tomilov, A.P.: Possible trends in the development of applied electrochemical synthesis of organic compounds (review). Russ. J. Electrochem. 51, 999–1020 (2015). https://doi.org/10.1134/s1023193515110191
Li, C., Bai, H., Shi, G.: Conducting polymer nanomaterials: electrosynthesis and applications. Chem. Soc. Rev. 38, 2397–2409 (2009). https://doi.org/10.1039/b816681c
Amar, I.A., Lan, R., Petit, C.T.G., et al.: Solid-state electrochemical synthesis of ammonia: a review. J. Solid State Electrochem. 15, 1845–1860 (2011). https://doi.org/10.1007/s10008-011-1376-x
Navarro, M.: Recent advances in experimental procedures for electroorganic synthesis. Curr. Opin. Electrochem. 2, 43–52 (2017). https://doi.org/10.1016/j.coelec.2017.03.004
Francke, R., Little, R.D.: Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 43, 2492–2521 (2014). https://doi.org/10.1039/c3cs60464k
Frontana-Uribe, B.A., Little, R.D., Ibanez, J.G., et al.: Organic electrosynthesis: a promising green methodology in organic chemistry. Green Chem. 12, 2099–2119 (2010). https://doi.org/10.1039/c0gc00382d
Schäfer, H.J.: Contributions of organic electrosynthesis to green chemistry. C. R. Chim. 14, 745–765 (2011). https://doi.org/10.1016/j.crci.2011.01.002
Martínez-Máñez, R., Sancenón, F.: Fluorogenic and chromogenic chemosensors and reagents for anions. Chem. Rev. 103, 4419–4476 (2003). https://doi.org/10.1021/cr010421e
Logan, B.E., Hamelers, B., Rozendal, R., et al.: Microbial fuel cells: methodology and technology. Environ. Sci. Technol. 40, 5181–5192 (2006). https://doi.org/10.1021/es0605016
Djerioui, A., Houari, A., Zeghlache, S., et al.: Energy management strategy of supercapacitor/fuel cell energy storage devices for vehicle applications. Int. J. Hydrog. Energy 44, 23416–23428 (2019). https://doi.org/10.1016/j.ijhydene.2019.07.060
Alcaide, F., Cabot, P.L., Brillas, E.: Fuel cells for chemicals and energy cogeneration. J. Power Sources 153, 47–60 (2006). https://doi.org/10.1016/j.jpowsour.2005.11.041
Stoukides, M.: Methane conversion to C2 hydrocarbons in solid electrolyte membrane reactors. Res. Chem. Intermed. 32, 187–204 (2006). https://doi.org/10.1163/156856706777346499
Xie, S.J., Lin, S.Q., Zhang, Q.H., et al.: Selective electrocatalytic conversion of methane to fuels and chemicals. J. Energy Chem. 27, 1629–1636 (2018). https://doi.org/10.1016/j.jechem.2018.03.015
Fan, Y., Wang, Q., Li, J.Q., et al.: Research progress in ethane dehydrogenation to cogenerate power and value-added chemicals in solid oxide fuel cells. J. Electrochem. 26, 243–252 (2020). https://doi.org/10.13208/J.ELECTROCHEM.191145
Abdelkareem, M.A., Lootah, M.A., Sayed, E.T., et al.: Fuel cells for carbon capture applications. Sci. Total. Environ. 769, 144243 (2021). https://doi.org/10.1016/j.scitotenv.2020.144243
Wang, W., Su, C., Wu, Y.Z., et al.: Progress in solid oxide fuel cells with nickel-based anodes operating on methane and related fuels. Chem. Rev. 113, 8104–8151 (2013). https://doi.org/10.1021/cr300491e
Liu, K.L., Qin, R.X., Zheng, N.F.: Insights into the interfacial effects in heterogeneous metal nanocatalysts toward selective hydrogenation. J. Am. Chem. Soc. 143, 4483–4499 (2021). https://doi.org/10.1021/jacs.0c13185
Langer, S.H., Landi, H.P.: Electrogenerative hydrogenation. J. Am. Chem. Soc. 85, 3043–3044 (1963). https://doi.org/10.1021/ja00902a052
Linebarger, C.E.: Text-book of physical chemistry. J. Am. Chem. Soc. 20, 389–392 (1898). https://doi.org/10.1021/ja02067a016
Lelièvre, T., Rousset, M., Stoltz, G.: Free Energy Computations. Imperial College Press, London (2010). https://doi.org/10.1142/p579
Srinivasan, S.: Fuel Cells: From Fundamentals to Applications. Springer, Berlin (2006). https://doi.org/10.1016/S1369-7021(06)71654-6
Sharaf, O.Z., Orhan, M.F.: An overview of fuel cell technology: fundamentals and applications. Renew. Sustain. Energy Rev. 32, 810–853 (2014). https://doi.org/10.1016/j.rser.2014.01.012
Logan, J.D., Joern, A., Wolesensky, W.: Chemical reactor models of optimal digestion efficiency with constant foraging costs. Ecol. Model. 168, 25–38 (2003). https://doi.org/10.1016/S0304-3800(03)00202-3
Fletcher, P.D.I., Haswell, S.J., Pombo-Villar, E., et al.: Micro reactors: principles and applications in organic synthesis. Tetrahedron 58, 4735–4757 (2002). https://doi.org/10.1016/S0040-4020(02)00432-5
Fogler, H.S.: Elements of chemical reaction engineering. Chem. Eng. Sci. 42, 2493 (1987). https://doi.org/10.1016/0009-2509(87)80130-6
Mathers, R.T., Magenau, A.J.D., Schröder, K., et al.: Overview of controlled/living polymerization methods of vinyl monomers. In: Reed, W.F., Alb, A.M. (eds.) Monitoring Polymerization Reactions: From Fundamentals to Applications, pp. 29–44. Wiley, New Jersey (2014)
Lazaridis, N., Alexopoulos, A.H., Kiparissides, C.: Semi-batch emulsion copolymerization of vinyl acetate and butyl acrylate using oligomeric nonionic surfactants. Macromol. Chem. Phys. 202, 2614–2622 (2001). https://doi.org/10.1002/1521-3935(20010801)202:12%3c2614::AID-MACP2614%3e3.0.CO;2-E
Liu, S., Chuang, K., Luo, J.: Double-layered perovskite anode with in situ exsolution of a Co-Fe alloy to cogenerate ethylene and electricity in a proton-conducting ethane fuel cell. ACS Catal. 6, 760–768 (2016). https://doi.org/10.1021/acscatal.5b02296
Fu, X.Z., Luo, X.X., Luo, J.L., et al.: Ethane dehydrogenation over nano-Cr2O3 anode catalyst in proton ceramic fuel cell reactors to co-produce ethylene and electricity. J. Power Sources 196, 1036–1041 (2011). https://doi.org/10.1016/j.jpowsour.2010.08.043
Hua, B., Yan, N., Li, M., et al.: Novel layered solid oxide fuel cells with multiple-twinned Ni0.8Co0.2 nanoparticles: the key to thermally independent CO2 utilization and power-chemical cogeneration. Energy Environ. Sci. 9, 207–215 (2016). https://doi.org/10.1039/c5ee03017j
Hua, B., Li, M., Sun, Y.F., et al.: Biogas to syngas: flexible on-cell micro-reformer and NiSn bimetallic nanoparticle implanted solid oxide fuel cells for efficient energy conversion. J. Mater. Chem. A 4, 4603–4609 (2016). https://doi.org/10.1039/c6ta00532b
Wishart, D.S., Sykes, B.D., Richards, F.M.: Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222, 311–333 (1991). https://doi.org/10.1016/0022-2836(91)90214-Q
Lipari, G., Szabo, A.: Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104, 4546–4559 (1982). https://doi.org/10.1021/ja00381a009
Hirabayashi, J.: Chromatographic and mass spectrometric techniques. In: Cummings, R.D., Pierce, J.M. (eds.) Handbook of Glycomics, pp. 161–176. Academic Press, Salt Lake City (2009). https://doi.org/10.1016/B978-0-12-373600-0.00007-X
Lonberg-Holm, K.K.: Chromatographic and electrophoretic techniques. Volume II. Zone electrophoresis. J. Am. Chem. Soc. 83, 3354–3355 (1961). https://doi.org/10.1021/ja01476a058
