
Abstract
Purpose
Blue light has been reported to enhance the differentiation and mineralisation potential of mesenchymal stem cells. No studies have investigated the photobiomodulation effects of blue light on the odontogenic differentiation of dental pulp stem cell-containing populations. This study aimed to determine the effect of 405-nm wavelength on the mineralisation response in these cells.
Methods
Human dental pulp cells were irradiated using a calibrated 405 nm light source at an irradiance of 57.7 mW/cm2 for 35, 69, 90 and 104 s; delivering energy densities of 2, 4, 5 and 6 J/cm2. Cell proliferation was investigated on days 2 and 4. Alkaline phosphatase activity and dentine matrix protein-1 levels were investigated on days 3 and 5. Mineralised nodule formation was investigated on days 7, 4 and 21.
Results
The 405-nm blue light had significant inhibitory effects on dental pulp cell proliferation. Simultaneously, blue light irradiation enhanced the mineralisation response, as determined by the upregulation of odontogenic markers. Irradiated cultures also demonstrated a significant increase in mineralised nodule formation compared with the non-irradiated controls at all time points.
Conclusion
Data indicate that phototherapy utilising a 405-nm wavelength may have therapeutic potential in stimulating odontogenic responses of pulp cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dental caries is an extremely common oral health disease which affects the global population and remains a significant healthcare and economic burden. Untreated caries in the permanent dentition affects over 2 billion people worldwide. Disease progression can cause significant pain and can ultimately lead to tooth loss [1, 2]. The dental pulp comprises a heterogeneous cell population which also contains self-renewing mesenchymal stem cells (MSCs) which have the potential of differentiation into odontoblast-like cells for tooth repair [3,4,5,6,7]. A key aspect for conservative disease management is the induction of the regenerative odontogenic processes in the dental pulp cell (DPC) population. Consequently, the tertiary or repair dentine deposited acts as a barrier against carious disease progression and can retain pulp vitality. Current vital pulp therapy protocols involve removal of infected tissue, followed by application of materials that aid the initiation of the reparative responses of the pulp (i.e. calcium hydroxide and mineral trioxide aggregate). However, there are challenges regarding the delivery and dosage control when using materials. Therefore, enhancement of the endogenous reparative responses using light potentially represents a useful adjunct to current dental procedures [8,9,10].
Photobiomodulation (PBM) has shown a range of effects in eukaryotic cells including increased cellular proliferation, differentiation and mineralisation responses as well as modulation of growth factor/cytokine activity [11]. Recently, the application of blue light (405–480 nm) delivered using lasers or light-emitting diodes (LEDs) has been shown to exert tissue repair-related effects [12,13,14,15,16]. The mode of action of blue light is understood to be mediated via relatively small increases in reactive oxygen species (ROS) levels. The mechanism underpinning this transduction is proposed to be elicited via light absorption by cellular flavins, such as nicotinamide adenine dinucleotide phosphate (NADPH) dehydrogenase, NAPDH oxidase and cryptochromes (CRY1 and CRY2) [17]. Notably, the cellular redox state has been reported to have a major influence on cell proliferation and mineralisation [18, 19].
The stimulatory effects of blue light on the mineralisation responses of MSCs have only recently been investigated [12,13,14,15,16]. Studies have, however, generally shown a lack of rigor in reporting irradiation parameters with inconsistent experimental set-ups providing issues with the results reported [14,15,16]. Another important confounding factor is that of temperature, and measurements following irradiation are not routinely reported even though hyperthermia on its own has been shown to enhance mineralisation in MSCs [17]. Despite these limitations, there appears to be a consensus that blue light irradiation is able to enhance osteogenic/odontogenic potential in MSCs at a range of energy densities from 1 to 6 J/cm2. The majority of previous studies have utilised a fixed irradiance of 100 mW/cm2 while varying the irradiation exposure time period [15, 16]. Notably, the literature has shown the biphasic effects of light irradiation and consequently the necessity to identify optimal irradiation parameters for a particular cell type. In addition, studies using blue light have reported that utilising multiple irradiations of cultures over a given time period also has the potential to enhance the cellular response [14,15,16,17]. Consequently, based on the previous literature, we hypothesised that blue light delivery could be used to stimulate the mineralisation processes in DPC cultures and that a multi-irradiation regimen would enhance this effect. We therefore designed blue light irradiation studies which aimed to test this hypothesis.
