
Abstract
The invasive ascomycete Hymenoscyphus fraxineus is the causative agent for ash dieback on the European species Fraxinus excelsior and Fraxinus angustifolia, and there is concern that it is going to replace the native, closely related and nonpathogenic Hymenoscyphus albidus. Fungal management in forests is limited, and alternative approaches for control are needed. Within the scope of the project “FraxForFuture”, several strategies are being investigated. One idea comprises the use of a viral hyperparasite, which can induce a reduced virulence in the fungal host H. fraxineus in an antagonist-like system. This phenomenon, the reduction of fungal virulence by a viral infection, is known as hypovirulence, and a similar method has already been established to control the Chestnut Blight in Europe. We examined 34 isolates of H. fraxineus for both their virulence and presence of a viral infection. Although a predominant number of isolates were found to be infected with Hymenoscyphus mitovirus 1 (HfMV1), no additional viruses were detected, and our data did not indicate a link to reduced virulence. The search for a viral infection was extended to one isolate of H. albidus in which we found and characterized a novel mycovirus. Based on phylogenetic analysis and sequence properties, it was assigned to the genus Victorivirus in the family of Totiviridae and was tentatively denominated as Hymenoscyphus albidus victorivirus 1. This novel and native mycovirus might be suitable for inducing hypovirulence in H. fraxineus as a biocide.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ascomycete Hymenoscyphus albidus (Gillet) W. Phillips is endemic in Europe, where it is nonpathogenic. Both Hymenoscyphus fraxineu and H. albidus are closely related, but H. albidus has steadily declined and has been rapidly replaced by the alien invasive pathogen H. fraxineus (T. Kowalski) (Hietala et al. 2013). Hymenoscyphus fraxineus affects the ash species Fraxinus excelsior L. and Fraxinus angustifolia Vahl and has led to severe decline of ash trees in the last three decades. Especially in Northern Europe, ash trees have already been significantly reduced (George et al. 2022).
Management of forest diseases is limited and differs from the management of fungal diseases affecting agricultural crops. Although fungicides in nurseries may be used to reduce selected pathogens, they cause a diverse effect on ectomycorrhizal fungi (Laatikainen and Heinonen-Tanski 2002) and only few fungicides have been approved for specific use in forests by the European Union.
As Prospero et al. (2021) stated, alternative approaches for controlling fungal diseases in forests are needed, and they discussed the prospects of biological control. Even though the potential of antagonistic bacteria or fungi has been tested to control H. fraxineus (Halecker et al. 2020; Kowalski and Bilański 2021; Ulrich et al. 2020), the closely related and nonpathogenic H. albidus is not suitable for the use as biocontrol by antagonistic traits (Gross and Sieber 2016). An example of a successful biological approach in Europe is the control of Chestnut Blight, which is caused by Cryphonectria parasitica (Murrill) M.E. Barr using the hyperparasite mycovirus Cryphonectria hypovirus 1 (CHV1) in an antagonist-like system (Prospero et al. 2021; Rigling and Prospero 2018). Most mycovirus infections remain cryptic; however, some mycoviruses can reduce the virulence of their fungal hosts. This phenomenon is called hypovirulence (Pearson et al. 2009). Xie and Jiang (2014) summarized the advantages of applying viruses as biocontrol agents. Once hypovirulence-associated mycoviruses are transmitted to a virulent strain of the pathogen, they quickly induce hypovirulent traits. Even if no transmission of the virus takes place, the mere growth of the hypovirulent strain in the host is likely to induce pathogen-associated molecular pattern (PAMP)-triggered immunity and/or to express effectors which produce a defense response to target the pathogen. Therefore, biological control of the pathogen based on a virus-infected hypovirulent H. fraxineus may be a promising approach.
One of the most simple mycoviruses is mitoviruses of the family Mitoviridae (Walker et al. 2022). While their genome is not protected by a virus-encoded capsid, a single polypeptide with an RNA-dependent RNA polymerase (RdRp) domain is translated from the positive sense single-stranded RNA (+ ssRNA) genome by the use of an internal UGA codon, which encodes tryptophan in fungal mitochondria, the compartment in which most mitoviruses replicate (Hillman and Cai 2013; Lefkowitz et al. 2018).
Schoebel et al. (2014) discovered the novel mitovirus Hymenoscyphus fraxineus mitovirus 1 (HfMV1) in H. fraxineus and determined its overall prevalence in European isolates to be about 80% with a high variation within different regions (Schoebel et al. 2017). In another study, they showed that HfMV1 is not only present in H. fraxineus but also in H. albidus, probably introduced by at least three cross-species transmission events (Schoebel et al. 2018). The conspecific HfMV1 was identified in about 50% of the screened H. albidus isolates (Schoebel et al. 2018).
The presence of HfMV1 in H. albidus is remarkable, since no extracellular stage has been reported for mycoviruses to date. They are transmitted almost exclusively by forming connections between fungal hyphae (anastomoses) of donor and acceptor, and only few reports suggest cross-species transmission (Arjona-Lopez et al. 2018; Vainio et al. 2017). Even though HfMV1 is present in high numbers in European H. fraxineus populations, it does not seem to alter host virulence (Lygis et al. 2017). No other viral infection in H. albidus has been described yet.
Shamsi et al. (2022) screened native Japanese H. fraxineus isolates using Illumina sequencing and found viruses in about 11% of the isolates with a high prevalence of the novel mitovirus Hymenoscyphus fraxineus mitovirus 2 (HfMV2). They examined its potential for inducing hypovirulence and, subsequently, for its use as a biological control agent after transmitting it to different isolates through co-culturing. This artificial introduction of HfMV2 to strains of H. fraxineus caused either no, increasing or decreasing effects on fungal growth in vitro. Only strains with reduced growth showed an altered virulence on ash saplings compared to their virus-free isogenic isolates (Shamsi et al. 2023). However, data from their bioassays are preliminary and must be confirmed.
According to the International Committee on Taxonomy of Viruses (ICTV), the family of Totiviridae, which is placed in the order of Ghabrivirales, is composed of five genera: Giardiavirus, Leishmaniavirus, Totivirus, Trichomonasvirus and Victorivirus. The genus Victorivirus is comprised of several members associated with fungal hosts (Ghabrial and Nibert 2009; ICTV 2011). The genome of victoriviruses consists of a single linear, bicistronic dsRNA segment, ranging from 4 to 6 kbp, which is encapsidated by virions with a diameter of around 40 nm. The 5ʹ-proximal open reading frame (ORF) is coding for the capsid protein with a alanine/glycine/proline-rich C-terminus, while the 3ʹ-proximal ORF encodes the viral RdRp. Both ORFs are connected by a − 1 ribosomal frameshift within the ORF-overlapping region containing the tetranucleotide AUGA (ICTV 2011).