Poole, C.F.: Chromatographic and spectroscopic methods for the determination of solvent properties of room temperature ionic liquids. J. Chromatogr. A 1037, 49–82 (2004). https://doi.org/10.1016/j.chroma.2003.10.127
Sombatmankhong, K., Yunus, K., Fisher, A.C.: Electrocogeneration of hydrogen peroxide: confocal and potentiostatic investigations of hydrogen peroxide formation in a direct methanol fuel cell. J. Power Sources 240, 219–231 (2013). https://doi.org/10.1016/j.jpowsour.2013.03.183
Drewnowski, A., Specter, S.: Poverty and obesity: the role of energy density and energy costs. Am. J. Clin. Nutr. 79, 6–16 (2004). https://doi.org/10.1093/ajcn/79.1.6
Chu, B., Zhou, X., Ren, K., et al.: A dielectric polymer with high electric energy density and fast discharge speed. Science 313, 334–336 (2006). https://doi.org/10.1126/science.1127798
Sulaiman, E., Kosaka, T., Matsui, N.: High power density design of 6-slot–8-pole hybrid excitation flux switching machine for hybrid electric vehicles. IEEE Trans. Magn. 47, 4453–4456 (2011). https://doi.org/10.1109/TMAG.2011.2140315
Lang, X.Y., Hirata, A., Fujita, T., et al.: Nanoporous metal/oxide hybrid electrodes for electrochemical supercapacitors. Nat. Nanotechnol. 6, 232–236 (2011). https://doi.org/10.1038/nnano.2011.13
Gutt, H.J., Grüner, A.: Definition of power density as a general utilization factor of electrical machines. Euro. Trans. Electr. Power 8, 305–308 (2007). https://doi.org/10.1002/etep.4450080414
Pacheco, C., Barbosa, R., Ordoñez, L.C., et al.: Design, simulation and experimental characterization of a high-power density fuel cell. Int. J. Hydrog. Energy 46, 26197–26204 (2021). https://doi.org/10.1016/j.ijhydene.2021.01.132
Wen, H.Q., Xiao, W.D., Wen, X.H., et al.: Analysis and evaluation of DC-link capacitors for high-power-density electric vehicle drive systems. IEEE Trans. Veh. Technol. 61, 2950–2964 (2012). https://doi.org/10.1109/TVT.2012.2206082
Murphy, O.J., Cisar, A., Clarke, E.: Low-cost light weight high power density PEM fuel cell stack. Electrochim. Acta 43, 3829–3840 (1998). https://doi.org/10.1016/S0013-4686(98)00143-1
Shao, Z., Haile, S.M., Ahn, J., et al.: A thermally self-sustained micro solid-oxide fuel-cell stack with high power density. Nature 435, 795–798 (2005). https://doi.org/10.1038/nature03673
Ren, X.M., Price, S.C., Jackson, A.C., et al.: Highly conductive anion exchange membrane for high power density fuel-cell performance. ACS Appl. Mater. Interfaces 6, 13330–13333 (2014). https://doi.org/10.1021/am503870g
Shao, Z.P., Zhang, C.M., Wang, W., et al.: Electric power and synthesis gas co-generation from methane with zero waste gas emission. Angew. Chem. Int. Ed. 50, 1792–1797 (2011). https://doi.org/10.1002/anie.201006855
Yamanaka, I., Onizawa, T., Takenaka, S., et al.: Direct and continuous production of hydrogen peroxide with 93% selectivity using a fuel-cell system. Angew. Chemie Int. Ed Engl. 42, 3653–3655 (2003). https://doi.org/10.1002/anie.200351343
Hua, B., Yan, N., Li, M., et al.: Anode-engineered protonic ceramic fuel cell with excellent performance and fuel compatibility. Adv. Mater. 28, 8922–8926 (2016). https://doi.org/10.1002/adma.201602103
Singhal, S.C.: Advances in solid oxide fuel cell technology. Solid State Ion. 135, 305–313 (2000). https://doi.org/10.1016/S0167-2738(00)00452-5
Shao, Z.P., Haile, S.M.: A high-performance cathode for the next generation of solid-oxide fuel cells. Nature 431, 170–173 (2004). https://doi.org/10.1038/nature02863
Minh, N.Q.: Solid oxide fuel cell technology: features and applications. Solid State Ion. 174, 271–277 (2004). https://doi.org/10.1016/j.ssi.2004.07.042
Yamamoto, O.: Solid oxide fuel cells: fundamental aspects and prospects. Electrochim. Acta 45, 2423–2435 (2000). https://doi.org/10.1016/S0013-4686(00)00330-3
Tao, S.W., Irvine, J.T.S.: A redox-stable efficient anode for solid-oxide fuel cells. Nat. Mater. 2, 320–323 (2003). https://doi.org/10.1038/nmat871
Wachsman, E.D., Lee, K.T.: Lowering the temperature of solid oxide fuel cells. Science 334, 935–939 (2011). https://doi.org/10.1126/science.1204090
Vernoux, P., Lizarraga, L., Tsampas, M.N., et al.: Ionically conducting ceramics as active catalyst supports. Chem. Rev. 113, 8192–8260 (2013). https://doi.org/10.1021/cr4000336
Sundmacher, K., Rihko-Struckmann, L.K., Galvita, V.: Solid electrolyte membrane reactors: status and trends. Catal. Today 104, 185–199 (2005). https://doi.org/10.1016/j.cattod.2005.03.074
Chalakov, L., Rihko-Struckmann, L.K., Munder, B., et al.: Reaction induced current generation by butane oxidation in high temperature electrochemical membrane reactor. Chem. Eng. J. 131, 15–22 (2007). https://doi.org/10.1016/j.cej.2006.11.018
Tagawa, T., Moe, K.K., Ito, M., et al.: Fuel cell type reactor for chemicals-energy co-generation. Chem. Eng. Sci. 54, 1553–1557 (1999). https://doi.org/10.1016/S0009-2509(99)00050-0
Vayenas, C.G., Farr, R.D.: Cogeneration of electric energy and nitric oxide. Science 208, 593–594 (1980). https://doi.org/10.1126/science.208.4444.593
Holler, F.J.: Software: integrated thermodynamic calculations. Anal. Chem. 68, 260A-261A (1996). https://doi.org/10.1021/ac961886l
Ji, B., Wang, J., Chu, W., et al.: Acrylic acid and electric power cogeneration in an SOFC reactor. Chem. Commun. 15, 2038–2040 (2009). https://doi.org/10.1039/b822300a
Alexander, B.R., Mitchell, R.E., Gür, T.M.: Steam-carbon fuel cell concept for cogeneration of hydrogen and electrical power. J. Electrochem. Soc. 158, B505–B513 (2011). https://doi.org/10.1149/1.3560475
Huang, T.J., Huang, M.C.: Temperature effect on electrochemical promotion of syngas cogeneration in direct-methane solid oxide fuel cells. J. Power Sources 175, 473–481 (2008). https://doi.org/10.1016/j.jpowsour.2007.09.061
Fu, X.Z., Lin, J.Y., Xu, S., et al.: CO2 emission free co-generation of energy and ethylene in hydrocarbon SOFC reactors with a dehydrogenation anode. Phys. Chem. Chem. Phys. 13, 19615–19623 (2011). https://doi.org/10.1039/c1cp22837d
Liu, S.B., Behnamian, Y., Chuang, K.T., et al.: A-site deficient La0.2Sr0.7TiO3−δ anode material for proton conducting ethane fuel cell to cogenerate ethylene and electricity. J. Power Sources 298, 23–29 (2015). https://doi.org/10.1016/j.jpowsour.2015.08.032
Chen, B., Xu, H.R., Zhang, Y., et al.: Combined methane reforming by carbon dioxide and steam in proton conducting solid oxide fuel cells for syngas/power co-generation. Int. J. Hydrog. Energy 44, 15313–15321 (2019). https://doi.org/10.1016/j.ijhydene.2019.02.244
Edwards, J.H., Maitra, A.M.: The chemistry of methane reforming with carbon dioxide and its current and potential applications. Fuel Process. Technol. 42, 269–289 (1995). https://doi.org/10.1016/0378-3820(94)00105-3