Materials and methods
Light source characterisation
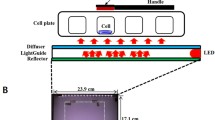
A 405-nm solid-state LED light source (AURA light engine®, Lumencor®, USA) was used in the current study. It was characterised and calibrated at the current experimental set-up according to a previously published standard protocol [20]. The light source was calibrated to accurately determine the irradiance—and hence energy density—delivered to the cell monolayer. In brief, the light source (7 mm) was placed in contact with the flat transparent underside of the well in a black 96-well plate (4titude®; UK), and the sensor/detector (5 mm) was in contact with the base of the well from the inside (positioned where the cell monolayer will be grown). Not only the diameter of the light source and individual well (6 mm) were comparable, but also the light source showed a homogenous flat-top beam profile [21]. This allowed even irradiation of the base of the well and therefore the cell culture monolayer. The light energy density was calculated using the formula ‘energy density (J/cm2) = irradiance (W/cm2) × time (s)’.
Dental pulp cell culture
Human DPCs (isolated from three individual donors) (Ethical approval BCHC Ref.: BCHCDent334.1533.TB) were retrieved from frozen stocks (− 80 °C). Cells were previously harvested from caries-free wisdom teeth according to a previously published protocol [22]. Cells were maintained and expanded at 37 °C in 5% CO2 (Heracell™ 150i, Thermo Fisher Scientific, USA) in minimum essential medium (MEM) with no phenol (Gibco, Thermo Fisher Scientific; USA) supplemented with 10% fetal bovine serum (FBS), 1% penicillin–streptomycin and 1% l-Glutamine (Sigma-Aldrich®, USA). For the preparation of mineralisation-inducing media, the MEM (10% FBS) was supplemented with 0.2 mM ascorbic acid, 10 mM beta-glycerophosphate and 100 nM dexamethasone (Sigma-Aldrich®, USA) [14]. All experiments were undertaken using cells at passages between 4 and 8 in mineralisation media.
Blue light irradiation of cell cultures
Cell monolayers at 70–100% confluence in 75 cm2 flasks were washed with sterile phosphate-buffered saline (PBS) (no calcium or magnesium) (Gibco, Thermo Fisher Scientific; USA) then detached using 0.05% trypsin (Sigma-Aldrich®, USA). Cells were seeded in black-walled 96-well plates at a density of 1 × 103 cells per well and incubated at 37 °C in 5% CO2 for 48 h to allow initial cell attachment to the cultureware. Media was removed and replaced by mineralisation-inducing media (200 µl) (described above) and blue light irradiation was delivered with the light source placed in direct contact with the base of the well. For all assays, the light source was operated in a continuous wave mode at an output power setting of 20 mW. Light was administrated using an irradiance of 57.7 mW/cm2 for 35, 69, 90 or 104 s, respectively, delivering energy densities of 2, 4, 5 and 6 J/cm2. Control wells received no light exposure. For assays of cell proliferation (BrdU), alkaline phosphatase (ALP) and dentine matrix protein-1 (DMP1) (all described below), cells either received one (day 1) or two irradiations (days 1 and 3). Proliferation was investigated 24 h post initial irradiation (day 2 for cells receiving one exposure), as well as 72 h post initial irradiation (day 4 for cells receiving two exposures). ALP and DMP1 levels were investigated at day 3 (one exposure) and day 5 (two exposures). For the mineralised nodule assay, cells were exposed to 405-nm blue light on alternate days for 5 days (i.e. 3 irradiations at days 1, 3 and 5). Mineralised nodule formation was determined at 7, 14 and 21 days. The effect of extended light irradiation (every other day beyond the initial 3 irradiations) on mineralised nodule formation was also investigated for the full duration of the 14 (6 irradiations) and 21 day (9 irradiations) time points. Media was changed every 2–3 days. Experiments were repeated three times and performed in duplicate.
Temperature measurements in culture media
A window was drilled in the lid of a 96-well plate to permit the entry of a K-type thermocouple (RS Components Ltd.©, UK) into the wells while irradiating 200 µl mineralisation-inducing MEM. The thermocouple was attached to a TC-08 data logger (Pico Technology; UK). The culture plates were removed from the incubator (37 °C) and the temperature readings immediately started and recorded for both control and irradiated samples. Measurements were undertaken at room temperature and were in triplicate. The temperature of the MEM cell culture media began decreasing once the 96-well plates were removed from the incubator; the average temperature decrease over 104 s (maximum irradiation time) was 7.22 °C. There was no significant difference in the temperature decrease between the non-irradiated control and the irradiated samples at any time point (see Fig. 1).