In the framework of the “FraxForFuture” project (Langer et al. 2022), funded by the German “Waldklimafonds”, the possibility to induce hypovirulence in H. fraxineus by a viral infection is being investigated. To find possible candidates for biocontrol, we followed two approaches: (i) the evaluation of virulence of HfMV1 and (ii) the identification of possible new candidates to induce hypovirulence after transfection. For that, we examined 34 H. fraxineus isolates for the presence of viruses and compared their presence with virulence of the H. fraxineus isolates to assess their potential as future biocontrol agents. We extended our search for viral infections to an isolate of H. albidus, where we detected a single dsRNA of about 5 kbp. Its sequence showed highest similarity to Corynespora cassiicola victorivirus 1 (CcVV1), and therefore, we tentatively named it Hymenoscyphus albidus victorivirus 1 (HaVV1). Additionally, a strain of HfMV1 was detected in H. albidus and was tentatively denominated as Hymenoscyphus fraxineus mitovirus 1 strain albidus (HfMV1alb).
Having identified the novel virus in the closely related and endemic H. albidus, successful replication of HaVV1 in the transfected host H. fraxineus seems possible and may serve as a potential agent for biocontrol of the as dieback disease.
Material and methods
Fungal isolates and plant material for virulence trials
Forest stands were sampled across Germany as part of the “FraxForFuture” project (Langer et al. 2022). Plant material was obtained from trees of a forest stand near Rhüden (32 U 579914 5757111) on February 22, 2021, and on March 01, 2021, as part of the “FraxPath” investigations (Peters et al. 2023). The F. excelsior trees had visible signs of necroses in the branches and/or root collar necroses, which are typically associated with H. fraxineus infections. Isolations were made onto 2% Malt Extract Agar (MEA) culture media and the identity of the isolates was confirmed with morphological and molecular techniques (Johansson et al. 2010). The H. albidus isolate 090812.3 was collected in 2009 from an ascocarp on a rachis of F. excelsior by O. Holdenrieder in Switzerland (Queloz et al. 2011).
For propagation of mycelium for dsRNA isolation or for particle extraction, the isolated strains of Hymenoscyphus sp. were cultivated on solid ash leaf medium (AMS) at RT in the dark as described in Lutz et al. (2023). For further analyses, mycelium was harvested from AMS covered with cellophane sheets.
Infection assay and symptom ranking
For the virulence trial of H. fraxineus isolates, two-year-old F. excelsior saplings (provenance 81102: Nordostdeutsches Tiefland) from Schlegel & Co. Gartenprodukte GmbH (Riedlingen, Germany) were repotted into 5.5 l plant pots using regular potting soil and acclimated in the greenhouse at 15 °C/20 °C night/day, and a minimum of 14 h of light to induce early flushing.
Inoculations were made in the greenhouse to the petiole of the F. excelsior saplings based on a random design in spring. The inoculations were conducted by first creating a superficial cut of approximately 1 cm in length from the lowest leaflets on the petiole toward the main stem using a sterile scalpel. A 3 mm plug of fresh mycelium of H. fraxineus grown on 2% MEA culture media was placed into the center of the wound before the wound was sealed using Parafilm (Amcor, Zurich, Switzerland). Three petiole inoculations were made per sapling. In total, each of the isolates was inoculated nine times across nine different saplings. In addition, 18 control inoculations were made across six saplings using sterile agar plugs.
The plants were monitored for the development of symptoms, including necrosis development on the petiole, leaf abscission, necrosis development on the stem, girdling of the stem and wilting of leaves starting two weeks after inoculations. Subsequent symptom development was monitored weekly for a total of eight weeks.
Based on the observed symptoms, a ranking was developed to provide insights into the variation in virulence between isolates. The isolates were ranked according to three factors. The presence/ absence of a stem necrosis was used as a primary indicator of virulence. The isolates indicating a capacity to cause a stem necrosis were further analyzed according to the number of observable infections compared to the number of inoculations (infection rate). These data were used to develop a confidence value for the observed infection rate. Finally, the ability of the isolate to cause mortality was inferred based on observations of wilting of the sapling’s leaves and girdling of the stem by H. fraxineus.
In summary, the ranking was performed according to presence/absence of a stem necrosis, infection rate and capacity to cause mortality. If no symptoms were observed during the monitoring period, the isolates were marked as not virulent (0). The remaining isolates were associated with symptoms and were further classified as having low virulence (1), intermediate virulence (2) or high virulence (3). To classify the isolates, infection rate was ranked according to these four categories relative to the highest observed infection rate in this trial. The capacity to cause mortality was then added to the ranking. Isolates that had a low infection rate and a capacity to cause girdling and/or wilting were more highly ranked than those with the same low infection rate and no observable capacity to cause mortality. For example, isolate “RH02_T8_B2_2” was ranked with a low virulence (1) based on infection rate alone. However, as this isolate was also observed as being capable of causing mortality, the isolate was assessed as having an intermediate virulence (2) in the final ranking. This adjustment was made to differentiate isolates capable of causing disease from those capable of causing mortality, which is critical in assessments of virulence and pathogenicity.
Double-stranded RNA extraction and screening for HfMV1 by RT-PCR
Double-stranded RNA (dsRNA) was extracted from mycelium or purified viral particles using the dsRNA Extraction kit (iNtRON Biotechnology, Seongnam-Si, South Korea) and was analyzed by 1% (w/v) agarose gel electrophoresis and ethidium bromide staining.
By reverse transcriptase PCR (RT-PCR), using the primer pair Cf_4F_1 and Cf_4R_3 (Schoebel et al. 2014), the isolates of H. fraxineus were screened for the presence of HfMV1. Briefly, 5 µl isolated dsRNA in a total volume of 14.5 µL was denatured in the presence of 20 pmol of reverse primer Cf_4R_3, 0.5 mM dNTPs and 11% v/v DMSO at 98 °C for 10 min and cooled down in a NaCl/ice-water mixture for 2 min. Viral dsRNA was reverse transcribed using 200 U Maxima H Minus Reverse Transcriptase (ThermoFisher Scientific, Waltham, MA, USA), 4 µl 5 × RT buffer and 20 U RiboLock RNase Inhibitor (ThermoFisher Scientific) in a final volume of 20 µl for 30 min at 60 °C. The reaction was terminated by heating at 85 °C for 5 min.
