
Abstract
Magnetic activated carbon (MG-AC), a solid product made by dispersing magnetic substrates on AC, is gaining attention for the removal of heavy metals from wastewater due to its favorable physico-chemical properties such as enhanced surface area and magnetic properties, respectively. However, the effects of two contrasting substrates, i.e., metal solution and metal particles, for the synthesis of MG-AC to obtain enhanced magnetic property have not been considered. The MG-AC was prepared by incorporating Fe3O4 into the AC from two different sources of iron: Fe3O4 extracted from electric arc furnace slag and from a ferric chloride/ferrous sulfate solution, to produce magnetic palm kernel shell from slag (MG-PKSS) and magnetic palm kernel shell from iron suspension (MG-PKSF), respectively. The adsorbent samples were characterized using Fourier transform infrared spectroscopy (FTIR), thermogravimetric analysis, X-ray diffraction, electron microscopy, i.e., SEM and FESEM, energy-dispersive X-ray, nitrogen adsorption, vibrating sample magnetometer. The results showed that the MG-PKSF had a greater BET surface area of 257 m2 g−1, a pore volume of 0.1124 cc g−1 and higher magnetic properties with a magnetic saturation of 49.55 emu g−1 relative to the MG-PKSS. The FTIR spectrum of the MG-PKSF illustrated the intense OH bending at 1629 cm−1 which can be attributed to the presence of oxygen in the samples. The absorption bands at 1093 and 579 cm−1 indicated the presence of C–O stretching and metal–oxygen (M–O) bands due to the interaction of iron and oxygen. Therefore, the MG-PKSF presented better characteristics for heavy metal removal from wastewater relative to the MG-PKSS, thereby suggesting that the raw material (metal solution) for impregnation played a crucial role in enhancing the quality of the MG-AC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Palm kernel shells from the palm tree (Elaeis Guineensis) have been widely used in the production of AC due to its high carbon content and low organic content besides its availability in the Southeast Asia [1, 2]. Several investigators [3,4,5] have demonstrated that a high quality of synthesized activated carbon can be obtained using PKS waste. The raw material of AC is carbonized by thermal decomposition or pyrolysis followed by the activation process at higher temperatures in a furnace. The developed AC usually exhibits high BET surface area, greater pore volume and smaller pore size which may enhance its adsorption capacity; hence, an excellent adsorption of adsorbate on the adsorbent surface can be achieved [6]. Large sorption capacity in micropores can be achieved by chemical or physical activation which can also result in wider micropore openings.
In particular, magnetic activated carbon (MG-AC) adsorbents exhibit magnetic characteristics with great efficiency for the adsorption of contaminants from aqueous solutions [7]. The MG-AC is developed by either chemically or physically dispersing the magnetic particles onto the surface of the porous AC [8, 9]. The high specific surface areas of MG-AC indicate the presence of micropores which enhance the adsorption efficiency of organic compounds and heavy metal ions onto the surface [10]. Further, adsorbents containing micropores exhibit higher surface area and hence contribute toward the increase in adsorption capacity. Interestingly, a magnetic separator can be utilized to separate the MG-AC from a highly concentrated reaction solution. Various types of magnetic adsorbents such as magnetic ion exchange resins [11], magnetic zeolites [12], magnetic activated carbon fibers [13,14,15] and much more have been used to remove contaminants, especially from water [2].
Conversely, commercial activated carbons (CAC) are expensive due to the utilization of coal which is an expensive starting material [16]. Palm kernel shells are rather cheaper compared to some other agricultural by-products. Further, in recent years, there is an increased interest in the production of AC from renewable sources, with a substantial supply of cost-saving industrial and agricultural by-products such as almond shells [17], bamboo [18], apricot shells [19], sugarcane lignin (Ribeiro et al. 2006), Brewer’s spent grain lignin [20] and oil palm shell [21].
Since the palm kernel shells have been widely used as AC, the product can be modified and converted to MG-AC. The MG-AC exhibits a magnetic characteristic and has demonstrated to be effective for adsorption in dilute solutions. Additionally, MG-AC is reported to exhibit a high specific surface area due to the presence of microporous structure and hence contributes to a high adsorption of soluble organic compounds and heavy metals [22]. Despite performing a normal adsorption procedure, the magnetic separation technology has been developed to manifest a rapid separation to separate the adsorbents from an aqueous medium.
In this study, MG-AC was prepared by reacting AC from palm kernel shell with two substrates. Substrate A consisted of particles from electric arc furnace (EAF) slag, and substrate B consisted of a metal solution prepared by using ferrous sulfate/ferric chloride suspension. The raw EAF slag can be used as an inexpensive absorbing agent in the treatment process, especially for wastewaters burdened with metallic ions [23]. On the other hand, studies have used raw electric furnace slag, which was not supported on AC for adsorption studies [23], thereby having the potential to result in secondary pollution [24, 25]. Again the studies mentioned above did not look into the magnetic properties of their electric furnace slag. Similarly, a previous study [26] had reported the characterization of municipal solid waste and biochar [27]; however, the characterization of synthesized palm kernel shell AC in tandem with magnetization from two substrates displaying varying degrees of magnetic properties for reuse in wastewater treatment has not been elucidated. In this study, the PKSAC was synthesized and converted into a MG-AC (MG-PKSF and MG-PKSS) by impregnating the MG-AC with two different sources of iron to result in greater dispersion and magnetization for wastewater purification.
Therefore, it is hypothesized that the MG-AC synthesized will present better characteristics for the greater adsorption of contaminants relative to the non-magnetic activated carbon due to its strong magnetism and colloidal property which enables it to be well dispersed in a solution [28]. The objectives of the study are: (a) to synthesize a high surface area AC using palm kernel shells, (b) to prepare MG-AC for the removal of heavy metals from water and (c) to characterize the physical and chemical properties of the prepared AC and MG-AC.
Materials and methods
Chemicals and reagents
In this study, the reagents used to pre-treat and impregnate the raw materials were phosphoric acid, H3PO4 (85 wt%), purchased from Merck. For the preparation of magnetic activated carbon, iron (III) chloride (>96%) was purchased from Sigma-Aldrich, iron(II) sulfate (99.5%) from Qrec.
Preparation of activated carbon
The raw palm kernel shells used in this study were obtained from an oil palm estate located at Jalan Sawah, Pekan Nenas, Johor. The palm kernel shells (100 g) were utilized for the preparation of the activated carbon. The raw material (50 g) was ground and sieved to particle sizes between 75 and 250 μm, soaked and impregnated with 10 mL of 30 wt% of phosphoric acid, H3PO4, at room temperature as a pre-treatment procedure with the ratio of PKS to acid as 1:1. The purpose of the pre-treatment was to destroy the lignin and cellulose present in the raw PKS [29]. The sample was left impregnated for 24 h, washed with distilled water and dried at room temperature.
A weighed amount (10 g) of the pre-treated palm kernel shells were transferred into five conical flasks containing different concentrations of H3PO4, i.e., 10% wt/wt., 20% wt/wt., 30% wt/wt., 40% wt/wt. and 50% wt/wt., respectively [29]. The pre-treated PKS was again treated using dilute acid (10, 20 and 30% wt/wt H3PO4) to determine the resulting surface area at which if the surface area obtained using dilute acid is considered high, thus the utilization of concentrated acid can be reduced and the experiment will be more cost-effective. The palm kernel shells in the five conical flasks were impregnated with an aqueous solution of H3PO4 at a ratio of 1:1. This ratio implied the use of 10 g of raw palm kernel shells soaked in 10 g of H3PO4 for 24 h. The excess acid was then filtered, and the soaked palm kernel shells were placed in a muffle furnace and heated at 200 °C for 30 min to initiate the carbonization process.
The temperature of the furnace was increased to the range of 400–550 °C and held for 2 h followed by cooling to room temperature. A total of four activation temperatures were studied, i.e., 400, 450, 500 and 550 °C. The samples were thoroughly washed and rinsed using vacuum filtration with hot distilled water to remove all the excess acid until the pH of the filtrate was approximately 7. The samples were dried in the oven at a temperature of 110 °C for 24 h. The activated carbons were then stored in desiccators for further characterization studies.