Mean temperature change (ΔT) (n = 3) measured in both the irradiated (57.7 mW/cm2) and non-irradiated controls at different time points; 35, 69, 90 and 104 s
Cell assays
BrdU cell proliferation assay
BrdU Cell Proliferation ELISA Kit (Abcam®, UK) was used according to the manufacturer’s instructions. Media containing the BrdU reagent was added immediately following light irradiation, either on day 1 or day 3, and the plates were incubated for 24 h at 37 °C. The cells were fixed, and incubated with the anti-BrdU monoclonal detector antibody for 1 h at room temperature. Peroxidase anti-mouse IgG conjugate was added to the cells and incubated for 30 min. Subsequently, the tetramethylbenzidine peroxidase substrate was incubated for 30 min in the dark. Finally, the stop solution was added and the optical density was read using a microplate reader (ELx800™—BioTek™, USA) at an absorbance of 450 nm.
Alkaline phosphatase assay
The ALP assay Kit (Abcam®, UK) was used according to the manufacturer’s instructions. The protocol uses p-nitrophenylphosphate (pNPP) as an ALP substrate. Briefly, cell lysates were transferred to fresh 96-well plates; then, pNPP was added to each sample and incubated for 1 h in the dark at room temperature. The reaction was stopped by adding 3 N sodium hydroxide, and the optical density of the samples was measured at an absorbance of 405 nm. ALP activity was calculated in U/ml using a standard curve (8.3 × 10–4–0.004 U/ml), and values were expressed relative to the control (non-irradiated) wells in each plate.
Dentine matrix protein-1 assay
Human DMP-1 DuoSet ELISA and DuoSet ELISA Ancillary Reagent Kit 2 (R & D systems©, USA) were used. All assay incubations were undertaken in sealed plates at room temperature. Twenty-four hours before the intended detection time, 96-well plates were coated with the capture antibody and incubated. The wells were blocked by adding 1% bovine serum albumin and incubated for 1 h. Subsequently, the sample supernatants were added and incubated for 2 h. The detection antibody was incubated for a further 2 h, followed by streptavidin horseradish peroxidase incubation for 20 min in the dark. Finally, the substrate solution was added to each well and incubated for 20 min in the dark, and 2 N sulphuric acid was added to stop the reaction. Absorbance (450 nm) of the samples was measured using the microplate reader. DMP1 levels (pg/ml) were retrieved from the calibration curve (156–10,000 pg/ml DMP1); then, values were expressed relative to the control (non-irradiated) wells in each plate.
Alizarin red assay
Cultures were washed twice with PBS, fixed with neutral buffered formalin (Sigma-Aldrich®, USA), re-washed and incubated with alizarin red stain solution (Sigma-Aldrich®, USA) with gentle agitation (DENLEY orbital mixer) for 30 min. Afterwards, cells were washed 3–6 times until all excess stain was removed. Macroscopic images of the stained wells were obtained using a SX620 HS camera (Canon, Japan). Microscopic images were captured at a magnification of 10X using an inverted phase contrast light microscope (Eclipse TE300, Nikon, Japan) along with a digital camera (D5100, Nikon, Japan). To extract the stain for quantitative data, 10% acetic acid (Sigma-Aldrich®, USA) was added to the samples and incubated at room temperature with gentle agitation. The cells along with the acetic acid were transferred to Eppendorf tubes, heated in a water bath (85 °C) for 10 min, transferred to ice for 5 min, then centrifuged at 14,000 RPM for 15 min (Jouan, UK). Subsequently, the supernatants were transferred to fresh Eppendorf tubes, and the pH was neutralized by adding 10% ammonium hydroxide (Sigma-Aldrich®, USA). All samples were transferred to a fresh 96-well plate, and the absorbance was read at 405 nm. A calibration curve using a range of concentrations of alizarin red (0.47–30 µM) was generated to enable quantification of the amount of stain present within each sample (in µM).
Statistical analysis
The Kruskal–Wallis test was used to determine significant differences between groups. The Bonferroni test was applied for pairwise comparisons. The significance value was set at P ≤ 0.05. Data were analyzed using SPSS 17 (IBM®, USA).
Blue light arrests proliferation and induces mineralisation in dental pulp cell cultures
Since odontogenic differentiation is associated with an arrest in proliferation [23], the BrdU assay was undertaken to monitor the effect of 405-nm blue light on DPC cultures. Results showed that blue light irradiation inhibited DPC proliferation rates. At day 2, the energy densities of 4, 5 and 6 J/cm2 significantly decreased cell proliferation rates compared with the non-irradiated control (P = 0.039, P = 0.003 and P = 0.001 respectively). At day 4, the blue light effects were replicated, and the 4, 5 and 6 J/cm2 exposure groups were significantly lower than the control group (P < 0.001). Notably, for the 2 J/cm2 exposure group, proliferation rates continued to increase from day 2 to day 4, as opposed to the 4, 5 and 6 J/cm2 groups whose proliferation rates were similar at both of these time points (see Fig. 2).