Complementary DNA (cDNA) was amplified with 0.625 U Dream-Taq™ Polymerase (ThermoFisher Scientific) in a 25 µl reaction using 10 pmol of each primer Cf4_R3 and Cf_4F_1 and 0.2 mM dNTPS with 30 cycles (95 °C for 30 s, 57 °C for 30 s, 72 °C for 60 s), starting with 3 min denaturation at 95 °C and a final synthesis at 72 °C for 10 min to obtain a 537 bp fragment covering a part of the RdRp ORF.
Virus-like particle purification and protein analysis
Virus-like particles (VLPs) were enriched according to Aoki et al. (2009), as described in Lutz et al. (2021) with modifications. One to three g of mycelium was crushed by means of liquid nitrogen. The powder was resuspended in 100 ml 0.1 M sodium phosphate (pH 7), and coarse material was removed by centrifugation (8000×g, 20 min). The supernatant was clarified once with 20% (v/v) chloroform/n-butanol (1:2). The upper phase was stirred in the presence of 8% (w/v) polyethylene glycol (PEG6000) and 1% (w/v) NaCl overnight at 4 °C. The precipitate was sedimented (10,000×g, 20 min) and resuspended in 0.05 M sodium phosphate (pH 7), which was layered on top of 20% (w/v) sucrose in 0.05 M sodium phosphate (pH 7) and centrifuged (105,000×g, 2 h). The pellet was resuspended in 0.05 M sodium phosphate (pH 7) and stored at − 70 °C.
Proteins of purified particles were separated by SDS-PAGE according to Laemmli (1970) and stained with Coomassie Brilliant Blue (Merril 1990). Peptides were sequenced with LC–MS/MS by a nano-liquid chromatography system (Dionex UltiMate™ 3000 RSLCnano, ThermoFisher Scientific) and analyzed by means of the Proteome Discoverer 2.0 (ThermoFisher Scientific) by the Universitätsklinikum Hamburg-Eppendorf (UKE, Hamburg, Germany).
Sequencing and sequence analysis
Isolated dsRNA of H. albidus isolate 090812.3 was submitted to next-generation sequencing. The libraries were prepared according to Nextera XT DNA Library Preparation Kit (Illumina Inc., San Diego, CA, USA) and run on a NextSeq2000 (Illumina Inc.) instrument at the Leibniz Institute DSMZ (Braunschweig, Germany) as pair-end reads (2 × 151). De novo assembly and contigs were analyzed using Geneious Prime software (Biomatters, Auckland, New Zealand, Version 2021.2.2). The extreme 5ʹ- and 3 ʹ-termini were determined by SPAT (single primer amplification technique) as it was described in Zhong et al. (2016), using an oligonucleotide (5′-PO4-tctcttcgtgggctcttgcg-23ddC-3′) with a phosphorylated 5′-terminus and a 2′,3′-dideoxyC-group (23ddC) at the 3′-terminus as a blocker to prevent self-ligation. From the ligated dsRNA, the 5ʹ-terminus was amplified with primer pair 5ʹ-atattggacttgtagcggcgg-3ʹ/5ʹ-cgcaagagcccacgaagaga-3ʹ and the 3ʹ-terminus was amplified with primer pair 5ʹ-ataccaagaccgagcgaggc-3ʹ/5ʹ-cgcaagagcccacgaagaga-3’ after cDNA synthesis using primer 5ʹ-cgcaagagcccacgaagaga-3ʹ following the protocol of Maxima H Minus Reverse Transkriptase (ThermoFisher Scientific) as described above. Amplicons were subcloned into pGEM®-T Vector (Promega Corporation, Madison, WI, USA) and sequenced.
Nucleic acid sequences and ORFs were analyzed by SnapGene (GSL Biotech, San Diego, CA, USA) and BLAST on the NCBI website (Altschul et al. 1997). Sequence alignments and phylogenetic analysis were performed using MEGA X (version 10.2.4) (Kumar et al. 2018), Clustal Omega (Goujon et al. 2010; McWilliam et al. 2013; Sievers et al. 2011) and Muscle (Edgar 2004a, 2004b; Li et al. 2015; McWilliam et al. 2013) in default settings.
Conserved protein domains were identified by conserved domain database (CDD) search on the NCBI website (Marchler-Bauer et al. 2011, 2015, 2017; Marchler-Bauer and Bryant 2004).
Phylogenetic analysis
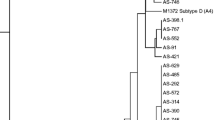
The evolutionary history was inferred by using the maximum likelihood method and the Le and Gascuel (2008) model. The tree with the highest log likelihood (− 43,385.71) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (five categories (+ G, parameter = 0.9076)). The rate variation model allowed for some sites to be evolutionarily invariable ([+ I], 13.02% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 39 amino acid sequences. There were a total of 1206 positions in the final dataset.
Phylogenetic analysis was carried out after sequence alignment of the putative RdRp of HaVV1 with the respective proteins of putative or approved members of species within the Totiviridae family found by BLASTp with an E-value of 0.0. As an outgroup, RdRps of the Magnaporthe oryze chrysovirus 1 D/B (MoCV1-D/B) (Higashiura et al. 2019; Urayama et al. 2014) of the Chrysoviridae family were added.
Figure generating and editing
Figures were generated and edited by Unipro UGENE (ugene.net, version 1.32.0), INKSCAPE (inkscape.org, version 1.1) and SnapGene.
Results
Virus characterization and phylogenetic relationship
Initial dsRNA screening of the H. albidus isolate 090812.3 revealed a distinct band at around 5 kbp after examination by agarose gel electrophoresis (Fig. 1a). For further investigations, VLPs were isolated.