Preparation of magnetic palm kernel shell activated carbon
The preparation of magnetic palm kernel shell activated carbon (MG-PKSAC) was achieved by utilizing two sources of iron: a suspension of ferric chloride/ferrous sulfate and iron oxide extracted from electric arc furnace (EAF) slag. A graphical overview of the procedures employed to prepare the MG-AC is represented in Fig. 1.

Schematic representation of the procedures employed to prepare the MG-AC
Preparation of magnetic palm kernel shell activated carbon from ferric chloride/ferrous sulfate solution
The palm kernel shells activated carbon (PKSAC) (50 g) was suspended in 500 mL distilled water. A ferric chloride solution (FeCl3·6H2O) was freshly prepared by adding 18 g ferric chloride into 1300 mL distilled water, while a ferrous sulfate solution (FeSO4·7H2O) was prepared by adding 20 g ferrous sulfate into 150 mL distilled water. Both solutions were mixed in a 2-L beaker and heated to a temperature of between 60 and 70 °C followed by vigorous stirring using a magnetic stirrer. The suspension formed from ferric chloride/ferrous sulfate solution was added to the previous aqueous suspension of PKSAC and was stirred slowly at room temperature for 30 min to ensure proper mixing. After mixing, 10 M of NaOH solution was added dropwise into the suspension until it attained a pH of between 10 and 11 and was thereafter left for approximately 60 min. The activated carbon suspension was aged without stirring at room temperature for 24 h, and the suspended materials obtained from the reaction between PKSAC and iron suspension were repeatedly washed with distilled water and ethanol until the pH of the filtrate was approximately 7. The MG-PKSF produced was vacuum filtered and dried at 50 °C in the oven overnight.
Preparation of magnetic activated carbon from electric arc furnace (EAF) slag
The electric arc furnace slag used was ground to a particle size between 75 and 250 µm. The powdered slag obtained (10 g) was mixed with 150 mL of 6 M NH4Cl in a 250-mL round-bottom flask. Ten grams of the powdered slag was used to obtain approximately 3.73 g of iron oxide. The solution was refluxed for 4 h in a fume cupboard and was left to cool at room temperature. The 50 mL of refluxed solution was transferred into a conical flask containing PKSAC, followed by the addition of 30 mL of 5 M NaOH. The mixture was then vacuum filtered to remove water. The solid obtained denoted as MPKSS was then calcined at temperatures ranging from 800 to 900 °C for 2 h in the carbolite muffle furnace. Additionally, the same procedure was repeated with the absence of PKSAC to obtain the iron oxide from two different sources (FeOS and FeOF) which were solely for the characterization purpose.
Characterization of activated carbon and magnetic activated carbon
The resulting AC and MG-AC were characterized by using the following instruments: Fourier transform infrared spectroscopy (FTIR), thermogravimetric analysis (TGA), X-ray diffraction (XRD), nitrogen adsorption analysis, electron microscopy (SEM and FESEM) and energy-dispersive X-ray spectroscopy (EDX). The magnetic saturation of iron oxide and synthesized magnetic activated carbon were determined using vibrating sample magnetometer (VSM), while the determination of particle size of iron oxide was conducted by particle size analyzer.
Fourier transform infrared spectroscopy (FTIR)
The functional groups present in the AC and MG-AC were determined using PerkinElmer Spectrum One FTIR Spectrometer. The samples were scanned in the range of 4000–400 cm−1. In this analysis, KBr pellets of the samples were prepared by mixing KBr powder with the samples at the ratio of 1:10. The mixture was then placed in a holder, and it was then compressed by applying a pressure of 10 tonnes for 2 min. Then, the transparent KBr pellet obtained was placed into the sample compartment for analysis.
Thermogravimetric analysis (TGA)
The thermogravimetric analysis was used to identify the mass change of the sample as a function of temperature. This analysis was conducted using PerkinElmer STA 8000 Simultaneous Thermal Analyzer equipped with Pyris software for data analysis. In this analysis, a small sample size was used in the range of 10 mg to 200 mg, which was heated in a small furnace capable of operating at temperatures ranging from 25 to 1600 °C. The weight sensor in the analyzer provides the detection in microgram level for weight change due to the sample loss or the oxidative gain. The purge gas used in this analysis was nitrogen at a flow rate of 100 mL/min. The analysis was carried out at a low decomposition rate which was at 20 °C/min.
X-ray diffraction (XRD)
X-ray diffraction analyzer (Bruker Advance D8) was used to characterize the crystalline properties of the PKSAC and both magnetic samples, i.e., MG-PKSF and MG-PKSS. The samples were ground into powder forms (75 μm) and then tightly packed into the sample holder for the analysis. The samples were scanned from 20.0° to 100.0° at a scan speed of 2° min−1. Then, the XRD diffractogram patterns for PKSAC and both magnetic activated carbons were obtained from the system.
Particle size analysis
The particle size analysis of the samples was carried out using Malvern Zetasizer Nano ZS90 at Technology Park, Bukit Jalil, Kuala Lumpur, Malaysia. The analyzer uses the dynamic light scattering technique to measure the size of materials in a range of 1 nm up to 3 microns. The procedure according to SOP indicates the use of a disposable polystyrene cuvette to place 1 mL or 1 g of the sample using distilled water as the solvent with 10-min sonication time. The particle size analysis of each iron oxide compounds was conducted based on their refractive index.
Nitrogen adsorption analysis
The specific surface area of prepared PKSAC and both MG-PKSAC samples was characterized using multipoint BET surface area. Before the analysis, two outgassing procedures which are at 90 °C (1 h) and 350 °C (4 h) were used to remove volatiles and vapors that may be adsorbed on the sample surfaces which may interrupt the measurement. The outgassing procedure is very important to obtain the required precision and accuracy of the specific surface area analysis. After the outgassing, the sample tube was removed, the stopper was then inserted, and the weight of the sample was recorded. The sample tube was attached to the volumetric apparatus, and the sample was evacuated, down to the specific pressure of between 2 Pa and 10 Pa.
The PKSAC and both MG-PKSAC were analyzed by nitrogen adsorption at 77 K using Quantachrome Autosorb Automated Gas Sorption system which is a surface area analyzer. An amount of 0.06 g of sample was transferred into the sample holder and the nitrogen gas flowed through the sample. Then, at 77 K, the gas was changed to liquid nitrogen and being adsorbed by the sample. From this analysis, some important information which was the multipoint surface area, pore volume, type of pore and pore size distribution was obtained.
Electron microscopy
The samples obtained from the development of AC and MG-AC procedures were analyzed using two types of electron microscopes which are scanning electron microscopy (SEM) and field emission scanning electron microscopy (FESEM).
Scanning electron microscopy (SEM)
Scanning electron microscope is widely used to identify the microstructure of organic and inorganic materials. The morphology of the PKSAC and MG-AC samples was examined with a Phenom G2 Pro SEM using secondary electron detector at Technology Park, Bukit Jalil, Kuala Lumpur, Malaysia. The samples were placed onto carbon tape which was affixed onto an aluminum stub followed by gold coating. The sample was loaded into the SEM holder, and then, the sample holder stubs were placed into the mounting holes and the monitor was turned on. The working distance was adjusted using focus knob, and the selected image was captured. In this study, the magnifications used for the analysis of activated carbon and magnetic activated carbon were in range of 1550× up to 13000×.
Field emission scanning electron microscopy (FESEM)
Field emission scanning electron microscopy (FESEM) was used to illustrate the surface morphology of the samples analyzed. The micrographs of the sample were obtained from the analysis of samples using field emission scanning electron microscopy Zeiss Supra 35 VP. The AC and MG-AC were well dried and ground to powdered forms. The samples were then placed onto aluminum stubs by means of carbon tape placed on the stub. The PKSAC and MG-PKSF were gold coated to prevent charging. The stubs were then placed into the vacuum chamber of the instrument and analyzed at magnifications in the ranges of 1500× to 5000×.