Data showing the effect of 405 nm light on proliferation rates of DPCs. The BrdU assay was performed after 24 h (day 2/ cells receiving 1 exposure) and 72 h (day 4/cells receiving 2 exposures), and absorbance for samples was read at 450 nm. Asterisks represent statistically significant differences compared to the non-irradiated controls at each time point (*P < 0.05) (**P < 0.001) (n = 3) (mean + / − SD). Experiments were performed in duplicate. Significance level set at P ≤ 0.05
To evaluate the effects of 405-nm blue light on the mineralised nodule formation of DPCs, cultures were exposed on alternate days for 5 days (i.e. 3 irradiations); alizarin red staining was performed at days 7, 14 and 21. Alizarin red staining showed that 3 irradiations on alternate days were sufficient to stimulate mineralised nodule formation at all energy densities investigated. At day 7, the energy densities of 2, 4 and 6 J/cm2 produced similar responses compared with the 5 J/cm2 exposure which lead to the lowest level of mineralisation induction following irradiation. At day 14, the 2 J/cm2 group resulted in the highest mineralised nodule formation, followed by 6, 4 and 5 J/cm2. Alizarin red staining of cultures exposed to 2 J/cm2 was statistically significantly higher than the non-irradiated control at days 7 and 14 (P = 0.039 and P = 0.01 respectively). At day 21, 2 and 4 J/cm2 resulted in the highest mineralised nodules formation, which were significantly different compared with the control (P < 0.001 and P = 0.002 respectively). Extended blue light irradiation (6 irradiations) showed relatively low levels of stimulation of the mineralised nodules deposited at day 14 for all energy densities studied. This stimulation was considerably lower compared with cells receiving only 3 irradiations. Conversely, extended irradiation resulted in inhibitory effects at all energy densities at day 21 (9 irradiations), and the energy density of 6 J/cm2 resulted in statistically significant lower mineralised nodule formation compared with the control group (P = 0.027) (see Fig. 3).

a Data showing the effect of 3 irradiations (on alternate days for 5 days) of blue light in stimulating mineralised nodule formation in DPCs assayed at days 7, 14 and 21. b Bar chart showing the effect of 405 nm light irradiation on mineralised nodule formation in DPCs cultures at days 14 and 21 when extended light irradiation was undertaken (every other day beyond the initial 3 irradiations). Cultures were exposed to blue light every other day for 14 days (6 irradiations) or 21 days (9 irradiations). Cultures were stained with alizarin red stain, after which the stain extraction and quantification (μM) was performed (n = 3) (mean + / − SD). Studies were performed in duplicate. Asterisks represent statistically significant differences compared with the respective non-irradiated control (*P < 0.05) (**P < 0.001). Significance level set at P ≤ 0.05. c Representative macroscopic images of wells (black walled 96-well plate) stained with alizarin red at day 21 after cells received 3 irradiations. d Representative light microscope images of DPCs mineralised nodules stained with alizarin red at day 21 of odontogenic differentiation after cells received only 3 irradiations. Control wells were non-irradiated, while wells receiving 405 nm light irradiation were exposed to 2, 4, 5 and 6 J/cm2. All irradiated samples showed dense mineralised nodule foci compared to the control sample. Scale bar is 400 μm
ALP and DMP1 levels were studied to ascertain the 405-nm blue light enhancement of DPC odontogenic differentiation. For ALP activity, at day 3, all energy densities investigated resulted in an increase in levels compared with the non-irradiated control, except for 2 J/cm2. The stimulatory effects of 5 and 6 J/cm2 were both significantly higher than that of 2 J/cm2 (P = 0.029 and P = 0.004 respectively). At day 5, samples irradiated with blue light maintained higher ALP levels relative to the non-irradiated control, although the stimulation level was lower than that observed at day 3. ALP levels in samples irradiated with 4 J/cm2 were comparable to the control group, whereas 6 J/cm2 resulted in the greatest simulation (20% higher than the control) (see Fig. 4(a)). Similarly, blue light irradiation enhanced DMP1 levels at day 3, and the energy densities of 2 and 6 J/cm2 lead to the highest stimulation compared with 4 and 5 J/cm2. The effects of 2 J/cm2 were highly significant compared with the 4 J/cm2 (P = 0.024) exposure, while there was no significant differences between DMP1 levels in the 4, 5 and 6 J/cm2 exposure groups. At day 5, the stimulation level was lower than that detected for day 3 at all energy densities applied, except for the 6 J/cm2 group where DMP1 levels continued to increase and were statistically significantly higher compared with the group irradiated with 4 J/cm2 (P = 0.002) (see Fig. 4(b)).