Rosen, M.A.: Thermodynamic investigation of hydrogen production by steam-methane reforming. Int. J. Hydrog. Energy 16, 207–217 (1991). https://doi.org/10.1016/0360-3199(91)90003-2
Zhu, H.Y., Kee, R.J., Pillai, M.R., et al.: Modeling electrochemical partial oxidation of methane for cogeneration of electricity and syngas in solid-oxide fuel cells. J. Power Sources 183, 143–150 (2008). https://doi.org/10.1016/j.jpowsour.2008.04.076
Xu, H.R., Chen, B., Liu, J., et al.: Modeling of direct carbon solid oxide fuel cell for CO and electricity cogeneration. Appl. Energy 178, 353–362 (2016). https://doi.org/10.1016/j.apenergy.2016.06.064
Quddus, M.R., Zhang, Y., Ray, A.K.: Multi-objective optimization in solid oxide fuel cell for oxidative coupling of methane. Chem. Eng. J. 165, 639–648 (2010). https://doi.org/10.1016/j.cej.2010.09.041
Borup, R., Meyers, J., Pivovar, B., et al.: Scientific aspects of polymer electrolyte fuel cell durability and degradation. Chem. Rev. 107, 3904–3951 (2007). https://doi.org/10.1021/cr050182l
Yuan, X., Ma, Z., Bueb, H., et al.: Cogeneration of electricity and organic chemicals using a polymer electrolyte fuel cell. Electrochim. Acta 50, 5172–5180 (2005). https://doi.org/10.1016/j.electacta.2005.02.080
Danilov, V.A., Denayer, J.F.M.: A modified tank in series model for cogeneration of hydroxylamine and electricity in the nitric oxide/hydrogen fuel cell. Int. J. Hydrog. Energy 37, 9863–9872 (2012). https://doi.org/10.1016/j.ijhydene.2012.03.106
Agladze, G., Nikoleishvili, P., Tsurtsumia, G., et al.: DMFC with hydrogen peroxide cogeneration. J. Electrochem. Soc. 157, E140–E147 (2010). https://doi.org/10.1149/1.3461161
Cai, R., Song, S., Ji, B., et al.: Phenol cogeneration with electricity by using in situ generated H2O2 in a H2–O2 PEMFC reactor. Catal. Today 104, 200–204 (2005). https://doi.org/10.1016/j.cattod.2005.03.052
Yuan, X.Z., Ma, Z.F., Jiang, Q.Z., et al.: Cogeneration of cyclohexylamine and electrical power using PEM fuel cell reactor. Electrochem. Commun. 3, 599–602 (2001). https://doi.org/10.1016/S1388-2481(01)00226-0
Alvarez-Gallego, Y., Dominguez-Benetton, X., Pant, D., et al.: Development of gas diffusion electrodes for cogeneration of chemicals and electricity. Electrochim. Acta 82, 415–426 (2012). https://doi.org/10.1016/j.electacta.2012.06.096
Zhang, H.M., Xu, W., Wu, Z.C., et al.: Removal of Cr(VI) with cogeneration of electricity by an alkaline fuel cell reactor. J. Phys. Chem. C 117, 14479–14484 (2013). https://doi.org/10.1021/jp400896w
Otsuka, K., Sawada, H., Yamanaka, I.: A hydrogen-nitric oxide cell for the synthesis of hydroxylamine. J. Electrochem. Soc. 143, 3491–3497 (1996). https://doi.org/10.1149/1.1837242
Tauszik, G.R., Crocetta, P.: Production of hydroxylamine from nitrogen oxide: a short review. Appl. Catal. 17, 1–21 (1985). https://doi.org/10.1016/S0166-9834(00)82699-8
Yuan, X.Z., Ma, Z.F., He, Q.G., et al.: Electro-generative hydrogenation of allyl alcohol applying PEM fuel cell reactor. Electrochem. Commun. 5, 189–193 (2003). https://doi.org/10.1016/S1388-2481(03)00016-X
Wang, J., Zhang, X., Wang, G., et al.: Sustainable 2,5-furandicarboxylic synthesis by a direct 5-hydroxymethylfurfural fuel cell based on a bifunctional PtNiSx catalyst. Chem. Commun. 56, 13611–13614 (2020). https://doi.org/10.1039/d0cc06087a
Yuan, J.X., Liu, S.J., Jia, L.J., et al.: Co-generation system of bioethanol and electricity with microbial fuel cell technology. Energy Fuels 34, 6414–6422 (2020). https://doi.org/10.1021/acs.energyfuels.0c00749
Kondaveeti, S., Kim, I.W., Otari, S., et al.: Co-generation of hydrogen and electricity from biodiesel process effluents. Int. J. Hydrog. Energy 44, 27285–27296 (2019). https://doi.org/10.1016/j.ijhydene.2019.08.258
Chen, H., Prater, M.B., Cai, R., et al.: Bioelectrocatalytic conversion from N2 to chiral amino acids in a H2/α-keto acid enzymatic fuel cell. J. Am. Chem. Soc. 142, 4028–4036 (2020). https://doi.org/10.1021/jacs.9b13968
Xiao, X., McGourty, K.D., Magner, E.: Enzymatic biofuel cells for self-powered, controlled drug release. J. Am. Chem. Soc. 142, 11602–11609 (2020). https://doi.org/10.1021/jacs.0c05749
Logan, B.E., Rabaey, K.: Conversion of wastes into bioelectricity and chemicals by using microbial electrochemical technologies. Science 337, 686–690 (2012). https://doi.org/10.1126/science.1217412
McLean, G.F., Niet, T., Prince-Richard, S., et al.: An assessment of alkaline fuel cell technology. Int. J. Hydrog. Energy 27, 507–526 (2002). https://doi.org/10.1016/S0360-3199(01)00181-1
Stoica, D., Ogier, L., Akrour, L., et al.: Anionic membrane based on polyepichlorhydrin matrix for alkaline fuel cell: synthesis, physical and electrochemical properties. Electrochim. Acta 53, 1596–1603 (2007). https://doi.org/10.1016/j.electacta.2007.03.034
Cai, P., Li, Y., Wang, G., et al.: Alkaline-acid Zn-H2O fuel cell for the simultaneous generation of hydrogen and electricity. Angew. Chem. Int. Ed 57, 3910–3915 (2018). https://doi.org/10.1002/anie.201712765
De Schepper, P., Danilov, V., Denayer, J.F.M.: A tank in series model for alkaline fuel cell in cogeneration of hydrogen peroxide and electricity. Int. J. Hydrog. Energy 37, 17258–17267 (2012). https://doi.org/10.1016/j.ijhydene.2012.08.065
Yamanaka, I., Nabae, Y., Otsuka, K.: Electrochemical studies of the alkene-NOx fuel cell for organic synthesis. J. Electrochem. Soc. 150, D129–D133 (2003). https://doi.org/10.1149/1.1579036
Daems, N., Sheng, X., Alvarez-Gallego, Y., et al.: Iron-containing N-doped carbon electrocatalysts for the cogeneration of hydroxylamine and electricity in a H2–NO fuel cell. Green Chem. 18, 1547–1559 (2016). https://doi.org/10.1039/c5gc02197a
Selman, J.R.: Molten-salt fuel cells: technical and economic challenges. J. Power Sources 160, 852–857 (2006). https://doi.org/10.1016/j.jpowsour.2006.04.126
Vallet, C.E., Braunstein, J.: Steady-state composition profiles in mixed molten salt electrochemical devices: II. Molten carbonate fuel cell analogs. J. Electrochem. Soc. 126, 527–534 (1979). https://doi.org/10.1149/1.2129080
Brouwer, J., Jabbari, F., Leal, E.M., et al.: Analysis of a molten carbonate fuel cell: numerical modeling and experimental validation. J. Power Sources 158, 213–224 (2006). https://doi.org/10.1016/j.jpowsour.2005.07.093
Chen, L., Yuh, C.Y.: Hardware materials in molten carbonate fuel cell: a review. Acta Metall. Sin. 30, 289–295 (2017). https://doi.org/10.1007/s40195-017-0547-x
Malhotra, S., Datta, R.: Feasibility studies of a fuel cell for cogeneration of homogeneously catalyzed acetaldehyde and electricity from ethanol. J. Electrochem. Soc. 143, 3058–3065 (1996). https://doi.org/10.1149/1.1837164