Energy-dispersive X-ray (EDX) analysis
Energy-dispersive X-ray analysis was used to examine the elemental analysis on the surface of the samples analyzed previously using FESEM. The FESEM and EDX analysis used the same instrument, but both analyses have different roles in which FESEM illustrated the image of the sample morphology, while EDX gave the specific elements that existed on the surface of the samples analyzed.
Vibrating sample magnetometer (VSM)
A vibrating sample magnetometer (VSM) was used to measure the magnetic saturation of iron oxide, MG-PKSF and MG-PKSS. In this analysis, the magnetic moment of the samples should be larger than 10−6 emu in order to be detected by the VSM analyzer. A small sample size with vertical dimension provided more accurate result compared to the baseline dimension of the detection coils. It is recommended that the samples be located at an offset of 35 mm. The height of the coil set should be 40 mm so that the offset was placed at the end of the sample holder, 5 mm above the puck surface. The sample was placed in a sample holder located in the optical magnetomechanical device (OMMD). The power supply was turned on, and the voltage was adjusted to 3 V.
Results and discussion
Physical properties
Surface area analysis
The iron oxide extracted from both the raw slag and iron suspension showed very low surface areas of 6.58 and 8.73 m2 g−1, respectively. Similarly, the raw palm kernel shells also exhibited a small surface area of 6.84 m2 g−1. After activation, the surface area of PKSAC increased to 1225 m2 g−1 at 50% phosphoric acid (H3PO4) concentration. Figure 2 shows that as the concentration of acid decreased from 40 to 10%, the surface area of PKSAC produced was reduced to 901.83 to 319.86 m2 g−1. Thus, the concentration of acid used is significant in the production of high surface area activated carbon. The pre-treatment step using H3PO4 impregnation in the production of AC is crucial in order to destroy the lignin structure in the raw PKS and reduce the crystallinity of cellulose and the acid which is also capable of increasing the porosity of the raw PKS itself.

Comparison surface area of raw PKS and PKSAC with 10, 20, 30, 40 and 50% acid concentration
As shown in Table 1, the surface area of commercial AC (CAC) is 846.15 m2 g−1. The magnetic CAC obtained upon magnetization using ferric chloride/ferrous sulfate solution shows a reduction to 833.73 m2 g−1. However, the reduction of the surface area was drastic to 9.64 m2 g−1 for magnetic CAC produced upon incorporation with EAF slag (MG-CACS). The surface areas of both magnetic samples, MG-CACS and MG-CACF, were lower than that of the CAC due to the coverage of iron oxide on the surface and pore blocking during the magnetization process. Additionally, MG-CACS used EAF slag as the source of iron oxide. Previous studies on the extraction of EAF slag by Yildirim, Prezzi [30], had reported that apart from the extraction of iron oxide, other oxide compounds (CaO, SiO2, Al2O3, MgO, SO3, MnO, Cr2O3, TiO2, P2O5, Na2O, K2O, Zn) were also extracted. Thus, the presence of these elements possibly contributed to pore blocking on MG-CACS surface.
Figure 2 shows the surface area of all PKS products developed in this study. PKSAC shows a very large surface area of 1225 m2 g−1 compared to raw PKS (6.84 m2 g−1). Upon magnetization, the surface areas of PKS impregnated with EAF slag (MG-PKSS) and the PKS impregnated with ferric chloride/ferrous sulfate solution (MG-PKSF) decreased to 79.18 and 257.86 m2 g−1, respectively. This can be attributed to the covering on the surface of the activated carbon produced by iron oxide during the impregnation process.
Pore size distribution analysis
The multipoint BET analysis was used to determine the specific surface area and the type of pores present on the surface of the adsorbents. The results obtained from the nitrogen adsorption multipoint BET analysis are tabulated in Table 2. The PKSAC exhibited a large specific surface area and total pore volume, but however displayed a lower average pore diameter and average pore size as compared to MG-PKSF. The large pore volume exhibited by PKSAC was produced upon the chemical activation process using 50% wt/wt. of phosphoric acid at a temperature of 500 °C. As reported by Liu et al. [31], the utilization of any chemical activation and thermal treatment on a carbon raw material has a potential to promote an effective adjustment on the porosity of the resulting activated carbon.
Studies have reported that the development of pores on activated carbon surface is also promoted by the high activation temperature at 500 °C, thus enhancing the reaction between the diffused phosphoric acid with activated carbon [32]. Hence, the development of more pores on activated carbon surface contributed to a larger specific surface area of the adsorbent, simultaneously. Meanwhile, the magnetized sample of activated carbon, MG-PKSF, exhibited a smaller specific surface area, total pore volume with larger average pore diameter and pore size. The specific surface area of MG-PKSF decreased to about 78.95% compared to the surface area of PKSAC. This may be due to the pore blockage owing to the iron oxide particles that entered or reside in the pore of the MG-PKSF which resulted to the inaccessibility of nitrogen gas into the micropores on MG-PKSF (Fig. 3).

Surface area of raw PKS, PKSAC, MG-PKSS and MG-PKSF
The activated carbon, PKSAC, all exhibited various sizes and diameters of pores developed on its surface. From the multipoint BET analysis results, the average type of pores formed in the activated carbon was found to be micropores, while mesopores also existed. The average pore diameter confirms that both PKSAC and MG-PKSF adsorbents exhibited mesopores as evidenced by the pore diameter in the size range of 2–50 nm. Thus, the mesopores in PKSAC and MG-PKSF are in the size range of 2.05 and 3.39 nm, respectively.
To understand the type of pores obtained after the activation process, Fig. 4a shows the nitrogen adsorption/desorption isotherm for PKSAC, which illustrates Type 1 adsorption isotherm pattern. The adsorption process for the adsorbent shows a rapid initial increase and a long nearly flat region as the relative pressure increased and is considered as a micropore filling mechanism on the PKSAC. At a lower pressure, the adsorbate molecules encounter an overlapping adsorption pattern on the porous walls, hence indicating a greater quantity of adsorbate up to a relative pressure of 0.4 which indicates the monolayer adsorption of gas molecules on the surface of PKSAC. The adsorption at higher pressure elucidates the pore filling by the adsorbed or condensed adsorbate which is attributed to the plateau, indicating an insignificant or no further adsorption attaining a maximum with respect to micropore filling [33]. Therefore, the adsorption process for both adsorbents was found to follow the Langmuir adsorption isotherm and authenticates the presence of micropores and small mesopores with the average pore diameter of 2.05 nm (Fig. 4b).


a Nitrogen adsorption/desorption isotherm of PKSAC. b BJH desorption pore size distribution graph of PKSAC. c Nitrogen adsorption/desorption isotherm of MG-PKSF. d BJH desorption pore size distribution graph of MG-PKSF. e BET adsorption plot of PKSAC. f BET adsorption plot of MG-PKSF. g The t-plot of PKSAC. h The t-plot of MG-PKSF
To understand the type of pores obtained after the magnetization process, the nitrogen adsorption/desorption isotherm and pore size distribution of MG-PKSF adsorbent are illustrated in Fig. 4c, d, respectively. The curve portrays Type IV isotherm which implies an adsorption on a porous adsorbate with pore diameter greater than micropores. The pore size distribution of MG-PKSF elucidated in Fig. 4d reveals the average pore diameter to be 3.39 nm, which can be classified as mesopores. This result is in agreement with Fig. 4c which illustrates that the adsorption onto MG-PKSF was in the mesopores with the completion of first adsorbed monolayer at a relative pressure around 0.1 followed by second and higher adsorption level at higher relative pressure until the maximum saturation of adsorbed layer is achieved [33].
To explain the greater adsorption by the PKSAC, the graphs of the adsorption data in Fig. 4e, f were plotted using the BET equation (equation not shown). The linear relationship between 1/Q[(Po/P) − 1] and P/Po provides the information on the quantity of nitrogen gas adsorbed onto the surfaces of both PKSAC and MG-PKSF. The slope and intercept values are used to determine the quantity of nitrogen adsorbed in a monolayer by using BET equation (equation not shown). Thus, this implied that the PKSAC with larger BET surface area exhibited a greater adsorption capacity on the surface compared to the MG-PKSF.