Data showing the effect of 405 nm light on a alkaline phosphatase (ALP) activity and b dentine matrix protein-1 (DMP1) levels in DPCs. ALP and DMP1 levels were investigated at day 3 (cultures receiving 1 exposure) and day 5 (cultures receiving 2 exposure). ALP activity was calculated in (unit/ml), and DMP1 activity was calculated in (pg/ml); values are expressed relative to the control (non-irradiated) wells in each plate (n = 3) (mean + / − SD). Analyses were performed in duplicate. Asterisks show statistically significant differences in ALP or DMP1 levels (*P < 0.05). Significance level set at P ≤ 0.05
Discussion
Results from the current investigation demonstrated that blue light (using an irradiance of 57.7 mW/cm2) inhibited DPC proliferation; however, these conditions were found to enhance odontogenic differentiation processes. An upregulation in ROS levels and oxidative status is reportedly essential for cellular differentiation. Nonetheless, PBM effects show biphasic responses, which defies reciprocity, meaning that if the light energy density is kept constant while changing other settings (irradiance and irradiation time), outcomes will not be of similar magnitudes [17]. The biphasic effects occur due to the different levels and types of ROS produced in each experimental condition. Indeed, this is evident when comparing results of the current study with those from previous investigations [24].
In the current study, delivering an energy density of 2 J/cm2 reduced the proliferation rates of DPCs, while higher energy densities (4, 5 and 6 J/cm2) resulted in a total arrest. Studies utilising an irradiance of 100 mW/cm2 [15, 16] have also demonstrated an inhibition in cell proliferation responses; however, compared with the current investigation, there are differences in the magnitude. For example, a 2-J/cm2 irradiation resulted in the lowest proliferation rates compared with exposures of 1, 4 and 6 J/cm2. To evaluate the effects of 405-nm blue light irradiation on the differentiation potential of DPCs, the levels of ALP and DMP1 were subsequently investigated. These markers are regarded as being important in odontogenic differentiation of DPCs [25]. Results showed that 5 and 6 J/cm2 irradiation resulted in higher ALP levels compared with the 2 and 4 J/cm2 energy densities applied. Previous studies have demonstrated similar outcomes, yet different magnitudes of stimulation were reported. Indeed, Zhu et al. [15] irradiated gingival MSCs with an irradiance of 100 mW/cm2 and it was reported that 2 and 4 J/cm2 exposure resulted in the induction of the highest ALP levels compared with the 1 and 6 J/cm2 irradiances. When using the same irradiance, Yang et al. [16] reported that 3 and 4 J/cm2 yielded the highest ALP levels in relation to 1 and 2 J/cm2 exposures when they irradiated stem cells from the apical papilla. These data are in agreement with results from the current study, i.e. the higher the light energy density, the greater the ALP level stimulated. Notably, the previous study also indicated that a blue light energy density of 4 J/cm2 significantly stimulated DMP1 levels. The current study showed that irradiating DPCs at 2 and 6 J/cm2 resulted in the highest DMP1 increase, and levels continued to increase from day 3 to day 5 in the 6 J/cm2 exposure group. However, further comparison between the studies is not possible as the only energy density previously investigated was 4 J/cm2.
Alizarin red staining showed that irradiating DPCs with 405 nm light significantly enhanced the mineralised nodule formation at days 7, 14 (2 J/cm2) and 21 (2 and 4 J/cm2). Interestingly, when MSCs were irradiated (at energy densities of 1, 2, 4 and 6 J/cm2) using an irradiance of 100 mW/cm2, a dose–response trend was observed at day 28. Moreover, when cells were irradiated at 1, 2, 3 and 4 J/cm2—using the same irradiance—the 3 J/cm2 exposure group resulted in the highest mineralised nodule formation followed by the 2 and 4 J/cm2 exposure groups. These previous studies have reported that irradiation was performed every other day, leading to a cumulative energy density of 10–80 J/cm2 at day 28 [15, 16]. Additionally, only 5 irradiations every two days (3 J/cm2) at 16 mW/cm2 (cumulative energy density of 15 J/cm2) resulted in similar outcomes at day 21 [14]. In the current study, the cumulative blue light energy density from 3 irradiations on alternate days amount to between 6 and 18 J/cm2. The extended irradiation on alternate days (6 irradiations) resulted in a non-significant increase in mineralised nodule formation at day 14—delivering a cumulative energy density of 12–36 J/cm2. Contrarily, 9 irradiations (cumulative energy density 18–54 J/cm2) resulted in an inhibitory effect on mineralised nodule formation. When Yuan et al. [26] irradiated bone marrow MSCs daily with an energy density of 12 J/cm2 (cumulative energy density 70–80 J/cm2) at an irradiance of 20 mW/cm2, data indicated statistically significantly reduction in calcific nodule formation at day 7. Collectively, these data suggest that when delivering blue light energy densities within the same range but at lower irradiance parameters, cells may require less frequent exposures although at longer irradiation times. Interestingly, the literature suggests that blue laser exposure does not follow this regulatory response. Indeed, MSCs only required a single exposure at multiple irradiance settings (50, 100, 150, 200, 300 mW/cm2) and a fixed irradiation time (180 s). Continuous wave 405 nm laser light delivery at considerably higher energy densities of 9, 18, 27, 36 and 54 J/cm2 enhanced bone marrow MSC mineralisation responses in a dose-dependent manner [12, 13].