Freni, S., Cavallaro, S.: Catalytic partial oxidation of methane in a molten carbonate fuel cell. Int. J. Hydrog. Energy 24, 75–82 (1999). https://doi.org/10.1016/S0360-3199(98)00031-7
Ferrigno, R., Stroock, A.D., Clark, T.D., et al.: Membraneless vanadium redox fuel cell using laminar flow. J. Am. Chem. Soc. 124, 12930–12931 (2002). https://doi.org/10.1021/ja020812q
Kjeang, E., Djilali, N., Sinton, D.: Microfluidic fuel cells: a review. J. Power Sources 186, 353–369 (2009). https://doi.org/10.1016/j.jpowsour.2008.10.011
Atobe, M., Tateno, H., Matsumura, Y.: Applications of flow microreactors in electrosynthetic processes. Chem. Rev. 118, 4541–4572 (2018). https://doi.org/10.1021/acs.chemrev.7b00353
Goulet, M.A., Kjeang, E.: Co-laminar flow cells for electrochemical energy conversion. J. Power Sources 260, 186–196 (2014). https://doi.org/10.1016/j.jpowsour.2014.03.009
Xuan, J., Leung, M.K.H., Leung, D.Y.C., et al.: Hydrodynamic focusing in microfluidic membraneless fuel cells: breaking the trade-off between fuel utilization and current density. Int. J. Hydrog. Energy 36, 11075–11084 (2011). https://doi.org/10.1016/j.ijhydene.2011.05.150
Hasegawa, S., Shimotani, K., Kishi, K., et al.: Electricity generation from decomposition of hydrogen peroxide. Electrochem. Solid-State Lett. 8, A119–A121 (2005). https://doi.org/10.1149/1.1849112
Cohen, J.L., Westly, D.A., Pechenik, A., et al.: Fabrication and preliminary testing of a planar membraneless microchannel fuel cell. J. Power Sources 139, 96–105 (2005). https://doi.org/10.1016/j.jpowsour.2004.06.072
Cohen, J.L., Volpe, D.J., Westly, D.A., et al.: A dual electrolyte H2/O2 planar membraneless microchannel fuel cell system with open circuit potentials in excess of 1.4 V. Langmuir 21, 3544–3550 (2005). https://doi.org/10.1021/la0479307
Wouters, B., Hereijgers, J., de Malsche, W., et al.: Electrochemical characterisation of a microfluidic reactor for cogeneration of chemicals and electricity. Electrochim. Acta 210, 337–345 (2016). https://doi.org/10.1016/j.electacta.2016.05.187
Godoi, C.M., Santos, M.C.L., Silva, A.J., et al.: Methane conversion to higher value-added product and energy co-generation using anodes of PdCu/C in a solid electrolyte reactor: alkaline fuel cell type monitored by differential mass spectroscopy. Res. Chem. Intermed. 47, 743–757 (2021). https://doi.org/10.1007/s11164-020-04296-4
Xia, C., Xia, Y., Zhu, P., et al.: Direct electrosynthesis of pure aqueous H2O2 solutions up to 20% by weight using a solid electrolyte. Science 366, 226–231 (2019). https://doi.org/10.1126/science.aay1844
Law, K.Y.: Effect of dye aggregation on the photogeneration efficiency of organic photoconductors. J. Phys. Chem. 92, 4226–4231 (1988). https://doi.org/10.1021/j100325a046
Clarke, T.M., Durrant, J.R.: Charge photogeneration in organic solar cells. Chem. Rev. 110, 6736–6767 (2010). https://doi.org/10.1021/cr900271s
Zong, X., Han, J., Seger, B., et al.: An integrated photoelectrochemical-chemical loop for solar-driven overall splitting of hydrogen sulfide. Angew. Chem. Int. Ed 53, 4399–4403 (2014). https://doi.org/10.1002/anie.201400571
Zong, X., Chen, H.J., Seger, B., et al.: Selective production of hydrogen peroxide and oxidation of hydrogen sulfide in an unbiased solar photoelectrochemical cell. Energy Environ. Sci. 7, 3347–3351 (2014). https://doi.org/10.1039/c4ee01503g
Shi, X.J., Zhang, Y.R., Siahrostami, S., et al.: Light-driven BiVO4–C fuel cell with simultaneous production of H2O2. Adv. Energy Mater. 8, 1801158 (2018). https://doi.org/10.1002/aenm.201801158
Tang, T.T., Li, K., Ying, D.W., et al.: High efficient aqueous-film rotating disk photocatalytic fuel cell (RDPFC) with triple functions: cogeneration of hydrogen and electricity with dye degradation. Int. J. Hydrog. Energy 39, 10258–10266 (2014). https://doi.org/10.1016/j.ijhydene.2014.04.134
Guo, Y., Yang, Q., Wang, D., et al.: A rechargeable Al-N2 battery for energy storage and highly efficient N2 fixation. Energy Environ. Sci. 13, 2888–2895 (2020). https://doi.org/10.1039/d0ee01241f
Xu, C., Huang, J., Ma, J.: Green, cheap and rechargeable Al-N2 battery with efficient N2 fixation. Rare Met. 40, 1–2 (2021). https://doi.org/10.1007/s12598-020-01626-8
Xie, J., Liu, Q., Huang, Y., et al.: A porous Zn cathode for Li-CO2 batteries generating fuel-gas CO. J. Mater. Chem. A 6, 13952–13958 (2018). https://doi.org/10.1039/c8ta02771d
Qiao, Y., Yi, J., Guo, S.H., et al.: Li2CO3-free Li-O2/CO2 battery with peroxide discharge product. Energy Environ. Sci. 11, 1211–1217 (2018). https://doi.org/10.1039/c7ee03341a
Xu, S.M., Lu, Y.Y., Wang, H.S., et al.: A rechargeable Na-CO2/O2 battery enabled by stable nanoparticle hybrid electrolytes. J. Mater. Chem. A 2, 17723–17729 (2014). https://doi.org/10.1039/c4ta04130e
Zhang, Z., Zhang, Q., Chen, Y., et al.: The first introduction of graphene to rechargeable Li-CO2 batteries. Angew. Chem Int. Ed. 54, 6550–6553 (2015). https://doi.org/10.1002/anie.201501214
Liu, Y.L., Wang, R., Lyu, Y.C., et al.: Rechargeable Li/CO2–O2 (2:1) battery and Li/CO2 battery. Energy Environ. Sci. 7, 677–681 (2014). https://doi.org/10.1039/c3ee43318h
Xie, J., Wang, X., Lv, J., et al.: Reversible aqueous zinc-CO2 batteries based on CO2-HCOOH interconversion. Angew. Chem. Int. Ed 57, 16996–17001 (2018). https://doi.org/10.1002/anie.201811853
Al Sadat, W.I., Archer, L.A.: The O2-assisted Al/CO2 electrochemical cell: a system for CO2 capture/conversion and electric power generation. Sci. Adv. 2, e1600968 (2016). https://doi.org/10.1126/sciadv.1600968
Jena, A., Tong, Z.Z., Chang, H., et al.: Capturing carbon dioxide in Na-CO2 batteries: a route for green energy. J. Chin. Chem. Soc. 68, 421–428 (2021). https://doi.org/10.1002/jccs.202000477
Ding, P., Zhang, J., Han, N., et al.: Simultaneous power generation and CO2 valorization by aqueous Al-CO2 batteries using nanostructured Bi2S3 as the cathode electrocatalyst. J. Mater. Chem. A 8, 12385–12390 (2020). https://doi.org/10.1039/d0ta03761c
Yang, R., Peng, Z., Xie, J., et al.: Reversible hybrid aqueous Li-CO2 batteries with high energy density and formic acid production. Chemsuschem 13, 2621–2627 (2020). https://doi.org/10.1002/cssc.201903297
Xie, J.F., Zhou, Z., Wang, Y.B.: Metal-CO2 batteries at the crossroad to practical energy storage and CO2 recycle. Adv. Funct. Mater. 30, 1908285 (2020). https://doi.org/10.1002/adfm.201908285
Hibino, T., Ushiki, K., Kuwahara, Y.: New concept for simplifying SOFC system. Solid State Ion. 91, 69–74 (1996). https://doi.org/10.1016/S0167-2738(96)00437-7
Hibino, T.: A novel cell design for simplifying SOFC system. Solid State Ion. 81, 1–3 (1995). https://doi.org/10.1016/0167-2738(95)00197-e
Raj, A., Rudkin, R.A., Atkinson, A.: Cogeneration of HCN in a solid oxide fuel cell. J. Electrochem. Soc. 157, B719–B725 (2010). https://doi.org/10.1149/1.3330704