The t-plot analysis
The micropore volume and micropore surface area are determined using the t-plot analysis. In this study, the quantity of nitrogen gas adsorbed was plotted against the statistical thickness of the adsorbed nitrogen film, t graph is plotted for both PKSAC and MG-PKSF adsorbents as shown in Fig. 4g, h, respectively. The values of the statistical thickness are calculated using BET equation (equation not shown). As illustrated in both graphs, the straight line is drawn to indicate the linearity of the thickness value, t, which is in the range of 0.31–0.49 nm for PKSAC and in the range of 0.31–0.50 nm for MG-PKSF. From the straight line, the slope is the value used for the external surface area, while the y-intercept provides the value for the calculation of micropore volume.
The values of the micropore volume, micropore surface area, external surface area, percentage of mesoporosity and percentage of microporosity were evaluated, and the results obtained are tabulated in Table 3. Previously, the average pore diameter of both PKSAC and MG-PKSF were shown to be in the range of 20–30 Å. This implied that macropores are deemed to be absent in both samples. Table 3 shows that the micropore surface area, SMP, for PKSAC (593.33 m2 g−1) is smaller than its external surface area, Sext (631.67 m2 g−1), while the SMP (181.56 m2 g−1) of MG-PKSF is much higher than its Sext (76.30 m2 g−1).
A higher SMP value compared to Sext indicates the presence of a predominantly microporous surface [34]. Thus, this indicates that the micropore dominates the surface of the MG-PKSF upon the incorporation of Fe3O4 iron oxide with MG-PKSF compared to the untreated PKSAC. In addition, a lower micropore volume of MG-PKSF (0.09 cc/g) may infer that the pores of MG-PKSF are nearly filled by Fe3O4 as compared to the untreated PKSAC (0.18 cc/g).
Zetasizer particle size analysis
The particle sizes of the samples were analyzed using Malvern Zetasizer Nano Series. The selected samples which are iron oxide extracted from ferric chloride/(FeOS) and magnetic PKS activated carbon using ferric chloride/ferrous sulfate solution (MG-PKSF) were studied in order to determine the size distribution of the elements in the samples.
Figure 5a illustrates the presence of three peaks which indicates three different sizes of Fe3O4 with a domain particle size of 162.3 nm followed by 966.4 and 4719.0 nm. It shows that the average size distribution of Fe3O4 particle in FeOF is 245.7 nm as determined by Zetasizer. Based on XRD analysis, the crystallite size of Fe3O4 was found to be 14.41 nm, as determined by Scherrer equation calculations. The particle size determined by Zetasizer indicates that the Fe3O4 particles in FeOF are agglomerated, indicating interactions with distilled water used during the analysis which must have resulted from magnetic and hydration forces [35]. The magnetic force exerts the attraction between Fe3O4 particles, while the hydration force serves as a repulsion of the hydrogen bond between water molecules and the surface of Fe3O4, which can be attributed to the agglomeration of Fe3O4 particles in the aqueous media.


a Size distribution graph of Fe3O4 particles in FeOF. b Size distribution graph of γ-Fe2O3 particles in FeOF. c Size distribution graph of α-Fe2O3 particles in FeOS. d Size distribution graph of γ-Fe2O3 particles in FeOS. e Size distribution graph of Fe3O4 particles in FeOS. f Size distribution graph of carbon, Fe3O4, γ-Fe2O3 and α-Fe2O3 in MG-PKSF
Figure 5b illustrates the graph for γ-Fe2O3 in FeOF which shows two significant peaks at 384.8 and 4021.0 nm, respectively. The average size distribution of γ-Fe2O3 particle is 236.4 nm. From both figures, it shows that the size distribution of Fe3O4 particles is slightly larger compared to particles of γ-Fe2O3. However, the difference in size distribution between these compounds is not too significant.
The size distribution for α-Fe2O3 in FeOS portrayed in Fig. 5c shows two significant peaks at 788.8 and 130.8 nm with the average size of 940.3 nm. Figure 5d shows the size distribution graph for γ-Fe2O3 in FeOS which illustrates three sharp peaks assigned for 266.8, 1198.0 and 5391.0 nm, respectively, with the average size of 1045 nm. In addition, Fig. 5e demonstrates that the Fe3O4 particles in the FeOS present in the sizes of 153.8, 1131.0 and 5337.0 nm, respectively. The results implied that the particle sizes in FeOS sample are significantly larger that in FeOF. This suggests that the aggregation of compounds in FeOS, i.e., α-Fe2O3, γ-Fe2O3 and Fe3O4, is deemed substantial compared to the FeOF, i.e., γ-Fe2O3 and Fe3O4.
Figure 5f shows the size distribution of carbon, Fe3O4, γ-Fe2O3 and α-Fe2O3 which are present in MG-PKSF sample. The average particle size for carbon, Fe3O4, γ-Fe2O3 and α-Fe2O3 is 160.7, 158.3, 168.4 ad 157.3 nm, respectively. Hence, from the cumulative value obtained, the particles present in MG-PKSF are in homogeneous size which is in a range of 157.3 to 168.4 nm. Similarly, it also illustrates that MG-PKSF sample was probably well dispersed in water during the analysis despite the agglomeration of the particles which is due to the action of interaction forces between the particles and the aqueous reaction medium.
Electron microscopy analysis
Scanning electron microscopy (SEM) and field emission scanning electron microscopy (FESEM) analysis was used to illustrate the surface morphology of the samples in the production of activated carbon and magnetic activated carbon. SEM analysis was used to illustrate the morphology of raw PKS, iron oxide extracted from raw slag (FeOS), iron oxide extracted from ferric chloride/ferrous sulfate solution (FeOF), MG-PKSS and also MG-CACS. Meanwhile, palm kernel shell activated carbon (PKSAC) and magnetic activated carbon prepared by using ferric chloride/ferrous sulfate solution (MG-PKSF) are characterized using FESEM. The micrographs obtained from both analyses show the surface morphology and also the porosity of the samples analyzed.
Figure 6 depicts the image of raw palm kernel shell (PKS) before the activation process under magnification (a) 1000× and (b) 3800×. The sample portrays an even and flat surface without any presence of pores upon analysis at higher magnification. Figure 6c shows the morphology of PKS after the activation process with 50 wt% phosphoric acid at 500 °C to produce activated carbon (PKSAC) under magnification of 1000x. As seen on the micrograph, there are abundant pores developed on the surface of activated carbon. The pores are developed randomly on the surface of the activated carbon with various diameters and width of external pores. Hence, PKSAC displayed a larger surface area and pore volume as more pores were created on its surfaces compared to the raw PKS.


Micrograph of raw PKS under magnification a ×1000 and b ×3800. c Micrograph of PKSAC at magnification of ×1000. Micrograph of FeOS under magnification d ×5400 and e ×13000. Micrograph of FeOF under magnification f ×5400 and g ×10000. h Micrograph of MG-PKSS under magnification ×5000. i Micrograph of MG-PKSF under magnification ×5000. j Micrograph of MG-CACS under magnification ×3000x. k Micrograph of MG-CACS under magnification 2700x
This implies that the pores on PKSAC are either micropore or mesopore as corroborated by nitrogen adsorption data with 51.56% mesoporosity compared to its microporosity (48.44%). However, there are some small white particles scattered on the surface and in the pores of activated carbon which presumably can be assigned to the impurities of ash formed during the carbonization process that was not washed away completely during the washing process.
