
Abstract
Introduction
The aim of this study was to evaluate the efficacy and safety of a new hard-boiled lozenge formulation containing ambroxol 20 mg versus placebo for the relief of sore throat in patients with acute pharyngitis.
Methods
This was a phase 3, randomized, double-blind, placebo-controlled, parallel-group multicenter trial conducted between June and September 2018 in South Africa. Patients with a diagnosis of acute pharyngitis, onset ≤ 72 h, and sore throat pain of at least moderate intensity were randomized to receive either ambroxol 20 mg or placebo hard-boiled lozenges. The primary efficacy endpoint was the normalized time-weighted sum of pain intensity differences (SPID) from baseline over 3 h following administration of the first lozenge (SPIDnorm,0–3h). Secondary efficacy endpoints included SPID 24 h after the first lozenge intake (SPIDnorm,0–24h) and patient assessment of efficacy at 3 and 24 h after the first lozenge.
Results
Of 422 patients from 11 centers, 390 were randomized to one of the two treatment groups (n = 196, ambroxol; n = 194, placebo) and 388 were analyzed (modified intention-to-treat). The mean ± standard deviation SPIDnorm,0–3h values were −0.386 (0.259) and −0.366 (0.243) in the ambroxol and placebo groups, respectively, and the adjusted mean ± standard error SPIDnorm0–3h difference between ambroxol and placebo was −0.020 (0.025) (p = 0.443). Comparable results between treatment groups were also found for SPIDnorm,0–24h and patient assessment of efficacy at 3 and 24 h after the first lozenge. The incidence of treatment-emergent adverse events (TEAEs) was similar between treatment groups (11.7% for ambroxol versus 9.3% for placebo).
Conclusion
Although marked pain relief was observed over the first 3 h of treatment, superiority of ambroxol 20 mg hard-boiled lozenges versus placebo was not demonstrated in this study.
Trial Registration
NCT03583658.
Funding
Sanofi-Aventis Group.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Acute pharyngitis is a common condition that usually recedes spontaneously within a few days, but discomfort can be such that most patients seek treatment. |
The efficacy of previous lozenge formulations (white compressed lozenges) containing ambroxol, a secretolytic agent with local anesthetic properties, has been extensively demonstrated for the symptomatic treatment of sore throat. |
The current study evaluated the efficacy and safety of a new hard-boiled lozenge formulation containing ambroxol 20 mg versus a matched placebo for the relief of sore throat in patients with acute pharyngitis. |
What was learned from the study? |
Comparable results were observed between treatment groups; the sore throat pain relief was 39% with ambroxol hard-boiled lozenges versus 37% with placebo over the first 3 h of treatment. |
This study failed to demonstrate the superiority of the new hard-boiled lozenges containing ambroxol for the treatment of sore throat pain. |
Potential explanations for this lack of superiority, including patient noncompliance, analgesic properties of the treatments outside the pharmacological action of ambroxol, and organoleptic characteristics related to hard-boiled versus compressed lozenges, are discussed. |
Introduction
Acute pharyngitis is a common condition characterized by inflammation of the upper respiratory tract, with symptoms including sore throat and dysphagia [1]. Although the inflammation usually recedes spontaneously within a few days, the discomfort can be such that most patients seek treatment [2]. Viral infections account for about 80% of acute pharyngitis cases [3], making antibiotics an ineffective option.
Symptomatic treatments are widely used and recommended [4]. Acute sore throat and fever can be managed with oral analgesics such as nonsteroidal anti-inflammatory drugs (NSAIDs) or paracetamol, but their use is limited by the risk of systemic adverse events and the low concentration of drug substance present in the affected area [5, 6]. However, the oropharyngeal cavity is easily accessible, and pharmaceutical agents can be directly administered to the target site.
Multiple topical formulations are available for the treatment of sore throat, but lozenges present several advantages compared with other pharmaceutical forms such as oral rinses. These include ease of administration and prolonged contact time at the site of action [4, 7]. Moreover, by relying on salivary transport, lozenges can reach the lower pharyngeal cavity and tonsils [6].