Zhu, T.L., Yang, Z.B., Han, M.F.: Performance evaluation of solid oxide fuel cell with in situ methane reforming. Fuel 161, 168–173 (2015). https://doi.org/10.1016/j.fuel.2015.08.050
Liu, K.F., Zhao, J., Zhu, D., et al.: Oxidative coupling of methane in solid oxide fuel cell tubular membrane reactor with high ethylene yield. Catal. Commun. 96, 23–27 (2017). https://doi.org/10.1016/j.catcom.2017.03.010
Ishihara, T.: Oxidative reforming of methane using solid oxide fuel cell with LaGaO3-based electrolyte. Solid State Ion. 79, 371–375 (1995). https://doi.org/10.1016/0167-2738(95)00090-s
Hiei, Y., Ishihara, T., Takita, Y.: Partial oxidation of methane for internally reformed solid oxide fuel cell. Solid State Ion. 86(87/88), 1267–1272 (1996). https://doi.org/10.1016/0167-2738(96)00299-8
Zhang, L., Yang, C.H., Frenkel, A.I., et al.: Co-generation of electricity and chemicals from propane fuel in solid oxide fuel cells with anode containing nano-bimetallic catalyst. J. Power Sources 262, 421–428 (2014). https://doi.org/10.1016/j.jpowsour.2014.04.009
Yentekakis, I.V., Debenedetti, P.G., Costa, B.: A novel fused metal anode solid electrolyte fuel cell for direct coal gasification: a steady-state model. Ind. Eng. Chem. Res. 28, 1414–1424 (1989). https://doi.org/10.1021/ie00093a022
Nakagawa, N., Ishida, M.: Performance of an internal direct-oxidation carbon fuel cell and its evaluation by graphic exergy analysis. Ind. Eng. Chem. Res. 27, 1181–1185 (1988). https://doi.org/10.1021/ie00079a016
Semin, G.L., Belyaev, V.D., Demin, A.K., et al.: Methane conversion to syngas over Pt-based electrode in a solid oxide fuel cell reactor. Appl. Catal. A Gen. 181, 131–137 (1999). https://doi.org/10.1016/S0926-860X(98)00388-3
Shi, H.G., Su, C., Yang, G.M., et al.: Fabrication and operation of flow-through tubular SOFCs for electric power and synthesis gas cogeneration from methane. AIChE J. 60, 1036–1044 (2014). https://doi.org/10.1002/aic.14312
Tagawa, T., Kyaw Moe, K., Hiramatsu, T., et al.: Design of electrode for solid oxide fuel cells reactor. Solid State Ion. 106, 227–235 (1998). https://doi.org/10.1016/S0167-2738(97)00497-9
Steele, B.C.H., Kelly, I., Middleton, H., et al.: Oxidation of methane in solid state electrochemical reactors. Solid State Ion. 28(29/30), 1547–1552 (1988). https://doi.org/10.1016/0167-2738(88)90417-1
Yentekakis, I.V., Jiang, Y., Makri, M., et al.: Ethylene production from methane in a gas recycle electrocatalytic reactor separator. Ionics 1, 286–291 (1995). https://doi.org/10.1007/BF02390209
Otsuka, K., Suga, K., Yamanaka, I.: Oxidative coupling of methane applying a solid oxide fuel cell system. Catal. Today 6, 587–592 (1990). https://doi.org/10.1016/0920-5861(90)85055-S
Appamana, W., Charojrochkul, S., Assabumrungrat, S., et al.: Synthesis of Na2WO4-Mn supported YSZ as a potential anode catalyst for oxidative coupling of methane in SOFC reactor. Eng. J. 19, 13–20 (2015). https://doi.org/10.4186/ej.2015.19.1.13
Tagawa, T., Kuroyanagi, K., Goto, S., et al.: Selective oxidation of methane in an SOFC-type reactor: effect of applied potential. Chem. Eng. J. 93, 3–9 (2003). https://doi.org/10.1016/S1385-8947(02)00105-5
Wiyaratn, W., Appamana, W., Charojrochkul, S., et al.: Au/La1−xSrxMnO3 nanocomposite for chemical-energy cogeneration in solid oxide fuel cell reactor. J. Ind. Eng. Chem. 18, 1819–1823 (2012). https://doi.org/10.1016/j.jiec.2012.04.011
McKenna, E., Stoukides, M.: Modeling of HCN synthesis in a solid electrolyte fuel cell. Chem. Eng. Sci. 47, 2951–2956 (1992). https://doi.org/10.1016/0009-2509(92)87157-L
Kiratzis, N., Stoukides, M.: The synthesis of hydrogen cyanide in a solid electrolyte fuel cell. J. Electrochem. Soc. 134, 1925–1929 (1987). https://doi.org/10.1149/1.2100791
Farr, R.D., Vayenas, C.G.: Ammonia high temperature solid electrolyte fuel cell. J. Electrochem. Soc. 127, 1478–1483 (1980). https://doi.org/10.1149/1.2129934
McKenna, E.A., Othoneos, A., Kiratzis, N., et al.: Synthesis of hydrogen cyanide in a solid-electrolyte-cell reactor. Ind. Eng. Chem. Res. 32, 1904–1913 (1993). https://doi.org/10.1021/ie00021a014
Wei, G.L., Liu, M., Luo, J.L., et al.: Influence of gas flow rate on performance of H2S/air solid oxide fuel cells with MoS2-NiS-Ag anode. J. Electrochem. Soc. 150, A463–A469 (2003). https://doi.org/10.1149/1.1559062
Cheng, Z., Zha, S.W., Aguilar, L., et al.: A solid oxide fuel cell running on H2S/CH4 fuel mixtures. Electrochem. Solid-State Lett. 9, A31–A33 (2006). https://doi.org/10.1149/1.2137467
Hayakawa, T., Tsunoda, T., Orita, H., et al.: Partial oxidation of propene by active oxygen generated electrochemically on gold through yttria-stabilized zirconia. J. Chem. Soc. Chem. Commun 12, 961–962 (1986). https://doi.org/10.1039/c39860000961
McIntosh, S., Vohs, J.M., Gorte, R.J.: An examination of lanthanide additives on the performance of Cu-YSZ cermet anodes. Electrochim. Acta 47, 3815–3821 (2002). https://doi.org/10.1016/S0013-4686(02)00352-3
Sun, W., Zhang, S., Liu, W.: Co-generation of electric power and carbon nanotubes from dimethyl ether (DME). Fuel Cells 14, 561–565 (2014). https://doi.org/10.1002/fuce.201300268
Michaels, J.N., Vayenas, C.G.: Styrene production from ethylbenzene on platinum in a zirconia electrochemical reactor. J. Electrochem. Soc. 131, 2544–2550 (1984). https://doi.org/10.1149/1.2115356
Michaels, J.N., Vayenas, C.G.: Kinetics of vapor-phase electrochemical oxidative dehydrogenation of ethylbenzene. J. Catal. 85, 477–487 (1984). https://doi.org/10.1016/0021-9517(84)90236-7
Fu, X.Z., Luo, J.L., Sanger, A.R., et al.: Fabrication of bi-layered proton conducting membrane for hydrocarbon solid oxide fuel cell reactors. Electrochim. Acta 55, 1145–1149 (2010). https://doi.org/10.1016/j.electacta.2009.10.010
Cavani, F., Ballarini, N., Cericola, A.: Oxidative dehydrogenation of ethane and propane: how far from commercial implementation? Catal. Today 127, 113–131 (2007). https://doi.org/10.1016/j.cattod.2007.05.009
Baerns, M., Buyevskaya, O.: Simple chemical processes based on low molecular-mass alkanes as chemical feedstocks. Catal. Today 45, 13–22 (1998). https://doi.org/10.1016/S0920-5861(98)00231-4
Azizi, Y., Petit, C., Pitchon, V.: Formation of polymer-grade ethylene by selective hydrogenation of acetylene over Au/CeO2 catalyst. J. Catal. 256, 338–344 (2008). https://doi.org/10.1016/j.jcat.2008.04.003
Fabbri, E., Pergolesi, D., Traversa, E.: Materials challenges toward proton-conducting oxide fuel cells: a critical review. Chem. Soc. Rev. 39, 4355–4369 (2010). https://doi.org/10.1039/b902343g
Guo, Y.M., Lin, Y., Ran, R., et al.: Zirconium doping effect on the performance of proton-conducting BaZryCe0.8−yY0.2O3−δ (0.0 \(\leqslant\) y \(\leqslant\) 0.8) for fuel cell applications. J. Power Sources 193, 400–407 (2009). https://doi.org/10.1016/j.jpowsour.2009.03.044