The mixed cell population used here provides a representation of the in-vivo environment [3,4,5,6,7]. However, this heterogeneity and the differences in experimental set-ups provide further possible explanations for the diversity in results obtained compared with the literature. In previous studies [15, 16], it was reported that the light source spot size was 35 mm in diameter. However, proliferation was investigated in 96-well plates, and ALP and mineralised nodule formation studies were performed in 35 mm dishes. The cell culture plates used for the associated molecular investigations were not described. It was however reported that irradiation was performed from above the cultures at a 1-cm distance, and the standard heights of both plates were different; i.e. 14–17 mm for 96-well plates and 12–14 mm for 35-mm dishes. Moreover, the spot size was 5–6 times larger in diameter than the well’s diameter in a 96-well plate (6 mm). In Wang et al.’s [14] study, all plates were covered in aluminum foil except for a 4 cm2 area/light spot size area (22.5 mm diameter). Irradiance was maintained at 16 mW/cm2 by varying the distance between the light source and the cell culture plates. Molecular investigations were performed in 96-well plates, while mineralised nodule studies were undertaken in 6-well plates. The location of the irradiation was not reported; however, again, differences exist between the heights of 96-well plates (14–17 mm) and that of the 6-well plates (20–23 mm). Additionally, there are discrepancies between the spot size (22.5 mm) and the well in a 96-well plate (6 mm), as well as the 6-well plate diameter (35 mm). There were also no details reported regarding how the light source was characterised, or where the sensor/detector was positioned during light characterisation [14,15,16]. These reports and inconsistencies highlight the importance of including both detailed explanation of each experimental set-up and how light is characterised, especially as blue light exhibits the highest Rayleigh scattering compared with longer wavelengths [27]. In the current investigation, the flat-top light source (7 mm diameter) was in direct contact with the black walled 96-well plate bottom and the attached cell monolayer. The experimental set-up used here ensured precision in the following: (i) light delivery, (ii) light characterisation, determination of the delivered energy density at the cell monolayer level and (iii) that all cells and cultures received a consistent energy density by matching the light spot size to the size of the well [20].
Another important variable which should be considered is the potential effect of temperature change following light irradiation. Indeed, none of the previous studies has reported measuring temperature variations [12,13,14,15,16]. Heat dissipation in cultures depends on their thermal relaxation time as well as the irradiance, exposure time, pulse frequency and pulse duration of the light source [28]. Not only can hyperthermia increase mitochondrial ROS [29], but also hyperthermia is reported to enhance the mineralisation processes in MSCs. These effects are reportedly mediated via heat shock protein (HSP70), as its knockout alleviates the positive effects of hyperthermia [30,31,32]. In the current study, there was no significant differences in the temperature change between the non-irradiated controls and the irradiated samples—at all energy densities investigated. This, subsequently, reduces the likelihood that any hyperthermic effects initiated the DPCs responses which were detected here. Noteworthy, PBM does not enhance differentiation in cultures maintained in media devoid of mineralisation-inducing supplements and blue light exposure inhibits cellular proliferation regardless of the type of media used [17]. Accordingly, all investigations in the current study—including proliferation—were carried out in FBS and mineralising supplemented media. Notably, the concentrations of these supplements have not always been reported [15, 16] and this may also be a source of differences in the outcomes reported here compared with the literature.