Gel electrophoresis of dsRNA and proteins of HaVV1. The sizes of the markers are given on the left of each figure. a Agarose gel electrophoresis (1% w/v) of dsRNA, isolated from VLPs. M, GeneRuler 1 kb plus ladder (Thermo Fisher Scientific). b VLPs separated by SDS-PAGE (10% w/v) and visualized by Coomassie Brilliant Blue staining. M, PageRuler Prestained Protein Ladder (Thermo Fisher Scientific). Two distinct bands are visible between 70 and 100 kDa, corresponding to the RdRp and the CP
The dsRNA extracted from VLPs was submitted to next-generation sequencing and completed by SPAT as described. The complete genome is 5143 bp in length with an overall GC content of 59.45%. On the genomic plus strand, two ORFs between 295 bp at the 5ʹ-terminus and 5060 bp at the 3ʹ-terminus are encoded. The 5ʹ-proximal ORF (ORF 1) encodes a protein of 758 aa with a predicted MW of 79.5 kDa. The protein clusters within the Totivirus_coat protein super family (pfam 05518; acc. ID: cl25797, E-value: 0e + 00) from amino acids (aa) 70 to 706. As is typical for victoriviruses, P1 harbors a C-terminal alanine-, glycine- and proline-rich region (13.46% alanine, 23.08% glycine, 30.77% proline in 52 aa) (Supplementary Fig. S1). A BLASTp search of the deduced protein of ORF 1 showed the highest degree of similarity (71.87% identical aa, E-value 0.0) to the putative coat protein (CP) of CcVV1 (acc. ID: UIB81488). The complete sequence was deposited in the GenBank database (acc. ID: OR209821).
The 3ʹ-proximal ORF (ORF 2) encodes a protein of 830 aa with a predicted MW of 90.5 kDa. Between aa 126 and aa 581, the motif of the RT_like superfamily was detected by CDD (acc. ID: pfam02123, E-value 6.65e-122). The start codon of ORF 2 overlaps with the stop codon of ORF 1 by the tetranucleotide AUGA (Fig. 2a).

Genomic organization of HaVV1 and its taxonomic position within the Totiviridae family. a Genome organization of HaVV1. The dsRNA segments are displayed as horizontal lines with their respective UTRs at each terminus. ORFs are represented as boxes with start and stop codon positions indicated above or underneath the boxes. Special features of the respective ORFs are highlighted. Note that the figure is not drawn to scale. b Maximum likelihood tree of HaVV1 and selected viruses with 1000 bootstrap replicates. Bootstrap values are displayed at the nodes. The scale bar (1) corresponds to the genetic distance. The dot indicates the novel virus HaVV1. The abbreviated names of viruses and dsRNA elements are as follows: AfV-S1 Aspergillus foetidus slow virus 1, BbVV Beauveria bassiana victorivirus 1, BbVV1 Beauveria bassiana victorivirus 1, BcVV1-3 Botrytis cinerea victorivirus 1–3, BdVV2(-like) Botryosphaeria dothidea victorivirus 2(-like), BfTV1 Botryotinia fuckeliana totivirus 1, BmVV1 Bipolaris maydis victorivirus 1, CchVV1 Cordyceps chanhua victorivirus 1, CcVV1 Corynespora cassiicola victorivirus 1, CeTV1 Colletotrichum eremochloae totivirus 1, CfVV1 Colletotrichum fructicola victorivirus 1, FaVV1 Fusarium asiaticum victorivirus 1, FTV2 Fushun totivirus 2, HaVV1 Hymenoscyphus albidus victorivirus 1, HTV3/4/5 Hangzhou totivirus 3/4/5, HVV-190S Helminthosporium victoriae virus 190S, MaVV1 M5 Metarhizium anisopliae M5 victorivirus 1, MoCV1B/D Magnaporthe oryzae chrysovirus 1 B/D, MoV1 Magnaporthe oryzae virus 1, NcVV1 Nigrospora chinensis victorivirus 1, NoVV2 Nigrospora oryzae victorivirus 2, NPVV1/3 Neofusicoccum parvum victorivirus 1/3, NsVV1 Nigrospora sphaerica victorivirus 1, PdTV2 Penicillium digitatum totivirus 2, RnVV1 Rosellinia necatrix victorivirus 1, ScVV1 Stagonosporopsis cucurbitacearum victorivirus 1, SnVV1 Sclerotinia nivalis victorivirus 1, SsV1 Sphaeropsis sapinea RNA virus 1, TcV1 Tolypocladium cylindrosporum virus 1, UvV13 Ustilaginoidea virens RNA virus 13
The BLASTp search of the deduced protein of ORF 2 showed the highest degree of similarity (63.61% identical aa, E-value 0.0) to the RdRp of CcVV1 (acc. ID UIB81489.1). Based on CDD search and the BLASTp results, ORF 2 is putatively coding for the viral RdRp.
On the basis of the aa sequence of the putative RdRp of HaVV1 and of RdRps of different related viruses, a maximum likelihood tree was constructed to analyze its phylogenetic position. The HaVV1 clusters with a high bootstrap value with CcVV1, Sclerotinia nivalis victorivirus 1 (SnVV1) and Aspergillus foetidus slow virus 1 (AfV-S1) (Fig. 2b).
Screening for the presence of HfMV1
All isolates were screened for the presence of HfMV1 by RT-PCR and in 23 out of 34 H. fraxineus isolates HfMV1 was detected (Fig. 3a). Additionally, HfMV1 was also detected in the isolate 090812.3 of H. albidus (Fig. 3b). Its sequence was determined by NGS and it was found that its protein, which is encoded on the only ORF, consists of 717 aa, has a calculated MW of 80.9 kDa and has 90.10% identity (E-value 0.0) with HfMV1 (acc. ID: AZG04296.1). It was named Hymenoscyphus fraxineus mitovirus 1 strain albidus (HfMV1alb). Due to the close relation of HfMV1alb to HfMV1, it was considered as a strain of HfMV1 and was therefore not further investigated. The incomplete nucleic acid sequence including the complete ORF was deposited in GenBank (acc. ID: OR224869).