Ambroxol hydrochloride is a well-described secretolytic agent that also has local anesthetic properties. It has been widely used for decades for the treatment of diseases of the respiratory tract including acute pharyngitis [2, 8]. Structurally, it is the active N-desmethyl metabolite of bromhexine, a synthetic derivative of vasicine [9], exhibiting local anesthetic action through sodium channel inhibition [10] along with anti-inflammatory activity [11]. In previous clinical trials, compressed lozenges containing 20 and 30 mg ambroxol provided a statistically significant reduction in sore throat pain compared with placebo [2]. The aim of the present study was to investigate the efficacy and safety of a new hard-boiled lozenge formulation containing 20 mg ambroxol compared with a matching placebo in adult patients with acute pharyngitis.
Methods
Patient Selection and Study Design
This was a phase 3, randomized, double-blind, placebo-controlled, parallel-group multicenter trial. The study was conducted between June and September 2018 at 11 centers in South Africa. Patients included in the trial were men and women, aged ≥ 18 years, diagnosed with sore throat due to acute pharyngitis with onset ≤ 72 h prior to visit 1 and a pain intensity score ≥ 6 on a 0–10-point numerical rating scale with increments of 1 (0 = “not sore”, 10 = “very sore”). Exclusion criteria included allergy and/or hypersensitivity to ambroxol hydrochloride or any other ingredients of the formulation, actual or suspected drug dependency and/or alcohol abuse, symptoms of primarily bacterial pharyngitis, and bacterial secondary infection. The full list of inclusion/exclusion criteria can be found in the Supplementary Appendix.
Following an enrollment evaluation, eligible patients were randomized to receive either ambroxol or placebo. At visit 1, after administration of the first lozenge, patients were required to remain at the trial site for the next 3 h, during which eating and drinking were prohibited. After this, patients were permitted to take a maximum of six lozenges per day for up to 3 consecutive days, on an as-needed basis with a minimum of 30 min between lozenges. As soon as possible after the patients had discontinued the lozenges (and no later than trial day 4), they were required to return to the trial center for a final assessment (visit 2). A follow-up visit (visit 3) was only required for patients who had an ongoing adverse event at the end of treatment or had a new adverse event in the 24 h after last trial drug administration (Fig. 1). During the study, concomitant treatments such as decongestants, antihistamines, beta-agonist medications, antibiotics, analgesics, anti-inflammatory agents, steroids for oral, inhaling or topical application, expectorants, and antitussives were prohibited. Similarly, the use of physical therapies such as throat compress or throat rinsing was not allowed. However, patients were not withdrawn from therapy or assessments in cases of study deviation (the full list of permanent discontinuation criteria can be found in the Supplementary Appendix). The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines as defined by the International Conference on Harmonization, and approved by the institutional review boards (IRB) and independent ethics committees for all participating centers (additional information can be found in the Supplementary Appendix). This study is registered on ClinicalTrials.gov (NCT03583658), and written informed consent was obtained from all patients prior to study enrollment.

Study design. AESI adverse event of special interest, SAE serious adverse event, V visit
Randomization and Blinding
Randomization was performed using either an interactive voice response system (IVRS) or interactive web response system (IWRS), whereby a unique identification code was generated for each eligible patient. Batches of ambroxol or placebo lozenges were identified with matching unique identification codes and provided to patients accordingly. Lozenges were otherwise indistinguishable by any other characteristics (color, shape, packaging, and labeling). Everyone involved in the conduct or analysis or with any other interest in the study remained blinded with regard to the randomized treatment assignments until after database lock.
Study Endpoints
The primary efficacy endpoint was the time-weighted sum of pain intensity differences from baseline (SPID) over the initial 3 h after taking the first lozenge, expressed as a ratio of the baseline score. The pain intensity (PI) was assessed by patients at baseline and at selected time intervals after the first dose (10, 15, 30, 45, 60, 90, 120, and 180 min) on a 0–10-point numerical rating scale. Pain intensity differences (PIDs) were then calculated between baseline and time points over the following 3 h, each one weighted for the time elapsed since previous assessment. The sum of PIDs (SPID) gave the area under the curve (AUC) or SPIDAUC, which was finally normalized by the complete observation period (180 min) and baseline PI score, resulting in SPIDnorm,0-3h. A negative value of SPIDnorm,0–3h thus indicated an overall pain reduction up to a maximum of −1, corresponding to complete relief of pain between baseline and 3 h after dosing. In other words, the pain relief experienced over 3 h after the first lozenge intake was described as a proportion of the maximum potential pain relief achievable over the same time period. Further details of the calculation can be found in the Supplementary Appendix. Secondary efficacy endpoints included the time-weighted sum of pain intensity differences from baseline 24 h after the first lozenge intake (with additional assessments of pain at 4, 6, 12, and 24 h), expressed as a ratio of the baseline score (SPIDnorm,0–24h), and patient assessment of efficacy 3 and 24 h after taking the first lozenge using a 0–4-point verbal rating scale (0 = poor; 1 = fair; 2 = good; 3 = very good; 4 = excellent). Additionally, exploratory efficacy endpoints included time course of PID from baseline over the first 24 h, daily number of lozenges taken, and final overall patient and investigator assessments of efficacy using a 0–4-point verbal rating scale (0 = poor; 1 = fair; 2 = good; 3 = very good; 4 = excellent). The safety endpoints were based on the reported adverse events (AEs) and other safety information, patient and investigator assessment of tolerability using a 0–4-point verbal rating scale (0 = poor; 1 = fair; 2 = good; 3 = very good; 4 = excellent), and physical examination.