Overall, based on results from this work, it can be hypothesised that under the current experimental set-up and irradiation parameters, blue light resulted in an arrest in proliferation rates due to promoting the cellular shift towards odontogenic differentiation. The difference in the magnitude of responses between the current study and previous investigations reported in the literature is likely due to differences in usage of cell types, light sources, irradiation parameters and exposure rates. In conclusion, blue light—typically accessible in every dental clinical setting via curing units—can potentially be applied therapeutically to promote hard tissue remineralisation. Furthermore, it is evident that blue light PBM studies should be aimed at standardizing the light sources, experimental set-ups and mineralising supplement concentrations.
References
Listl S, Galloway J, Mossey PA, Marcenes W (2015) Global economic impact of dental diseases. J Dent Res 94(10):1–7. https://doi.org/10.1177/0022034515602879
Frencken JE, Sharma P, Stenhouse L, Green D, Laverty D, Dietrich T (2017) Global epidemiology of dental caries and severe periodontitis – a comprehensive review. J Clin Periodontol 44(Suppl 18):S94–S105. https://doi.org/10.1111/jcpe.12677
Sloan AJ (2015) Biology of the dentin-pulp complex. In: Stem Cell Biology and Tissue Engineering in Dental Sciences. Elsevier, pp 371–378. https://doi.org/10.1016/B978-0-12-397157-9.00033-3
Levine S (2015) Endotontics. In: Dental Secrets (4th edition). Elsevier, pp 126–146. https://doi.org/10.1016/B978-0-323-26278-1.00007-6
Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A, DenBesten P, Robey PG, Shi S (2002) Stem cell properties of human dental pulp stem cells. J Dent Res 81(8):531–535. https://doi.org/10.1177/154405910208100806
Takeda T, Tezuka Y, Horiuchi M, Hosono K, Iida K, Hatakeyama D, Miyaki S, Kunisada T, Shibata T, Tezuka K (2008) Characterization of dental pulp stem cells of human tooth germs. J Dent Res 87(7):676–681. https://doi.org/10.1177/154405910808700716
Nanci A (2017) Ten Cate’s Oral Histology-Development, structure, and function (9th edition). Elsevier
Tziafas D, Smith AJ, Lesot H (2000) Designing new treatment strategies in vital pulp therapy. J Dent 28(2):77–92. https://doi.org/10.1016/s0300-5712(99)00047-0
Duncan HF, Cooper PR, Smith AJ (2019) Dissecting dentine-pulp injury and wound healing responses: consequences for regenerative endodontics. Int Endod J 52(3):261–266. https://doi.org/10.1111/iej.13064
Tziafas D (2019) Characterization of odontoblast-like cell phenotype and reparative dentin formation in vivo: a comprehensive literature review. J Endod 3:241–249. https://doi.org/10.1016/j.joen.2018.12.002
Hamblin MR, Demidova TN (2006) Mechanisms of low level light therapy. Proc SPIE 6140:614001. https://doi.org/10.1117/12.646294
Kushibiki T, Awazu K (2008) Controlling osteogenesis and adipogenesis of mesenchymal stromal cells by regulating a circadian clock protein with laser irradiation. Int J Med Sci 5(6):319–326. https://doi.org/10.7150/ijms.5.319
Kushibiki T, Awazu K (2009) Blue laser irradiation enhances extracellular calcification of primary mesenchymal stem cells. Photomed Laser Surg 27(3):493–498. https://doi.org/10.1089/pho.2008.2343
Wang Y, Huan YY, Wang Y, Lyu P, Hamblin MR (2016) Photobiomodulation (blue and green light) encourages osteoblastic-differentiation of human adipose-derived stem cells: Role of intracellular calcium and light-gated ion channels. Sci Rep 6:33719. https://doi.org/10.1038/srep33719
Zhu T, Wu Y, Zhou X, Yang Y, Wang Y (2019) Irradiation by blue light-emitting diode enhances osteogenic differentiation in gingival mesenchymal stem cells in vitro. Lasers Med Sci 34:1473–1481. https://doi.org/10.1007/s10103-019-02750-3
Yang Y, Zhu T, Wu Y, Shu C, Chen Q, Yang J, Luo X, Wang Y (2020) Irradiation with blue light-emitting diode enhances osteogenic differentiation of stem cells from the apical papilla. Lasers Med Sci 35:1981–1988. https://doi.org/10.1007/s10103-020-02995-3