RT-PCR screening for the presence of HfMV1 in the respective isolates of H. fraxineus (a) and H. albidus (b) with an expected band size of 537 bp
Infection study
To evaluate an influence of HfMV1 presence on the virulence of H. fraxineus isolates, infection assays with two-year-old saplings of F. excelsior were performed. According to the final ranking, there were 20 isolates that were considered as virulent because a necrosis developed on the stem. The isolates determined as virulent from this trial were not evenly distributed between the remaining classifications, i.e., having a high virulence (3), intermediate virulence (2), low virulence (1) or were avirulent (0). There were three isolates originating from three different trees which were considered as highly virulent. Sixteen isolates were considered as having an intermediate virulence based on having an intermediate infection rate and indicating a capacity to cause mortality in the saplings. Only one isolate (RH03-T1-B16-1) was considered as having a low virulence. This isolate caused a single necrosis that only became visible in the final week of monitoring, and thus was associated with a slow and smaller than average necrosis. However, it could still have potentially resulted in a larger necrosis given more time, and subsequent wilting and girdling of the stem could have been possible beyond the scope of the monitoring period. The remaining 14 isolates were considered as avirulent. In this case, the definition of avirulence includes the possibility that an infection developed on the petiole, but the infection remained localized and did not spread to the main stem. The results of the virulence assay are summarized in Table 1, and a detailed table is drawn in the supplementary material (Supplementary Table S1). There was no significant difference in virulence between HfMV1-positive and HfMV1-negative isolates (Mann–Whitney U test, p > 0.05).
Discussion
An alternative approach for controlling the invasive pathogen H. fraxineus is the use of viruses that cause hypovirulence to its host. While there is a similar system already established for the control of C. parasitica with the hyperparasite CHV1 on chestnut trees in Europe (Rigling and Prospero 2018), such a system has not been established for the control of the ash dieback disease due to the unavailability of a hypovirulent isolate of H. fraxineus to date. To find a strain of H. fraxineus which may be used as a biocontrol agent, 34 isolates were evaluated for their variation in virulence and its relation to the presence of viral infections. In addition, a strain of the closely related H. albidus was investigated for virus presence. Since it has been reported from several virus–fungus combinations that heterologous transfection resulted in a hypovirulence trait (Kanematsu et al. 2010; Lee et al. 2011; Sasaki et al. 2002; Yang et al. 2021), a virus isolated from H. albidus and transfected to H. fraxineus might be the perfect candidate to establish a hypovirulent isolate for use as a biocontrol agent in an antagonist-like system.
When testing 34 strains of H. fraxineus, which were isolated from ten different trees, the rate of necrosis development was very low compared to the total number of inoculations. There were only two instances where more than a third of inoculations resulted in a necrosis for a single isolate. These were for isolates “RH02-T7-B02-2” and “RH02-T8-B02-2”, where four out of nine and five out of nine inoculations resulted in necrosis, respectively. This is a limitation of using petiole inoculations compared to stem inoculations in virulence trials. However, stem inoculations arguably overestimate isolate virulence because the natural infection court of H. fraxineus is through the leaves and stem inoculations bypass numerous natural plant defenses (Haňáčková et al. 2017). It is also important to consider that H. fraxineus isolates are known to lose virulence over time (e.g., Kowalski and Bartnik 2012). Therefore, the low rate of necrosis development may be partly explained by the loss of virulence between the isolation of H. fraxineus and the initiation of the virulence trial.
To compare the data from the bioassays with the virome, all 34 isolates were screened for the existence of dsRNA as an indicator for a virus infection and were tested for the presence of HfMV1 by RT-PCR. In 23 out of 34 isolates, HfMV1, and in the H. albidus isolate 090812.3, a strain of HfMV1 (HfMV1alb) was detected. This high prevalence is in accordance with the findings of Schoebel et al. (2017) who found a mean prevalence of 80% for this virus in isolates of European H. fraxineus with variations between 24 and 92% in different regions. The presence of HfMV1 in H. albidus also supports the findings of Schoebel et al. (2018) who determined HfMV1 to be present in about 50% of H. albidus isolates. The absence of a relation between virus presence and virulence revealed that the presence of HfMV1 had no influence on the infection rate of different isolates of H. fraxineus in F. excelsior saplings. In our studies, isolates with a HfMV1 infection, ranged in their virulence from avirulent (0) to highly virulent (3) and likewise did non-infected isolates. This absence of correlation between HfMV1 infection and virulence confirmed the data of Schoebel et al. (2017). This is not surprising, since only few mitoviruses affecting host biology have been described so far (Jian et al. 1997; Khalifa and Pearson 2013; Wu et al. 2007, 2010; Xie and Ghabrial 2012; Xu et al. 2015).
Shamsi et al. (2022) described the introduction of HfMV2, which was discovered in a Japanese population of H. fraxineus (Shamsi et al. 2022), by co-cultivation to European isolates. In vitro plate assays, they observed growth reduction, growth increase and no effect in the infected strains compared to the virus-free, isogenic strains. They stated that these variable effects had also been described for mitovirus infections in other studies (Flores-Pacheco et al. 2018; Romeralo Tapia et al. 2012; Wu et al. 2007) and a similar effect of increase, decrease or no effect was described by Hyder et al. (2013). Therefore, mitoviruses may be improper for the use as biocontrol agents.
In addition to HfMV1, Čermáková et al. (2017) discovered a dsRNA band of about 4.5 kbp with a moderate incidence (15.7%) in European isolates of H. fraxineus, but the origin of these bands has remained unidentified. In a screening of 116 H. fraxineus isolates from pooled samples, Shamsi et al. (2022) found three viruses that were not mitoviruses, however they did not assign them to individual isolates of H. fraxineus. Therefore, the use of these viruses for the evaluation regarding their potential for biocontrol is limited.
The findings of Schoebel et al. (2018) that HfMV1 replicates in both, H. fraxineus and H. albidus, suggests that other viruses can also infect both of these species and may replicate in both hosts. Due to the lower grade of adaption, the probability to induce growth alterations and hypovirulence seems likely in the heterologous host. Therefore, we extended our search for viruses to the closely related H. albidus. Here, dsRNA with a size of about 5 kbp was detected and an additional infection with HfMV1 was confirmed by RT-PCR. Sequence analysis revealed that H. albidus 090812.3 harbored a dsRNA virus related to victoriviruses of the Totiviridae family and therefore we tentatively denominated it as Hymenoscyphus albidus victorivirus 1 (HaVV1). The sequence showed the typical coding strategy with a 5ʹ-proximal putative capsid gene which had an alanine/glycine/proline-rich C-terminus and similarities to the Totivirus_coat superfamily were detected by CDD search. The 3ʹ-proximal ORF codes for the RdRp and is initiated by the ORF-overlapping tetranucleotide AUGA which is in the -1 frame in relation to ORF 1.