The morphology of FeOS under magnification 5400× illustrated in Fig. 6f indicates a homogeneous surface with a significantly similar size of FeOS particles. FeOS particles are uniformly scattered on the carbon tape which implied that the particles were likely to be very fine particles, having tiny sizes of less than 0.5 μm. Moreover, under a higher magnification of 13000× demonstrated in Fig. 6g, the surface morphology of FeOS showed the presence of α-Fe2O3 which presented a needle-like shape observed between the scattering of FeOS particles. A similar observation has been reported by Kim et al. [36] whose study revealed that α-Fe2O3 exists in the worm-like shape. Even though the XRD diffractogram of the MG-AC using iron oxide extracted from EAF slag (FeOS) illustrates some peaks assigned to γ-Fe2O3 and Fe3O4, the micrograph shows that the structure and shape are quite similar to each other; hence, it is difficult to differentiate between them.
Figure 6f shows the surface morphology of FeOF at 5400×, while a micrograph obtained at a higher magnification of 10000× is illustrated in Fig. 6g. The micrographs show that the particle size of FeOF is not homogeneous with the presence of irregular particle shapes. Fe3O4 is known to exist in various crystal shapes such as cubic, octahedral, octahedron, dodecahedron, while the crystals are opaque [37]. Hence, the micrograph of FeOF implied that the presence of crystal shape with the opaque transparency can be assigned to Fe3O4.
In Fig. 6h, MG-PKSS developed from the incorporation of iron oxide extracted from slag with palm kernel shell activated carbon (PKSAC), shows the presence of needle-like flakes on the surface of activated carbon which can be assigned to the presence of α-Fe2O3 in the sample upon the analysis at magnification 5000×. This observation is in good agreement with the observation reported by Tadić et al. [38]. Furthermore, the presence of some opaque crystallites was also observed, which are assigned to Fe3O4 or γ-Fe2O3.
Similarly, Fig. 6i depicts the morphology of MG-PKSF produced from the reaction of palm kernel shell activated carbon (PKSAC) with ferric chloride/ferrous sulfate solution under the magnification of 5000×. It is observed that some pores are slightly occupied with Fe3O4 or γ-Fe2O3 particles during the magnetization procedure. The external pores of MG-PKSF are larger compared to the external pores of PKSAC, which may be attributable to the blockage of micropores upon incorporation with iron oxide, hence contributing to the presence of residual mesopores on MG-PKSF instead of the presence of micropores. The iron oxide particles scatter freely by either total or partial coverage on the porous PKSAC surface, hence resulting in the surface area of the MG-PKSF becoming smaller as well as its pore volume compared to that of the PKSAC.
Further analysis to observe the morphology of MG-CACS was carried out due to its very low surface area as stated previously in Table 1. The micrograph of MG-CACS obtained under magnification 3000× depicted in Fig. 6j portrays the coverage of iron oxide extracted from the slag (FeOS) on the surface of MG-CACS. As pointed out previously, the low surface area of MG-CACS may have resulted from the random scattering of FeOS over the pores of CAC. This assumption is corroborated by the micrograph which indicates the presence of FeOS particles on the surface of CAC. A micrograph with lower magnification (2700×) is illustrated in Fig. 6k to observe the coverage and scattering of FeOS over the surface of CAC upon the magnetization process which reduced the surface area of CAC.
Energy-dispersive X-ray analysis (EDX analysis)
Figure 7a, b shows the EDX spectrum and the summary for the elemental composition for both samples using energy-dispersive X-ray analysis (EDX) for PKSAC and MG-PKSF, respectively. The PKSAC spectrum revealed that the carbon content was greatest of 85.30%, followed by oxygen (13.32%) and phosphorus (1.37%). Meanwhile, the presence of phosphorus in the sample was attributed to the use of phosphoric acid during the chemical activation process. During activation, the activated carbon was washed repeatedly using distilled water to remove the excess phosphoric acid. However, the presence of phosphorus explains the presence of a residual phosphoric acid which was not washed completely.

a EDX spectrum for sample PKSAC. b EDX spectrum for sample MG-PKSF
The MG-PKSF spectrum indicates the presence of four main elements, namely carbon, oxygen, phosphorus and iron. The composition of iron was the highest of 49.45%, which was attributed to the iron oxide precipitated on the surface of activated carbon during the magnetization process. Hence, the composition of oxygen also increased simultaneously with the iron. In addition, the composition of carbon and phosphorus in MG-PKSF decreased as compared to PKSAC due to the coverage of iron oxide on its surface which reduced the composition of both elements.
Chemical properties
Fourier transform infrared spectroscopy (FTIR) analysis
The prepared activated carbon was characterized using FTIR spectroscopy. The FTIR spectroscopy analysis was used to obtain information on the characteristics of the sorbent and sorbent–sorbate interactions. The spectrum of Fe3O4, commercial activated carbon (CAC), palm kernel shells activated carbon (PKSAC) and magnetic CAC are shown in Fig. 8a. The spectrum of CAC shows some peaks around 3430, 2921 and 2851, 1596 cm−1 with a small shoulder at 1109 cm−1 due to OH stretching, C–H stretching, OH bending and C–O functional groups, respectively. For MG-CACS and MG-CACF, a broad peak at 3458 and 3428 cm−1, as well as at 1634 and 1631 cm−1, respectively, is related to the OH functional groups.


a FTIR spectrum of PKSAC, CAC, Fe3O4, MG-CACS and MG-CACF. b FTIR spectrum of raw PKS and PKSAC. c FTIR spectrum of raw PKS, PKSAC, MG-PKSS and MG-PKSF
Additionally, a broad peak centered at 1056 and 1114 cm−1 indicates the presence of C–O bond in both samples, i.e., MG-CACS and MG-CACF. This broad peak was obtained from the overlapping of peaks existing in this region (Mohan et al., 2011). Further, the FTIR spectrum of Fe3O4 particles in the figure shows an intense peak at 552 cm−1 due to M–O band. Thus, the small peak at 529 cm−1 in MG-CACS and at 616 cm−1 in MG-CACF spectrum can be assigned to the Fe3O4 particles on the surface (Mohan et al., 2011). The FTIR spectrum of activated carbon synthesized from palm kernel shells (PKSAC) showed some peaks at wave numbers of 3434, 2910, 1610 and 1171 cm−1 which indicated the presence of functional groups such as OH stretching, C–H, OH bending and C–O groups, respectively, which coincided with those revealed by the CAC spectrum.
Figure 8b depicts the FTIR spectra of the raw palm kernel shell (PKS) and palm kernel shell activated carbon (PKSAC) developed from the preliminary experiment to observe the effect of carbonization and activation process on the functional groups of raw PKS. The spectrum illustrates that the functional groups of the untreated PKS exhibited a complicated spectrum which shows many peaks belonging to different functional groups. The raw PKS resulted in a finely similar spectrum to the spectrum of PKSAC with a broad peak present at 3437 and 3441 cm−1, respectively, which can be assigned to O–H bonding due to the bonded hydroxyl on the sample surfaces. Studies have found that the hydrogen bond existing in the raw PKS was reduced by phosphoric acid resulting into a reduction of hydrogen bond in PKSAC [39].
A shoulder peak observed in both spectra at 2927 and 2920 cm−1 can be assigned to C–H stretching as well as to C–H stretching in methyl group [29] for PKSAC and raw PKS, respectively. An intense peak at 1637 and 1626 cm−1 for raw PKS as well as for PKSAC indicates C=C for aromatic compound present in both samples. In addition, a peak exists in the raw PKS spectrum around 1513 cm−1 which was ascribed to the presence of secondary amide group [40] and a fairly significant band at 1249 cm−1 as well as a broadband at 1034 cm−1 which is associated with the C–O stretching of carboxylic acid, alcohol, phenol, ester and ether [41]. Raw PKS spectrum also reveals some weak bands in the range of 762–535 cm−1 which can be assigned to C–C stretching.
Meanwhile, the PKSAC spectrum in Fig. 8b elucidates the presence of two weak bands at 3841 and 3741 cm−1 which can be assigned to O–H stretching mode of the hydroxyl group [39]. The spectrum also revealed the presence of an overlapping broad peak at 1175 cm−1 which indicate C–O stretching of ester, ether and phenol as well as an intense band at 501 cm−1 which can be attributed to C–C stretching. Hence, this implies that the spectrum of raw PKS and PKSAC validates the fact that the main functional groups present in the raw and carbon samples are a carbonyl, ether, ester, alcohol and also phenol [29].