Zuo, C., Zha, S., Liu, M., et al.: Ba(Zr0.1Ce0.7Y0.2)O3–δ as an electrolyte for low-temperature solid-oxide fuel cells. Adv. Mater. 18, 3318–3320 (2006). https://doi.org/10.1002/adma.200601366
Tao, S.W., Irvine, J.T.S.: A stable, easily sintered proton-conducting oxide electrolyte for moderate-temperature fuel cells and electrolyzers. Adv. Mater. 18, 1581–1584 (2006). https://doi.org/10.1002/adma.200502098
Zawodzinski, T.A., Derouin, C., Radzinski, S., et al.: Water uptake by and transport through Nafion® 117 membranes. J. Electrochem. Soc. 140, 1041–1047 (1993). https://doi.org/10.1149/1.2056194
Ren, X., Springer, T.E., Gottesfeld, S.: Water and methanol uptakes in nafion membranes and membrane effects on direct methanol cell performance. J. Electrochem. Soc. 147, 92–98 (2000). https://doi.org/10.1149/1.1393161
Osseo-Asare, K., Xue, T.: Donnan dialysis with Nafion® membranes: application of a rotating diffusion cell. J. Membr. Sci. 43, 5–17 (1989). https://doi.org/10.1016/S0376-7388(00)82349-2
Chen, S.L., Bocarsly, A.B., Benziger, J.: Nafion-layered sulfonated polysulfone fuel cell membranes. J. Power Sources 152, 27–33 (2005). https://doi.org/10.1016/j.jpowsour.2005.03.214
Skou, E., Kauranen, P., Hentschel, J.: Water and methanol uptake in proton conducting Nafion® membranes. Solid State Ion. 97, 333–337 (1997). https://doi.org/10.1016/S0167-2738(97)00033-7
Casciola, M., Alberti, G., Sganappa, M., et al.: On the decay of Nafion proton conductivity at high temperature and relative humidity. J. Power Sources 162, 141–145 (2006). https://doi.org/10.1016/j.jpowsour.2006.06.023
Sone, Y., Ekdunge, P., Simonsson, D.: Proton conductivity of Nafion 117 as measured by a four-electrode AC impedance method. J. Electrochem. Soc. 143, 1254–1259 (1996). https://doi.org/10.1149/1.1836625
Lee, S.J., Mukerjee, S., McBreen, J., et al.: Effects of nafion impregnation on performances of PEMFC electrodes. Electrochim. Acta 43, 3693–3701 (1998). https://doi.org/10.1016/S0013-4686(98)00127-3
Hofmann, D.W.M., Kuleshova, L.N., D’Aguanno, B.: Theoretical simulations of proton conductivity: basic principles for improving the proton conductor. J. Power Sources 195, 7743–7750 (2010). https://doi.org/10.1016/j.jpowsour.2009.10.019
Iwahara, H., Uchida, H., Morimoto, K., et al.: High-temperature C1-gas fuel cells using proton-conducting solid electrolytes. J. Appl. Electrochem. 19, 448–452 (1989). https://doi.org/10.1007/BF01015250
Iwahara, H., Uchida, H., Tanaka, S.: High temperature-type proton conductive solid oxide fuel cells using various fuels. J. Appl. Electrochem. 16, 663–668 (1986). https://doi.org/10.1007/BF01006916
Cui, S.H., Li, J.H., Luo, J.L., et al.: Co-generation of energy and ethylene in hydrocarbon fueled SOFCs with Cr3C2 and WC anode catalysts. Ceram. Int. 40, 11781–11786 (2014). https://doi.org/10.1016/j.ceramint.2014.04.007
Liu, S.B., Liu, Q.X., Fu, X.Z., et al.: Cogeneration of ethylene and energy in protonic fuel cell with an efficient and stable anode anchored with in situ exsolved functional metal nanoparticles. Appl. Catal. B Environ. 220, 283–289 (2018). https://doi.org/10.1016/j.apcatb.2017.08.051
Lin, J.Y., Shao, L., Si, F.Z., et al.: Co2CrO4 nanopowders as an anode catalyst for simultaneous conversion of ethane to ethylene and power in proton-conducting fuel cell reactors. J. Phys. Chem. C 122, 4165–4171 (2018). https://doi.org/10.1021/acs.jpcc.7b11680
Wan, T., Zhu, A., Guo, Y., et al.: Co-generation of electricity and syngas on proton-conducting solid oxide fuel cell with a perovskite layer as a precursor of a highly efficient reforming catalyst. J. Power Sources 348, 9–15 (2017). https://doi.org/10.1016/j.jpowsour.2017.02.074
Lee, B., Sakamoto, Y., Hirabayashi, D., et al.: Direct oxidation of methane to methanol over proton conductor/metal mixed catalysts. J. Catal. 271, 195–200 (2010). https://doi.org/10.1016/j.jcat.2010.01.011
Tomita, A., Nakajima, J., Hibino, T.: Direct oxidation of methane to methanol at low temperature and pressure in an electrochemical fuel cell. Angew. Chem. Int. Ed. 47, 1462–1464 (2008). https://doi.org/10.1002/anie.200703928
Lee, B., Hibino, T.: Efficient and selective formation of methanol from methane in a fuel cell-type reactor. J. Catal. 279, 233–240 (2011). https://doi.org/10.1016/j.jcat.2010.12.020
Otsuka, K., Hosokawa, K., Yamanaka, I., et al.: One-step oxidation of benzene to phenol applying a fuel cell system. Electrochim, Acta 34, 1485–1488 (1989). https://doi.org/10.1016/0013-4686(89)87192-0
Otsuka, K., Yamanaka, I.: One step synthesis of hydrogen peroxide through fuel cell reaction. Electrochim. Acta 35, 319–322 (1990). https://doi.org/10.1016/0013-4686(90)87004-L
Otsuka, K., Kunieda, M., Yamagata, H.: Direct synthesis of phenol from benzene during O2–H2 fuel cell reactions. J. Electrochem. Soc. 139, 2381–2386 (1992). https://doi.org/10.1149/1.2221235
Otsuka, K., Shimizu, Y., Yamanaka, I.: Selective synthesis of acetaldehyde using a fuel cell system in the gas phase. J. Chem. Soc. Chem. Commun. 18, 1272–1273 (1988). https://doi.org/10.1039/c39880001272
Otsuka, K., Shimizu, Y., Yamanaka, I., et al.: Wacker type and π-allyl type oxidations of propylene controlled by fuel cell system in the gas phase. Catal. Lett. 3, 365–369 (1989). https://doi.org/10.1007/BF00771264
Otsuka, K., Yamanaka, I., Hosokawa, K.: A fuel cell for the partial oxidation of cyclohexane and aromatics at ambient temperatures. Nature 345, 697–698 (1990). https://doi.org/10.1038/345697a0
Yamanaka, I., Otsuka, K.: The partial oxidations of cyclohexane and benzene on the FeCl3-embedded cathode during the O2–H2 fuel cell reactions. J. Electrochem. Soc. 138, 1033–1038 (1991). https://doi.org/10.1149/1.2085710
Breen, J.P., Burch, R., Coleman, H.M.: Metal-catalysed steam reforming of ethanol in the production of hydrogen for fuel cell applications. Appl. Catal. B Environ. 39, 65–74 (2002). https://doi.org/10.1016/S0926-3373(02)00075-9
Alexander, B.R., Mitchell, R.E., Gür, T.M.: Viability of coupled steam-carbon-air fuel cell concept for spontaneous co-production of hydrogen and electrical power. J. Electrochem. Soc. 159, F810–F818 (2012). https://doi.org/10.1149/2.032212jes
Gür, T.M., Homel, M., Virkar, A.V.: High performance solid oxide fuel cell operating on dry gasified coal. J. Power Sources 195, 1085–1090 (2010). https://doi.org/10.1016/j.jpowsour.2009.08.098
Lee, A.C., Mitchell, R.E., Gür, T.M.: Thermodynamic analysis of gasification-driven direct carbon fuel cells. J. Power Sources 194, 774–785 (2009). https://doi.org/10.1016/j.jpowsour.2009.05.039
Lee, A.C., Mitchell, R.E., Gür, T.M.: Feasibility of hydrogen production in a steam-carbon electrochemical cell. Solid State Ion. 192, 607–610 (2011). https://doi.org/10.1016/j.ssi.2010.05.034