The VLP enrichment from fungal mycelium was examined by SDS-PAGE and Coomassie Brilliant Blue staining and resulted in a distinct protein pattern showing bands corresponding to the calculated sizes of capsid and RdRp. The protein bands which were detected between 70 and 100 kDa were clearly assigned by protein sequencing to the capsid which is encoded on ORF 1 (79.44 kDa) and to the RdRp, encoded on ORF 2 (90.64 kDa). Additionally, viral dsRNA could be extracted from VLP preparations. The possibility to purify VLPs from H. albidus together with the protocol for the transformation of H. fraxineus (Lutz et al. 2023) may enable transfection experiments to obtain a hypovirulent isolate, which could help to control ash dieback disease in Europe in the future.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res 25:3389–3402. https://doi.org/10.1093/nar/25.17.3389
Aoki N, Moriyama H, Kodama M, Arie T, Teraoka T, Fukuhara T (2009) A novel mycovirus associated with four double-stranded RNAs affects host fungal growth in Alternaria alternata. Virus Res 140:179–187. https://doi.org/10.1016/j.virusres.2008.12.003
Arjona-Lopez JM, Telengech P, Jamal A, Hisano S, Kondo H, Yelin MD, Arjona-Girona I, Kanematsu S, Lopez-Herrera CJ, Suzuki N (2018) Novel, diverse RNA viruses from Mediterranean isolates of the phytopathogenic fungus, Rosellinia necatrix: insights into evolutionary biology of fungal viruses. Environ Microbiol 20:1464–1483. https://doi.org/10.1111/1462-2920.14065
Čermáková V, Eichmeier A, Herrero N, Botella L (2017) HfMV1 and another putative mycovirus in Central European populations of Hymenoscyphus fraxineus, the causal agent of ash dieback in Europe. Balt for 23:107–115
Edgar RC (2004a) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformat 5:113. https://doi.org/10.1186/1471-2105-5-113
Edgar RC (2004b) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Flores-Pacheco JA, Muñoz-Adalia EJ, Martínez-Álvarez P, Pando V, Díez-Casero JJ, Martín-García J (2018) Short communication: effect of mycoviruses on growth, spore germination and pathogenicity of the fungus Fusarium circinatum. Forest Syst 26:eSC07. https://doi.org/10.5424/fs/2017263-11060
George J-P, Sanders TGM, Timmermann V, Potočić N, Lang M (2022) European-wide forest monitoring substantiate the neccessity for a joint conservation strategy to rescue European ash species (Fraxinus spp.). Sci Rep 12:4764. https://doi.org/10.1038/s41598-022-08825-6
Ghabrial SA, Nibert ML (2009) Victorivirus, a new genus of fungal viruses in the family Totiviridae. Arch Virol 154:373–379. https://doi.org/10.1007/s00705-008-0272-x
Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucl Acids Res 38:W695–W699. https://doi.org/10.1093/nar/gkq313
Gross A, Sieber TN (2016) Virulence of Hymenoscyphus albidus and native and introduced Hymenoscyphus fraxineus on Fraxinus excelsior and Fraxinus pennsylvanica. Plant Pathol 65:655–663. https://doi.org/10.1111/ppa.12450
Halecker S, Wennrich J-P, Rodrigo S, Andrée N, Rabsch L, Baschien C, Steinert M, Stadler M, Surup F, Schulz B (2020) Fungal endophytes for biocontrol of ash dieback: the antagonistic potential of Hypoxylon rubiginosum. Fungal Ecol 45:100918. https://doi.org/10.1016/j.funeco.2020.100918
Haňáčková Z, Koukol O, Čmoková A, Zahradník D, Havrdová L (2017) Direct evidence of Hymenoscyphus fraxineus infection pathway through the petiole-shoot junction. For Pathol 47:e12370. https://doi.org/10.1111/efp.12370
Hietala AM, Timmermann V, BØrja I, Solheim H, (2013) The invasive ash dieback pathogen Hymenoscyphus pseudoalbidus exerts maximal infection pressure prior to the onset of host leaf senescence. Fungal Ecol 6:302–308. https://doi.org/10.1016/j.funeco.2013.03.008
Higashiura T, Katoh Y, Urayama S-I, Hayashi O, Aihara M, Fukuhara T, Fuji S-I, Kobayashi T, Hase S, Arie T, Teraoka T, Komatsu K, Moriyama H (2019) Magnaporthe oryzae chrysovirus 1 strain D confers growth inhibition to the host fungus and exhibits multiform viral structural proteins. Virology 535:241–254. https://doi.org/10.1016/j.virol.2019.07.014
Hillman BI, Cai G (2013) The family Narnaviridae: simplest of RNA viruses. Adv Virus Res 86:149–176. https://doi.org/10.1016/B978-0-12-394315-6.00006-4
Hyder R, Pennanen T, Hamberg L, Vainio EJ, Piri T, Hantula J (2013) Two viruses of Heterobasidion confer beneficial, cryptic or detrimental effects to their hosts in different situations. Fungal Ecol 6:387–396
ICTV (2011) Virus taxonomy: the classification and nomenclature of viruses: the 9th report of the ICTV. https://ictv.global/report_9th
Jian J, Lakshman DK, Tavantzis SM (1997) Association of distinct double-stranded RNAs with enhanced or diminished virulence in Rhizoctonia solani infecting potato. MPMI 10:1002–1009. https://doi.org/10.1094/MPMI.1997.10.8.1002
Johansson SBK, Vasaitis R, Ihrmark K, Barklund P, Stenlid J (2010) Detection of Chalara fraxinea from tissue of Fraxinus excelsior using species-specific ITS primers. For Pathol 40:111–115. https://doi.org/10.1111/j.1439-0329.2009.00614.x
Kanematsu S, Sasaki A, Onoue M, Oikawa Y, Ito T (2010) Extending the fungal host range of a partitivirus and a mycoreovirus from Rosellinia necatrix by inoculation of protoplasts with virus particles. Phytopathology 100:922–930. https://doi.org/10.1094/phyto-100-9-0922
Khalifa ME, Pearson MN (2013) Molecular characterization of three mitoviruses co-infecting a hypovirulent isolate of Sclerotinia sclerotiorum fungus. Virology 441:22–30. https://doi.org/10.1016/j.virol.2013.03.002