Mohamad SA, Milward MR, Hadis MA, Kuehne SA, Cooper PR (2021) Photobiomodulation of mineralisation in mesenchymal stem cells. Photochem Photobiol Sci 20:699–714. https://doi.org/10.1007/s43630-021-00047-5
Chen CT, Shih YRV, Kuo TK, Lee OK, Wei YH (2008) Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 26(4):960–968. https://doi.org/10.1634/stemcells.2007-0509
Lyublinskaya OG, Borisov YG, Pugovkina NA, Smirnova IS, Obidina JV, Ivanova JS, Zenin VV, Shatrova AN, Borodkina AV, Aksenov ND, Zemelko VI, Burova EB, Puzanov MV, Nikolsky NN (2015) Reactive oxygen species are required for human mesenchymal stem cells to initiate proliferation after the quiescence exit. Oxid Med Cell Longev 502105. https://doi.org/10.1155/2015/502105
Mohamad SA, Milward MR, Kuehne SA, Hadis MA, Palin WM, Cooper PR (2021) Potential for direct application of blue light for photo-disinfection of dentine. J Photochem Photobiol B 215:112123. https://doi.org/10.1016/j.jphotobiol.2021.112123
Shortall AC, Hadis MA, Palin WM (2021) On the inaccuracies of dental radiometers. PLoS ONE 16(1):e0245830. https://doi.org/10.1371/journal.pone.0245830
Hadis MA, Cooper PR, Milward MR, Gorecki P, Tarte E, Churm J, Palin WM. The effect of UV-Vis to near-infrared light on the biological response of human dental pulp cells. Proc. SPIE 2015; 9309, Mechanisms for Low-Light Therapy X, 930906. https://doi.org/10.1117/12.2077645
Robert AW, Marcon BH, Dallagiovanna B, Shigunov P (2020) Adipogenesis, osteogenesis, and chondrogenesis of human mesenchymal stem/stromal cells: a comparative transcriptome approach. Front Cell Dev Biol 8:561. https://doi.org/10.3389/fcell.2020.00561
Huang YY, Sharma SK, Carroll J, Hamblin MR (2011) Biphasic dose response in low level light therapy - an update. Dose Response 9(4):602–618. https://doi.org/10.2203/dose-response.11-009.Hamblin
Ching HS, Luddin N, Rahman IA, Ponnuraj KT (2017) Expression of odontogenic and osteogenic markers in DPSCs and SHED: a review. Curr Stem Cell Res Ther 12(1):71–79. https://doi.org/10.2174/1574888x11666160815095733
Yuan Y, Yan G, Gong R, Zhang L, Liu T, Feng C, du W, Wang Y, Yang F, Li Y, Guo S, Ding F, Ma W, Idiiatullina E, Pavlov V, Han Z, Cai B, Yang L (2017) Effects of blue light emitting diode irradiation on the proliferation, apoptosis and differentiation of bone marrow-derived mesenchymal stem cells. Cell Physiol Biochem 43(1):237–246. https://doi.org/10.1159/000480344
Acton JC, Dawson PL (2004) Proteins in food processing. Woodhead Publishing Limited
Glavaš H, Vukobratović M, Keser T (2018) Infrared thermography as control of handheld IPL device for home-use. J Cosmet Laser Ther 20(5):269–277. https://doi.org/10.1080/14764172.2017.1406607
Wang Z, Cai F, Chen X, Luo M, Hu L, Lu Y (2013) The role of mitochondria-derived reactive oxygen species in hyperthermia-induced platelet apoptosis. PLoS ONE 8(9):e75044. https://doi.org/10.1371/journal.pone.0075044
Shui C, Scutt A (2001) Mild heat shock induces proliferation, alkaline phosphatase activity, and mineralization in human bone marrow stromal cells and Mg-63 cells in vitro. J Bone Miner Res 16(4):731–741. https://doi.org/10.1359/jbmr.2001.16.4.731
Chen J, Shi ZD, Ji X, Morales J, Zhang J, Kaur N, Wang S (2013) Enhanced osteogenesis of human mesenchymal stem cells by periodic heat shock in self-assembling peptide hydrogel. Tissue Eng Part A 19(5–6):716–728. https://doi.org/10.1089/ten.tea.2012.0070
Li C, Sunderic K, Nicoll SB, Wang S (2018) Downregulation of heat shock protein 70 impairs osteogenic and chondrogenic differentiation in human mesenchymal stem cells. Sci Rep 8:553. https://doi.org/10.1038/s41598-017-18541-1
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
All procedures were approved by Birmingham and The Black Country Comprehensive Local Research Network, Research Management and Governance (BBC CLRN RM&G) Consortium Office (Approval number: BCHCDent334.1533.TB).
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohamad, S.A., Milward, M.R., Hadis, M.A. et al. Blue light photobiomodulation of dental pulp cells. Laser Dent Sci 6, 79–87 (2022). https://doi.org/10.1007/s41547-022-00152-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41547-022-00152-3