Sigal, C.T., Vayenas, C.G.: Ammonia oxidation to nitric oxide in a solid electrolyte fuel cell. Solid State Ion. 5, 567–570 (1981). https://doi.org/10.1016/0167-2738(81)90318-0
Sabbe, M.K., Reyniers, M.F., Reuter, K.: First-principles kinetic modeling in heterogeneous catalysis: an industrial perspective on best-practice, gaps and needs. Catal. Sci. Technol. 2, 2010–2024 (2012). https://doi.org/10.1039/c2cy20261a
Dietz, A.G., III., Schmidt, L.D.: Conditions for HCN synthesis and catalyst activation over Pt-Rh gauzes. Appl. Catal. A Gen. 180, 287–298 (1999). https://doi.org/10.1016/S0926-860X(98)00350-0
Trusov, N.V., Grin’, G.I., Prezhdo, V.V.: Industrial monitoring as a source of valuable information on the unit process. Theor. Found. Chem. Eng. 36, 505–510 (2002). https://doi.org/10.1023/A:1020690232025
Grasselli, R.K.: Advances and future trends in selective oxidation and ammoxidation catalysis. Catal. Today 49, 141–153 (1999). https://doi.org/10.1016/S0920-5861(98)00418-0
Li, W., Bonakdarpour, A., Gyenge, E., et al.: Design of bifunctional electrodes for co-generation of electrical power and hydrogen peroxide. J. Appl. Electrochem. 48, 985–993 (2018). https://doi.org/10.1007/s10800-018-1232-0
Chadderdon, D.J., Xin, L., Qi, J., et al.: Selective oxidation of 1,2-propanediol in alkaline anion-exchange membrane electrocatalytic flow reactors: experimental and DFT investigations. ACS Catal. 5, 6926–6936 (2015). https://doi.org/10.1021/acscatal.5b01085
Xin, L., Zhang, Z.Y., Wang, Z.C., et al.: Simultaneous generation of mesoxalic acid and electricity from glycerol on a gold anode catalyst in anion-exchange membrane fuel cells. ChemCatChem 4, 1105–1114 (2012). https://doi.org/10.1002/cctc.201200017
Zhang, Z.Y., Xin, L., Li, W.Z.: Electrocatalytic oxidation of glycerol on Pt/C in anion-exchange membrane fuel cell: cogeneration of electricity and valuable chemicals. Appl. Catal. B Environ. 119(120), 40–48 (2012). https://doi.org/10.1016/j.apcatb.2012.02.009
Prasanna, D., Selvaraj, V.: Development of non-covalent ternary polymer-CNT composites as a novel supporting material for electrooxidation of glycerol. RSC Adv. 5, 98822–98833 (2015). https://doi.org/10.1039/c5ra17172e
Marchionni, A., Bevilacqua, M., Bianchini, C., et al.: Electrooxidation of ethylene glycol and glycerol on Pd-(Ni-Zn)/C anodes in direct alcohol fuel cells. Chemsuschem 6, 518–528 (2013). https://doi.org/10.1002/cssc.201200866
Santos, M.C.L., Godoi, C.M., Kang, H.S., et al.: Effect of Ni content in PdNi/C anode catalysts on power and methanol co-generation in alkaline direct methane fuel cell type. J. Colloid Interface Sci. 578, 390–401 (2020). https://doi.org/10.1016/j.jcis.2020.06.017
Singh, A.K., Singh, S., Kumar, A.: Hydrogen energy future with formic acid: a renewable chemical hydrogen storage system. Catal. Sci. Technol. 6, 12–40 (2016). https://doi.org/10.1039/c5cy01276g
Benipal, N., Qi, J., Liu, Q., et al.: Carbon nanotube supported PdAg nanoparticles for electrocatalytic oxidation of glycerol in anion exchange membrane fuel cells. Appl. Catal. B Environ. 210, 121–130 (2017). https://doi.org/10.1016/j.apcatb.2017.02.082
Qi, J., Benipal, N., Liang, C.H., et al.: PdAg/CNT catalyzed alcohol oxidation reaction for high-performance anion exchange membrane direct alcohol fuel cell (alcohol = methanol, ethanol, ethylene glycol and glycerol). Appl. Catal. B Environ. 199, 494–503 (2016). https://doi.org/10.1016/j.apcatb.2016.06.055
Fashedemi, O.O., Miller, H.A., Marchionni, A., et al.: Electro-oxidation of ethylene glycol and glycerol at palladium-decorated FeCo@Fe core–shell nanocatalysts for alkaline direct alcohol fuel cells: functionalized MWCNT supports and impact on product selectivity. J. Mater. Chem. A 3, 7145–7156 (2015). https://doi.org/10.1039/c5ta00076a
Zhang, Z.Y., Xin, L., Qi, J., et al.: Selective electro-oxidation of glycerol to tartronate or mesoxalate on Au nanoparticle catalyst via electrode potential tuning in anion-exchange membrane electro-catalytic flow reactor. Appl. Catal. B Environ. 147, 871–878 (2014). https://doi.org/10.1016/j.apcatb.2013.10.018
Thia, L., Xie, M.S., Liu, Z.L., et al.: Copper-modified gold nanoparticles as highly selective catalysts for glycerol electro-oxidation in alkaline solution. ChemCatChem 8, 3272–3278 (2016). https://doi.org/10.1002/cctc.201600725
Zhang, Z.Y., Xin, L., Qi, J., et al.: Selective electro-conversion of glycerol to glycolate on carbon nanotube supported gold catalyst. Green Chem. 14, 2150–2152 (2012). https://doi.org/10.1039/c2gc35505a
Qi, J., Xin, L., Chadderdon, D.J., et al.: Electrocatalytic selective oxidation of glycerol to tartronate on Au/C anode catalysts in anion exchange membrane fuel cells with electricity cogeneration. Appl. Catal. B Environ. 154(155), 360–368 (2014). https://doi.org/10.1016/j.apcatb.2014.02.040
Wang, H.B., Thia, L., Li, N., et al.: Selective electro-oxidation of glycerol over Au supported on extended poly(4-vinylpyridine) functionalized graphene. Appl. Catal. B Environ. 166(167), 25–31 (2015). https://doi.org/10.1016/j.apcatb.2014.11.009
Shi, N., Xue, S., Xie, Y., et al.: Co-generation of electricity and olefin via proton conducting fuel cells using (Pr0.3Sr0.7)0.9Ni0.1Ti0.9O3 catalyst layers. Appl. Catal. B Environ. 272, 118973 (2020). https://doi.org/10.1016/j.apcatb.2020.118973
Garcia, L.M.S., Rajak, S., Chair, K., et al.: Conversion of methane into methanol using the [6,6′-(2,2′-bipyridine-6,6′-diyl)bis(1,3,5-triazine-2,4-diamine)](nitrate-O)copper (II) complex in a solid electrolyte reactor fuel cell type. ACS Omega 5, 16003–16009 (2020). https://doi.org/10.1021/acsomega.0c01363
Oliveira, V.L., Morais, C., Servat, K., et al.: Studies of the reaction products resulted from glycerol electrooxidation on Ni-based materials in alkaline medium. Electrochim. Acta 117, 255–262 (2014). https://doi.org/10.1016/j.electacta.2013.11.127
Xin, L., Zhang, Z.Y., Qi, J., et al.: Electricity storage in biofuels: selective electrocatalytic reduction of levulinic acid to valeric acid or γ-valerolactone. Chemsuschem 6, 674–686 (2013). https://doi.org/10.1002/cssc.201200765
Hua, B., Li, M., Pu, J., et al.: BaZr0.1Ce0.7Y0.1Yb0.1O3–δ enhanced coking-free on-cell reforming for direct-methane solid oxide fuel cells. J. Mater. Chem. A 2, 12576–12582 (2014). https://doi.org/10.1039/c4ta01989j
Hua, B., Li, M., Luo, J.L., et al.: Carbon-resistant Ni-Zr0.92Y0.08O2−δ supported solid oxide fuel cells using Ni-Cu-Fe alloy cermet as on-cell reforming catalyst and mixed methane-steam as fuel. J. Power Sources 303, 340–346 (2016). https://doi.org/10.1016/j.jpowsour.2015.11.029