Kowalski T, Bartnik C (2012) Morphologial variation in colonies of Chalara fraxinea isolated from ash (Fraxinus excelsior L.) stems with symptoms of dieback and effects of temperature on colony growth and structure. Acta Agrobot 63:99–106. https://doi.org/10.5586/aa.2010.012
Kowalski T, Bilański P (2021) Fungi detected in the previous year’s leaf petioles of Fraxinus excelsior and their antagonistic potential against Hymenoscyphus fraxineus. Forests 12:1412. https://doi.org/10.3390/f12101412
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Laatikainen T, Heinonen-Tanski H (2002) Mycorrhizal growth in pure cultures in the presence of pesticides. Microbiol Res 157:127–137. https://doi.org/10.1078/0944-5013-00139
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. https://doi.org/10.1038/227680a0
Langer GJ, Fuchs S, Osewold J, Peters S, Schrewe F, Ridley M, Kätzel R, Bubner B, Grüner J (2022) FraxForFuture—research on European ash dieback in Germany. J Plant Dis Prot 129:1285–1295. https://doi.org/10.1007/s41348-022-00670-z
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320. https://doi.org/10.1093/molbev/msn067
Lee K-M, Yu J, Son M, Lee Y-W, Kim K-H (2011) Transmission of Fusarium boothii mycovirus via protoplast fusion causes hypovirulence in other phytopathogenic fungi. PLoS ONE 6:e21629. https://doi.org/10.1371/journal.pone.0021629
Lefkowitz EJ, Dempsey DM, Hendrickson RC, Orton RJ, Siddell SG, Smith DB (2018) Virus taxonomy: the database of the International Committee on Taxonomy of Viruses (ICTV). Nucl Acids Res 46:D708–D717. https://doi.org/10.1093/nar/gkx932
Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S, Park YM, Buso N, Lopez R (2015) The EMBL-EBI bioinformatics web and programmatic tools framework. Nucl Acids Res 43:W580–W584. https://doi.org/10.1093/nar/gkv279
Lutz T, Petersen JM, Yanık C, de Oliveira C, Heinze C (2021) Processing of the capsid proteins of the Betachrysovirus Fusarium graminearum virus-China 9 (FgV-ch9). Virology 563:50–57. https://doi.org/10.1016/j.virol.2021.08.007
Lutz T, Hadeler B, Jaeckel M, Schulz B, Heinze C (2023) Stable overexpression and targeted gene deletion of the causative agent of ash dieback Hymenoscyphus fraxineus. Fungal Biol Biotechnol 10:1. https://doi.org/10.1186/s40694-023-00149-y
Lygis V, Prospero S, Burokiene D, Schoebel CN, Marciulyniene D, Norkute G, Rigling D (2017) Virulence of the invasive ash pathogen Hymenoscyphus fraxineus in old and recently established populations. Plant Pathol 66:783–791. https://doi.org/10.1111/ppa.12635
Marchler-Bauer A, Bryant SH (2004) CD-Search: protein domain annotations on the fly. Nucl Acids Res 32:W327–W331. https://doi.org/10.1093/nar/gkh454
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH (2011) CDD: a conserved domain database for the functional annotation of proteins. Nucl Acids Res 39:D225–D229. https://doi.org/10.1093/nar/gkq1189
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH (2015) CDD: NCBI’s conserved domain database. Nucl Acids Res 43:D222–D226. https://doi.org/10.1093/nar/gku1221
Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Geer LY, Bryant SH (2017) CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucl Acids Res 45:D200–D203. https://doi.org/10.1093/nar/gkw1129
McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R (2013) Analysis tool web services from the EMBL-EBI. Nucl Acids Res 41:W597-600. https://doi.org/10.1093/nar/gkt376
Merril CR (1990) Gel-staining techniques. Methods Enzymol 182:477–488. https://doi.org/10.1016/0076-6879(90)82038-4
Pearson MN, Beever RE, Boine B, Arthur K (2009) Mycoviruses of filamentous fungi and their relevance to plant pathology. Mol Plant Pathol 10:115–128. https://doi.org/10.1111/j.1364-3703.2008.00503.x
Peters S, Fuchs S, Bien S, Bußkamp J, Langer GJ, Langer EJ (2023) Fungi associated with stem collar necroses of Fraxinus excelsior affected by ash dieback. Mycol Progress 22:52. https://doi.org/10.1007/s11557-023-01897-2
Prospero S, Botella L, Santini A, Robin C (2021) Biological control of emerging forest diseases: How can we move from dreams to reality? For Ecol Manage 496:119377. https://doi.org/10.1016/j.foreco.2021.119377
Queloz V, Grünig CR, Berndt R, Kowalski T, Sieber TN, Holdenrieder O (2011) Cryptic speciation in Hymenoscyphus albidus. Forest Pathol 41:133–142. https://doi.org/10.1111/j.1439-0329.2010.00645.x
Rigling D, Prospero S (2018) Cryphonectria parasitica, the causal agent of chestnut blight: invasion history, population biology and disease control. Mol Plant Pathol 19:7–20. https://doi.org/10.1111/mpp.12542
Romeralo Tapia C, Botella L, Santamaría O, Diez J (2012) Effect of putative mitoviruses on in vitro growth of Gremmeniella abietina isolates under different laboratory conditions. For Syst 21:515. https://doi.org/10.5424/fs/2012213-02266
Sasaki A, Onoue M, Kanematsu S, Suzaki K, Miyanishi M, Suzuki N, Nuss DL, Yoshida K (2002) Extending chestnut blight hypovirus host range within diaporthales by biolistic delivery of viral cDNA. MPMI 15:780–789. https://doi.org/10.1094/mpmi.2002.15.8.780
Schoebel CN, Zoller S, Rigling D (2014) Detection and genetic characterisation of a novel mycovirus in Hymenoscyphus fraxineus, the causal agent of ash dieback. Infect Genet Evol 28:78–86. https://doi.org/10.1016/j.meegid.2014.09.001
Schoebel CN, Botella L, Lygis V, Rigling D (2017) Population genetic analysis of a parasitic mycovirus to infer the invasion history of its fungal host. Mol Ecol 26:2482–2497. https://doi.org/10.1111/mec.14048
Schoebel CN, Prospero S, Gross A, Rigling D (2018) Detection of a conspecific mycovirus in two closely related native and introduced fungal hosts and evidence for interspecific virus transmission. Viruses. https://doi.org/10.3390/v10110628