Figure 8c shows FTIR spectra of all PKS products including raw PKS, PKSAC and two magnetic products, MG-PKSS and MG-PKSF. The adsorption bands for all samples are summarized in Table 4. The spectra show that MG-PKSS and MG-PKSF revealed the O–H stretching band at 3441 and 3435 cm−1. This O–H group exists due to the presence of water molecules probably caused by semi-dried samples during the analysis.
The spectrum of MG-PKSS illustrates the presence a shoulder band at 2940 cm−1 which were assigned to the C–H alkanes bond stretching due to the presence of carbon and hydrogen groups in the untreated palm kernel shell itself. Besides that, the intense O–H bending is shown in both MG-PKSS and MG-PKSF spectra at 1634 and 1629 cm−1 and can be attributed to the presence of oxygen in the samples. The absorption bands at 1034 cm−1 for MG-PKSS and 1093 cm−1 for MG-PKSF both show the presence of C–O stretching. As portrayed in the spectra of MG-PKSS and MG-PKSF, a broad and intense band at 571 and 579 cm−1, respectively, could be assigned to M–O bands which may indicate the interaction between iron and oxygen in the samples.
Thermogravimetric analysis (TGA)
Thermogravimetric analysis is a technique used to determine the weight changes of a sample as a function of temperature under a controlled atmosphere. The thermal stability of a sample can also be observed using TG analysis. Figure 9 illustrates the thermogram of PKSAC at a starting temperature of 50 °C up to 1000 °C. Table 5 summarizes the temperature, percent weight loss and PKSAC residual after decomposition stage.

Thermogram of PKSAC at temperature up to 1000 °C
Figure 9 illustrates that PKSAC sample has good thermal stability property as it can withstand very high temperatures. At the early stage, a loss of 3.50% of its total weight was due to humidity up to a temperature of 151 °C. At a temperature around 200 °C, the sample experiences a slight weight loss due to desorption of the physisorbed water together with the rigidly bound water onto the PKSAC. Generally, the rigidly bound water can be assigned to the carboxylic group together with the phenolic hydroxyl groups. This is consistent with the findings of Haydar et al. [42] whose study reported that the desorption of physisorbed water was observed around 130 °C, while the rigidly bound water was observed around 250 °C. In addition, the sample was continuously experiencing weight loss, which started at a temperature of 500 °C until 850 °C. This weight loss was due to the decomposition of low volatile organic compounds which exist in the PKSAC. The decomposition process continued to occur within the temperature ranges of 424–924 and 924–1000 °C with weight loss percentages of 17.65 and 3.75%, respectively.
At temperatures above 900 °C, the decomposition of carbon starts to occur. Thus, it shows that at the temperature ranges of 924–1000 °C, PKSAC starts to lose its weight, of about 3.75% due to the decomposition of carbon from its sample. However, the residual weight obtained after this analysis is still high which is 71.45%. Hence, this analysis proves that the PKSAC sample is stable and can withstand high temperatures of up to 1000 °C and reliable to be used in higher-temperature activation.
X-ray diffraction (XRD) analysis
X-ray diffraction (XRD) is an analytical technique used to identify the crystalline phase of a material. In order to validate the crystalline phase of iron oxide in the porous carbon sample to be magnetized (Fe3O4), the samples were analyzed using powder X-ray diffraction analysis. Figure 10a illustrates the XRD diffractogram pattern of iron oxide extracted from ferric chloride/ferrous sulfate solution (FeOF). The pattern shows three intense peaks assigned to Fe3O4 (JCPDS #75-0033) at 2θ 30.25°, 35.70° and 57.20° and the presence of γ-Fe2O3 (JCPDS #39-1346) peaks at 2θ 43.45°, 53.85° and 63.00°. Similar observation was reported by Depci (2012) and El Ghandoor et al. (2012). This shows the crystalline nature of the Fe3O4.


a XRD diffractogram of FeOF. b XRD diffractogram of FeOS. c XRD diffractogram of CAC. d XRD diffractogram of MG-CACS. e XRD diffractogram of MG-CACF. f XRD diffractogram of raw PKS. g XRD diffractogram of PKSAC. i XRD diffractogram of MG-PKSF
The diffractogram of the iron oxide extracted from the electric arc furnace (EAF) slag (FeOS) is depicted in Fig. 10b. The diffractogram pattern illustrates the presence of three intense peaks at 2θ 30.15°, 35.65° and 57.20° which are assigned to Fe3O4, while three peaks at 2θ 43.25°, 54.15° and 62.65° are assigned to γ-Fe2O3. In addition, it displays four peaks which are compatible with the presence of α-Fe2O3 (JCPDS #80-2377) at 2θ 24.10°, 33.35°, 41.00° and 49.50° and a peak assigned to α-FeOOH (JCPDS #29-0713) is observed at 2θ 64.00°.
Figure 10c–e portrays the XRD diffractograms of CAC, MG-CACS and MG-CACF, respectively. As demonstrated in Fig. 10c, the amorphous nature of commercial activated carbon (CAC) is shown at peak about 2θ 24.46°. Meanwhile, Fig. 10d shows some intense peak which can be assigned to four different compounds which are Fe3O4, γ-Fe2O3, α-Fe2O3 and α-FeOOH. Fe3O4 shows four peaks at 2θ 30.05°, 31.35°, 35.85° and 56.95°. Meanwhile, three peaks at 2θ 42.75°, 54.20° and 62.75° are assigned to γ-Fe2O3. Further, α-Fe2O3 shows four obvious peaks at 2θ 24.45°, 33.20°, 41.05° and 49.45°, while the other two peaks at 2θ 26.85° and 64.15° were assigned to α-FeOOH. Figure 10e depicts the presence of Fe3O4 and γ-Fe2O3 in the MG-CACF diffractogram. Five peaks assigned to Fe3O4 are shown at 2θ 30.35°, 36.15°, 53.95°, 57.40° and 63.05°, while γ-Fe2O3 is represented by the presence of two peaks at 2θ 43.85° and 54.95°.
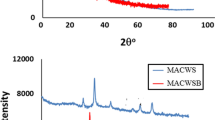
In addition, Fig. 10f, g both shows the diffractogram of raw PKS and PKSAC, respectively. The XRD diffractogram of raw PKS shows a broad peak at the range of 2θ 10° to 30° indicating the amorphous state of raw PKS during the analysis. However, three sharp peaks can be observed at 2θ 30.05°, 39.95° and 47.85° presumably due to the presence of some microcrystalline materials in the sample. Further, the XRD pattern of PKSAC illustrates a broad peak at 2θ 23.35° which may be assigned to the amorphous nature of the synthesized PKSAC. As illustrated in both figures, there was a disappearance of the crystalline materials, as the PKS was converted to PKSAC which may be due to the destruction of molecular order of cellulose and lignin crystalline structure which had occurred during pre-treatment [43].
Figure 10h, i indicates the MG-PKSS sample synthesized upon incorporation of PKSAC with iron oxide extracted from EAF slag. Figure 10h shows four peaks which can be assigned to Fe3O4 at 2θ 31.40°, 36.80°, 52.05° and 57.15°. There are three peaks assigned to γ-Fe2O3 which are at 2θ 43.50°, 45.80° and 62.95°. Additionally, the presence of α-Fe2O3 was depicted by the peaks at 2θ 23.95° and 24.25°, while the presence of α-FeOOH was depicted at 2θ 26.85°, 27.95° and 39.30°. Meanwhile, Fig. 10i illustrates four intense peaks assigned to Fe3O4 at 2θ 30.75°, 35.95°, 57.35° and 63.20°. Further, three peaks at 2θ 19.30°, 43.45°, 54.10° and a peak at 2θ 24.40° can be assigned to γ-Fe2O3 and α-Fe2O3, respectively.