Guerra, C., Lanzini, A., Leone, P., et al.: Optimization of dry reforming of methane over Ni/YSZ anodes for solid oxide fuel cells. J. Power Sources 245, 154–163 (2014). https://doi.org/10.1016/j.jpowsour.2013.06.088
Dimitriou, I., García-Gutiérrez, P., Elder, R.H., et al.: Carbon dioxide utilisation for production of transport fuels: process and economic analysis. Energy Environ. Sci. 8, 1775–1789 (2015). https://doi.org/10.1039/c4ee04117h
Centi, G., Quadrelli, E.A., Perathoner, S.: Catalysis for CO2 conversion: a key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 6, 1711–1731 (2013). https://doi.org/10.1039/c3ee00056g
Djinović, P., Črnivec, I.G.O., Pintar, A.: Biogas to syngas conversion without carbonaceous deposits via the dry reforming reaction using transition metal catalysts. Catal. Today 253, 155–162 (2015). https://doi.org/10.1016/j.cattod.2015.01.039
Wang, F., Wang, W., Ran, R., et al.: Aluminum oxide as a dual-functional modifier of Ni-based anodes of solid oxide fuel cells for operation on simulated biogas. J. Power Sources 268, 787–793 (2014). https://doi.org/10.1016/j.jpowsour.2014.06.087
Ma, J.J., Jiang, C.R., Connor, P.A., et al.: Highly efficient, coking-resistant SOFCs for energy conversion using biogas fuels. J. Mater. Chem. A 3, 19068–19076 (2015). https://doi.org/10.1039/c5ta06421j
Sengodan, S., Choi, S., Jun, A., et al.: Layered oxygen-deficient double perovskite as an efficient and stable anode for direct hydrocarbon solid oxide fuel cells. Nat. Mater. 14, 205–209 (2015). https://doi.org/10.1038/nmat4166
Garcia, A., Yan, N., Vincent, A., et al.: Highly cost-effective and sulfur/coking resistant VOx-grafted TiO2 nanoparticles as an efficient anode catalyst for direct conversion of dry sour methane in solid oxide fuel cells. J. Mater. Chem. A 3, 23973–23980 (2015). https://doi.org/10.1039/c5ta06453h
Ren, T., Patel, M.K., Blok, K.: Steam cracking and methane to olefins: energy use, CO2 emissions and production costs. Energy 33, 817–833 (2008). https://doi.org/10.1016/j.energy.2008.01.002
Lemonidou, A.A., Vasalos, I.A.: Preparation and evaluation of catalysts for the production of ethylene via steam cracking: effect of operating conditions on the performance of 12CaO-7Al2O3 Catalyst. Appl. Catal. 54, 119–138 (1989). https://doi.org/10.1016/S0166-9834(00)82360-X
Ren, T., Patel, M., Blok, K.: Olefins from conventional and heavy feedstocks: energy use in steam cracking and alternative processes. Energy 31, 425–451 (2006). https://doi.org/10.1016/j.energy.2005.04.001
Wang, S.Y., Luo, J.L., Sanger, A.R., et al.: Performance of ethane/oxygen fuel cells using yttrium-doped barium cerate as electrolyte at intermediate temperatures. J. Phys. Chem. C 111, 5069–5074 (2007). https://doi.org/10.1021/jp066690w
Fu, X.Z., Luo, J.L., Sanger, A.R., et al.: Y-doped BaCeO3−δ nanopowders as proton-conducting electrolyte materials for ethane fuel cells to co-generate ethylene and electricity. J. Power Sources 195, 2659–2663 (2010). https://doi.org/10.1016/j.jpowsour.2009.10.069
Shi, Z.C., Luo, J.L., Wang, S.Y., et al.: Protonic membrane for fuel cell for co-generation of power and ethylene. J. Power Sources 176, 122–127 (2008). https://doi.org/10.1016/j.jpowsour.2007.10.056
Ohe, T., Miyaura, N., Suzuki, A.: Palladium-catalyzed cross-coupling reaction of organoboron compounds with organic triflates. J. Org. Chem. 58, 2201–2208 (1993). https://doi.org/10.1021/jo00060a041
Arnarson, L., Schmidt, P.S., Pandey, M., et al.: Fundamental limitation of electrocatalytic methane conversion to methanol. Phys. Chem. Chem. Phys. 20, 11152–11159 (2018). https://doi.org/10.1039/c8cp01476k
Promoppatum, P., Viswanathan, V.: Identifying material and device targets for a flare gas recovery system utilizing electrochemical conversion of methane to methanol. ACS Sustain. Chem. Eng. 4, 1736–1745 (2016). https://doi.org/10.1021/acssuschemeng.5b01714
Li, J., Hou, J., Xi, X.A., et al.: Cogeneration of ethylene and electricity in symmetrical protonic solid oxide fuel cells based on a La0.6Sr0.4Fe0.8Nb0.1Cu0.1O3–δ electrode. J. Mater. Chem. A 8, 25978–25985 (2020). https://doi.org/10.1039/d0ta08974e
Langer, S.H., Sakellaropoulos, G.P.: Electrogenerative and voltameiotic processes. Ind. Eng. Chem. Proc. Des. Dev. 18, 567–579 (1979). https://doi.org/10.1021/i260072a001
Ross, J.: Ullman’s encyclopedia of industrial chemistry. Appl. Catal. 27, 403–404 (1986). https://doi.org/10.1016/S0166-9834(00)82943-7
Polizzi, S., Benedetti, A., Fagherazzi, G., et al.: Hydroxylamine production via hydrogenation of nitric oxide in aqueous sulfuric acid catalyzed by carbon-supported platinum. J. Catal. 106, 494–499 (1987). https://doi.org/10.1016/0021-9517(87)90262-4
Colucci, J.A., Foral, M.J., Langer, S.H.: The electro reduction of nitric oxide on bulk platinum in acid solutions. Electrochim. Acta 30, 521–528 (1985). https://doi.org/10.1016/0013-4686(85)80042-6
Campos-Martin, J.M., Blanco-Brieva, G., Fierro, J.L.G.: Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962–6984 (2006). https://doi.org/10.1002/anie.200503779
Forti, J.C., Rocha, R.S., Lanza, M.R.V., et al.: Electrochemical synthesis of hydrogen peroxide on oxygen-fed graphite/PTFE electrodes modified by 2-ethylanthraquinone. J. Electroanal. Chem. 601, 63–67 (2007). https://doi.org/10.1016/j.jelechem.2006.10.023
Agladze, G.R., Tsurtsumia, G.S., Jung, B.I., et al.: Comparative study of hydrogen peroxide electro-generation on gas-diffusion electrodes in undivided and membrane cells. J. Appl. Electrochem. 37, 375–383 (2007). https://doi.org/10.1007/s10800-006-9269-x
Agladze, G.R., Tsurtsumia, G.S., Jung, B.I., et al.: Comparative study of chemical and electrochemical Fenton treatment of organic pollutants in wastewater. J. Appl. Electrochem. 37, 985–990 (2007). https://doi.org/10.1007/s10800-007-9325-1
Luyben, W.L.: Design and control of the cumene process. Ind. Eng. Chem. Res. 49, 719–734 (2010). https://doi.org/10.1021/ie9011535
Acknowledgements
The National Natural Science Foundation of China (No. 21975163), the Natural Science Foundation of Guangdong Province of China (2020A1515011165), and the Shenzhen Science and Technology Program (Nos. KQTD20190929173914967, JCYJ20200109110416441, JCYJ20220531102408019).
Author information
Authors and Affiliations
Contributions
Jing-Li Luo: validation, resources, supervision. Xian-Zhu Fu: validation, resources, writing - review & editing, funding acquisition, project administration, supervision.
Corresponding authors
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Si, F., Liu, S., Liang, Y. et al. Fuel Cell Reactors for the Clean Cogeneration of Electrical Energy and Value-Added Chemicals. Electrochem. Energy Rev. 5 (Suppl 2), 25 (2022). https://doi.org/10.1007/s41918-022-00168-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41918-022-00168-0