Shamsi W, Kondo H, Ulrich S, Rigling D, Prospero S (2022) Novel RNA viruses from the native range of Hymenoscyphus fraxineus, the causal fungal agent of ash dieback. Virus Res 320:198901. https://doi.org/10.1016/j.virusres.2022.198901
Shamsi W, Mittelstrass J, Kondo H, Ulrich S, Rigling D, Prospero S (2023) Possible biological control of ash dieback using the parasitic Hymenoscyphus fraxineus mitovirus 2? bioRxiv https://doi.org/10.1101/2023.03.03.530786
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. https://doi.org/10.1038/msb.2011.75
Ulrich K, Becker R, Behrendt U, Kube M, Ulrich A (2020) A comparative analysis of ash leaf-colonizing bacterial communities identifies putative antagonists of Hymenoscyphus fraxineus. Front Microbiol 11:966. https://doi.org/10.3389/fmicb.2020.00966
Urayama S-I, Sakoda H, Takai R, Katoh Y, Le Minh T, Fukuhara T, Arie T, Teraoka T, Moriyama H (2014) A dsRNA mycovirus, Magnaporthe oryzae chrysovirus 1-B, suppresses vegetative growth and development of the rice blast fungus. Virology 448:265–273. https://doi.org/10.1016/j.virol.2013.10.022
Vainio EJ, Pennanen T, Rajala T, Hantula J (2017) Occurrence of similar mycoviruses in pathogenic, saprotrophic and mycorrhizal fungi inhabiting the same forest stand. FEMS Microbiol Ecol. https://doi.org/10.1093/femsec/fix003
Walker PJ, Siddell SG, Lefkowitz EJ, Mushegian AR, Adriaenssens EM, Alfenas-Zerbini P, Dempsey DM, Dutilh BE, García ML, Curtis Hendrickson R, Junglen S, Krupovic M, Kuhn JH, Lambert AJ, Łobocka M, Oksanen HM, Orton RJ, Robertson DL, Rubino L, Sabanadzovic S, Simmonds P, Smith DB, Suzuki N, van Doorslaer K, Vandamme A-M, Varsani A, Zerbini FM (2022) Recent changes to virus taxonomy ratified by the International Committee on Taxonomy of Viruses (2022). Arch Virol 167:2429–2440. https://doi.org/10.1007/s00705-022-05516-5
Wu MD, Zhang L, Li GQ, Jiang DH, Hou MS, Huang H-C (2007) Hypovirulence and double-stranded RNA in Botrytis cinerea. Phytopathology 97:1590–1599. https://doi.org/10.1094/PHYTO-97-12-1590
Wu M, Zhang L, Li G, Jiang D, Ghabrial SA (2010) Genome characterization of a debilitation-associated mitovirus infecting the phytopathogenic fungus Botrytis cinerea. Virology 406:117–126. https://doi.org/10.1016/j.virol.2010.07.010
Xie J, Ghabrial SA (2012) Molecular characterization of two mitoviruses co-infecting a hypovirulent isolate of the plant pathogenic fungus Sclerotinia sclerotiorum corrected. Virology 428:77–85. https://doi.org/10.1016/j.virol.2012.03.015
Xie J, Jiang D (2014) New insights into mycoviruses and exploration for the biological control of crop fungal diseases. Annu Rev Phytopathol 52:45–68. https://doi.org/10.1146/annurev-phyto-102313-050222
Xu Z, Wu S, Liu L, Cheng J, Fu Y, Jiang D, Xie J (2015) A mitovirus related to plant mitochondrial gene confers hypovirulence on the phytopathogenic fungus Sclerotinia sclerotiorum. Virus Res 197:127–136. https://doi.org/10.1016/j.virusres.2014.12.023
Yang S, Dai R, Salaipeth L, Huang L, Liu J, Andika IB, Sun L (2021) Infection of two heterologous mycoviruses reduces the virulence of Valsa mali, a fungal agent of apple Valsa Canker disease. Front Microbiol 12:659210. https://doi.org/10.3389/fmicb.2021.659210
Zhong J, Pang XD, Zhu HJ, Da Gao B, Huang WK, Zhou Q (2016) Molecular characterization of a trisegmented mycovirus from the plant pathogenic fungus Colletotrichum gloeosporioides. Viruses. https://doi.org/10.3390/v8100268
Acknowledgements
We would like to acknowledge the Northwest German Forest Research Institute (NW-FVA) for providing plant samples, Vanessa Reckemeyer for technical assistance in the isolation of H. fraxineus, as well as Nina Gruschwitz for assistance in the establishment and monitoring of saplings in the virulence trial. The authors also thank Waldklimafonds (WKF) of the Fachagentur für Nachwachsende Rohstoffe e.V. for financial support.
Funding
Open Access funding enabled and organized by Projekt DEAL. This project is financed by the Agency for Renewable Resources (FNR) in the program “Waldklimafonds” [Forest and Climate Fund] (2219WK22B4, 2219WK22F4 and 2219WK22G4) funded by the German Federal Ministry of Food and Agriculture and the German Federal Ministry for Environment, Nature Conservation and Nuclear Safety.
Author information
Authors and Affiliations
Contributions
Tobias Lutz, Maia Ridley, Barbara Schulz, Rasmus Enderle, Michael Steinert and Cornelia Heinze were involved in conceptualization; Tobias Lutz, Maia Ridley and Birgit Hadeler assisted with methodology; Tobias Lutz and Maia Ridley carried out formal analysis and investigation; Tobias Lutz, Maia Ridley, Rasmus Enderle and Cornelia Heinze wrote and prepared the original draft; Tobias Lutz, Maia Ridley, Barbara Schulz, Rasmus Enderle and Cornelia Heinze wrote—reviewed and edited—the manuscript; and Michael Steinert, Rasmus Enderle and Cornelia Heinze acquired the funding.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animal performed by any of the authors.
Consent to participate
Not applicable.
Consent for publication
I, the undersigned, give my consent for the publication of identifiable details, which can include photograph(s) and/or videos and/or case history and/or details within the text (“Material”) to be published in the above journal and article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lutz, T., Ridley, M., Hadeler, B. et al. Evaluation and identification of viruses for biocontrol of the ash dieback disease. J Plant Dis Prot (2023). https://doi.org/10.1007/s41348-023-00804-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41348-023-00804-x