A major presence of the Fe3O4 compound in all the magnetic samples proves that the MG-AC samples have the possibility to become good adsorbent due to the good magnetic property of Fe3O4 as compared to γ-Fe2O3, α-Fe2O3 and α-FeOOH compounds. The α-FeOOH compound is absent in MG-CACF and MG-PKSF probably because the iron oxide used in synthesizing both MG-CACF and MG-PKSF was obtained from the reaction suspension between ferric chloride and ferrous sulfate. The α-FeOOH compound likely exists in a certain natural source of iron. Hence, it implied that MG-CACS and MG-PKSS samples which were prepared using iron oxide extracted from electric arc furnace (EAF) slag exhibit the presence of α-FeOOH.
Vibrating sample magnetometer (VSM) analysis
The magnetic field and magnetic strength of the samples were analyzed using a vibrating sample magnetometer (VSM). The iron oxide samples from two different procedures and the MG-AC sample were selected for this analysis. Studies have reported that upon the magnetization process, the activated carbon acquires magnetic properties from the added magnetic materials, in addition to acquiring the ability to enhance the adsorption capacity [44]. Figure 11a, b shows the VSM magnetic hysteresis for iron oxide extracted from ferric chloride/ferrous sulfate solution (FeOF) and iron oxide extracted from electric arc furnace slag (FeOS), respectively, at room temperature.


a Magnetic hysteresis of FeOF at room temperature. b Magnetic hysteresis of FeOS at room temperature. c Image of magnet test on MG-PKSF. d Magnetic hysteresis of MG-PKSF at room temperature
Both hysteresis loops illustrate that the samples exhibited the typical superparamagnetic behavior. A previous study had defined superparamagnetic behaviour as a phenomenon usually displayed by a magnetic material/sample following the application of an external magnetic force, during which the particles of the magnetic material exhibit magnetism [45]. Further, it also explains the presence of strong magnetic properties in the samples [46].
In Fig. 11a, the magnetic hysteresis loop representing the magnetization saturation of FeOF was 62.848 emu g−1, which was higher than the magnetization saturation of FeOS, i.e., 21.156 emu g−1 as depicted by the magnetic hysteresis loop in Fig. 11b. This may be due to the presence of Fe3O4. A previous study had demonstrated that Fe3O4 possessed greater magnetic properties relative to other iron oxides such as γ-Fe2O3, α-Fe2O3 and α-FeOOH [47]. In this study, the VSM analysis was consistent with the fact that the magnetic properties were inherent in the FeOF, FeOS and MG-PKSF due to the predominance of Fe3O4 and other iron oxides, i.e., γ-Fe2O3 and α-Fe2O3 in the samples.
Consequently, owing to the higher magnetic saturation of FeOF as stated above, the ferric chloride/ferrous sulfate solution was used to produce the magnetic palm kernel shell activated carbon (MG-PKSF). Figure 11c illustrates the ability of a magnet to attract the MG-PKSF from aqueous solution, thus indicating that the magnetic saturation of the superparamagnetic MG-PKSF depends on the magnetism of the magnet. Subsequently, the magnetization value of MG-PKSF, which was 49.554 emu g−1 as illustrated by the magnetic hysteresis loop of MG-PKSF depicted in Fig. 11d, was lower compared to that of the FeOF impregnating solution, which had an initial higher magnetization value of 62. 848 emu g−1 as reported previously. This further confirms the fact that the FeOF had greater magnetic properties and hence the idea behind its selection as the impregnation solution.
Since the MG-PKSF has a potential to remove contaminants in the aqueous environment, the ease of removing MG-PKSF after being utilized becomes a concern. One of the reasons for magnetizing the MG-PKSF was to apply it in subsequent magnetic separation procedure to be removed from water medium after the treatment. In the magnetic separation procedure, the minimum magnetic saturation value was 16.3 emu g−1 for the procedure utilizing a conventional magnet [48]. In this study, the magnetic saturation of MG-PKSF was considered high. In this regard, the magnetic saturation values achieved by all the three magnetic samples are more than sufficient to be applied in the magnetic separation procedure.
Conclusions
This study revealed that the MG-PKSF presented better characteristics than the MG-PKSS and the PKSAC for the removal of heavy metals from wastewater. A highly porous PKSAC with a high BET surface area was developed by utilizing 50% wt/wt phosphoric acid activation at a temperature of 500 °C for 2 h in a furnace. The acid activation played an important role to initiate the bond breaking of functional groups in the starting material of the AC. The FTIR spectrum obtained for the PKSAC was observed to be similar to the spectrum of CAC, thereby implying that the PKSAC was successfully developed. The average pore size of the PKSAC was concluded to be small mesopores with the micropores still existing. The thermal stability of the PKSAC obtained from the TGA thermogram indicated that the PKSAC was significantly stable and can withstand a high temperature. The electron micrograph of PKSAC was used to show the formation of various sizes of pores on its surface compared to the micrograph of raw PKS.
Subsequently, the desired MG-AC was prepared by impregnating the porous palm kernel shell activated carbon (PKSAC) with both iron oxide extracted from ferric chloride/ferrous sulfate solution (MG-PKSF) and EAF slag (MG-PKSS), thereby resulting in the successful preparation of the magnetic samples. Both MG-PKSF and MG-PKSS exhibited magnetic properties; hence, the composites can be easily separated from the water medium by a simple magnetic separation procedure. Smaller BET surface areas were obtained for both magnetic samples compared to untreated PKSAC due to the incorporation of iron oxide, i.e., Fe3O4, γ-Fe2O3 and α-Fe2O3 over the porous surface of PKSAC upon the magnetization process. The presence of iron oxide in MG-PKSF and MG-PKSS was validated using XRD analysis. The diffractograms depicted the presence of various types of iron oxide, namely Fe3O4, γ-Fe2O3, α-Fe2O3 and α-FeOOH, in both magnetic samples. Electron micrographs illustrated that the surface morphology of MG-PKSF and MPKSS was significantly different compared to the untreated PKSAC due to the presence of iron oxide coverage over the porous surface. The magnetic samples analyzed using VSM revealed the high magnetic saturation properties attributable to the superparamagnetic behavior of the developed magnetic samples.
It can be concluded that the MG-PKSF presented better characteristics such as greater surface area and magnetic properties for the removal of heavy metals from water and can be easily separated from water containing contaminants using a simple magnetic separation compared to the PKSAC and MG-PKSS.
References
Daud WMAW, Ali WSW (2004) Comparison on pore development of activated carbon produced from palm shell and coconut shell. Biores Technol 93(1):63–69
Guo J, Lua AC (2003) Textural and chemical properties of adsorbent prepared from palm shell by phosphoric acid activation. Mater Chem Phys 80(1):114–119
Hussein M, Tarmizi R, Zainal Z, Ibrahim R, Badri M (1996) Preparation and characterization of active carbons from oil palm shells. Carbon 34(11):1447–1449
Budinova T, Petrov N, Razvigorova M, Parra J, Galiatsatou P (2006) Removal of arsenic (III) from aqueous solution by activated carbons prepared from solvent extracted olive pulp and olive stones. Ind Eng Chem Res 45(6):1896–1901
Adhoum N, Monser L (2002) Removal of cyanide from aqueous solution using impregnated activated carbon. Chem Eng Process 41(1):17–21
Stoeckli F, Centeno TA, Fuertes A, Muniz J (1996) Porous structure of polyarylamide-based activated carbon fibres. Carbon 34(10):1201–1206
Xu J, Gao N, Tang Y, Deng Y, Sui M (2010) Perchlorate removal using granular activated carbon supported iron compounds: synthesis, characterization and reactivity. J Environ Sci 22(11):1807–1813
Depci T (2012) Comparison of activated carbon and iron impregnated activated carbon derived from Gölbaşı lignite to remove cyanide from water. Chem Eng J 181:467–478
Nieto-Delgado C, Rangel-Mendez JR (2012) Anchorage of iron hydro (oxide) nanoparticles onto activated carbon to remove As (V) from water. Water Res 46(9):2973–2982
Daud WMAW, Ali WSW, Sulaiman MZ (2000) The effects of carbonization temperature on pore development in palm-shell-based activated carbon. Carbon 38(14):1925–1932
Slunjski M, Bourke M, O’Leary B (2000) MIEX® DOC process for removal of humics in water treatment. In: International humic substances society Australian chapter meeting Melbourne, Australia
Oliveira LC, Petkowicz DI, Smaniotto A, Pergher SB (2004) Magnetic zeolites: a new adsorbent for removal of metallic contaminants from water. Water Res 38(17):3699–3704
Ngarmkam W, Sirisathitkul C, Phalakornkule C (2011) Magnetic composite prepared from palm shell-based carbon and application for recovery of residual oil from POME. J Environ Manag 92(3):472–479
Bastami TR, Entezari MH (2012) Activated carbon from carrot dross combined with magnetite nanoparticles for the efficient removal of p-nitrophenol from aqueous solution. Chem Eng J 210:510–519
Zhong-liang S, F-m LIU, S-h YAO (2011) Adsorptive removal of phosphate from aqueous solutions using activated carbon loaded with Fe(III) oxide. New Carbon Mater 26(4):299–306
Attia AA, Girgis BS, Fathy NA (2008) Removal of methylene blue by carbons derived from peach stones by H 3 PO 4 activation: batch and column studies. Dyes Pigm 76(1):282–289
Mohan D, Sarswat A, Singh VK, Alexandre-Franco M, Pittman CU (2011) Development of magnetic activated carbon from almond shells for trinitrophenol removal from water. Chem Eng J 172(2):1111–1125
Hameed B, Din AM, Ahmad A (2007) Adsorption of methylene blue onto bamboo-based activated carbon: kinetics and equilibrium studies. J Hazard Mater 141(3):819–825
Karagozoglu B, Tasdemir M, Demirbas E, Kobya M (2007) The adsorption of basic dye (Astrazon Blue FGRL) from aqueous solutions onto sepiolite, fly ash and apricot shell activated carbon: kinetic and equilibrium studies. J Hazard Mater 147(1):297–306
Mussatto SI, Fernandes M, Rocha GJ, Órfão JJ, Teixeira JA, Roberto IC (2010) Production, characterization and application of activated carbon from brewer’s spent grain lignin. Biores Technol 101(7):2450–2457
Mohan D, Pittman CU (2007) Arsenic removal from water/wastewater using adsorbents—a critical review. J Hazard Mater 142(1):1–53
Nakahira A, Nishida S, Fukunishi K (2006) Adsorption of contaminants by MACs and magnetic separation. J Ceram Soc 114:135–137
Ćurković L, Cerjan-Stefanović Š, Rastovean-Mioe A (2001) Batch Pb2+ and Cu2+ removal by electric furnace slag. Water Res 35(14):3436–3440. doi:10.1016/S0043-1354(01)00037-9
Anyika C, Majid ZA, Isa AB, Ismail N, Zakaria MP, Yahya A (2016) Toxic and nontoxic elemental enrichment in biochar at different production temperatures. J Clean Prod 131:810–821
Li P, Lin K, Fang Z, Wang K (2017) Enhanced nitrate removal by novel bimetallic Fe/Ni nanoparticles supported on biochar. J Clean Prod 151:21–33
Miezah K, Obiri-Danso K, Kádár Z, Fei-Baffoe B, Mensah MY (2015) Municipal solid waste characterization and quantification as a measure towards effective waste management in Ghana. Waste Manag 46:15–27
Yargicoglu EN, Sadasivam BY, Reddy KR, Spokas K (2015) Physical and chemical characterization of waste wood derived biochars. Waste Manag 36:256–268
Do MH, Phan NH, Nguyen TD, Pham TTS, Nguyen VK, Vu TTT, Nguyen TKP (2011) Activated carbon/Fe3 O4 nanoparticle composite: fabrication, methyl orange removal and regeneration by hydrogen peroxide. Chemosphere 85(8):1269–1276
Yacob AR, Wahab N, Suhaimi NH, Mustajab MKAA (2013) Microwave induced carbon from waste palm kernel shell activated by phosphoric acid. Int J Eng Technol 5(2):214
Yildirim IZ, Prezzi M (2011) Chemical, mineralogical, and morphological properties of steel slag. Adv Civil Eng 2011:13. doi:10.1155/2011/463638
Liu Q-S, Zheng T, Li N, Wang P, Abulikemu G (2010) Modification of bamboo-based activated carbon using microwave radiation and its effects on the adsorption of methylene blue. Appl Surf Sci 256(10):3309–3315
Tan I, Hameed B, Ahmad A (2007) Equilibrium and kinetic studies on basic dye adsorption by oil palm fibre activated carbon. Chem Eng J 127(1):111–119
Lowell S, Shields JE (2013) Powder surface area and porosity, vol 2. Springer, Berlin
Noor ABM, Nawi MABM (2008) Textural characteristics of activated carbons prepared from oil palm shells activated with ZnCl2 and pyrolysis under nitrogen and carbon dioxide. J Phys Sci 19(2):93–104
Pensini E, Sleep BE, Yip CM, O’Carroll D (2013) Forces of interaction between fresh iron particles and iron oxide (magnetite): effect of water chemistry and polymer coatings. Colloids Surf A 433:104–110
Kim JY, Magesh G, Youn DH, Jang J-W, Kubota J, Domen K, Lee JS (2013) Single-crystalline, wormlike hematite photoanodes for efficient solar water splitting. Sci Rep 3:2681
Arakaki A, Masuda F, Amemiya Y, Tanaka T, Matsunaga T (2010) Control of the morphology and size of magnetite particles with peptides mimicking the Mms6 protein from magnetotactic bacteria. J Colloid Interface Sci 343(1):65–70
Tadić M, Čitaković N, Panjan M, Stojanović Z, Marković D, Spasojević V (2011) Synthesis, morphology, microstructure and magnetic properties of hematite submicron particles. J Alloys Compd 509(28):7639–7644
Yakout SM, Sharaf El-Deen G (2016) Characterization of activated carbon prepared by phosphoric acid activation of olive stones. Arab J Chem 9:S1155–S1162. doi:10.1016/j.arabjc.2011.12.002
Sugumaran P, Susan VP, Ravichandran P, Seshadri S (2012) Production and characterization of activated carbon from banana empty fruit bunch and Delonix regia fruit pod. J Sustain Energy Environ 3:125–132
Hesas RH, Arami-Niya A, Daud WMAW, Sahu J (2013) Preparation and characterization of activated carbon from apple waste by microwave-assisted phosphoric acid activation: application in methylene blue adsorption. BioResources 8(2):2950–2966
Haydar S, Moreno-Castilla C, Ferro-Garcıa M, Carrasco-Marın F, Rivera-Utrilla J, Perrard A, Joly J (2000) Regularities in the temperature-programmed desorption spectra of CO2 and CO from activated carbons. Carbon 38(9):1297–1308
Lai L-W, Idris A (2013) Disruption of oil palm trunks and fronds by microwave-alkali pretreatment. BioResources 8(2):2792–2804
Liu K, Xu L, Zhang F (2014) A new preparation process of coal-based magnetically activated carbon. Chin J Geochem 33(2):173–177
Wahajuddin SA (2012) Superparamagnetic iron oxide nanoparticles: magnetic nanoplatforms as drug carriers. Int J Nanomed 7:3445
Lunge S, Rayalu S (2012) Low cost magnetic iron oxide (MIO) potential adsorbent for arsenic removal. In: Paper presented at the international conference on chemical, civil and environment engineering
Laurenzi MA (2008) Transformation kinetics and magnetism of magnetite nanoparticles. ProQuest, Ann Arbor
Weber JWJ (1985) Adsorption theory, concepts and models. In: Slejko FL (ed) Adsorption technology: a step-by-step approach to process evaluation and application. Marcel & Dekker Inc., New York
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Anyika, C., Asri, N.A.M., Majid, Z.A. et al. Synthesis and characterization of magnetic activated carbon developed from palm kernel shells. Nanotechnol. Environ. Eng. 2, 16 (2017). https://doi.org/10.1007/s41204-017-0027-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41204-017-0027-6