Statistical Methods
Based on the findings of seven previous trials [12], the mean difference in SPIDnorm 0–3h between ambroxol 20 mg and placebo was expected to be equal to −0.114 and the common standard deviation to 0.343, with a standardized mean difference of −0.333. A sample size of approximately 190 patients in each treatment group was calculated to give a power of 90% to detect a difference of −0.333 between ambroxol and placebo using a two-sided test at 5% significance level. In order to adjust for possible dropouts, 195 patients were needed in each treatment group. Primary and secondary efficacy endpoints were tested using an analysis of variance (ANOVA), including treatments as fixed effect. Treatment differences were estimated by reference to the adjusted least square means (LS means) and the corresponding 95% confidence intervals (CI). A logistic regression model including the term treatment, with baseline PI as a covariate, was fitted to analyze the multinomial endpoints of 3- and 24-h patient assessment of efficacy. For the time course of PID over the first 24 h, restricted maximum likelihood estimation based on a mixed-effect model for repeated measures analysis was used to obtain adjusted means for the treatment effects. This model included treatment and time as discrete fixed effects, baseline PI as a continuous fixed effect, and interactions time-by-treatment and baseline PI-by-time. Efficacy analyses were performed only on the data from the modified intention-to-treat (mITT) population, consisting of all randomized patients who used at least one dose or part of a dose, and who had a baseline value and at least one post-baseline value for pain intensity within the first 3 h after the first intake of treatment. Efficacy was not planned to be assessed on a per-protocol population. Safety analyses were descriptive and based on the safety population (randomized population who received at least one dose or part of a dose of the double-blind treatment).
Results
Of 422 patients enrolled in the study, 390 were randomized either to the ambroxol (n = 196) or the placebo (n = 194) group. All of them received the study medication and were therefore included in the safety analysis. Six patients permanently discontinued from treatment (four in the ambroxol group and two in the placebo group) due to AEs, lack of efficacy or withdrawal by subject. Two patients in the ambroxol group were excluded from the mITT population as they had no baseline value for pain intensity, thus the mITT population was composed of 388 patients. All patients completed the study by attending the last visit (Fig. 2). Demographics and baseline characteristics were balanced between the two treatment groups (Table 1). Overall, 65.1% of the patients were females and 34.9% males, with a mean ± standard deviation (SD) age of 36.90 (12.88) years. Baseline pain intensity mean (SD) scores in ambroxol and placebo groups were 7.09 (0.98) and 6.93 (0.92), respectively, and the mean duration of sore throat was 2.56 and 2.61 days. A total of 117 patients were treated with concomitant medications during the study (n = 63 ambroxol, n = 54 placebo); of those patients, 33 took at least one prohibited medication during the course of the study. Among them, nine patients met exclusion criteria due to the use of analgesic/antipyretic within 4 h preceding the first administration of the study drug (n = 2 ambroxol, n = 4 placebo), or using any cold medication within 8 h from study entry (n = 1 ambroxol, n = 2 placebo). Additionally, three patients in the ambroxol group took at least one additional medicine for the treatment of sore throat during the first 3 h after the first lozenge intake (benzydamine hydrochloride spray, iodized throat lozenge and Difflam-C). Overall, the most frequently used prohibited medications were analgesics (n = 12 ambroxol, n = 11 placebo), with paracetamol reported in five and nine patients, in the ambroxol and placebo group, respectively; anti-inflammatory agents and antihistamines were also used. Nevertheless, none of the patients were excluded from the analysis due to these deviations as per study design.

Patient disposition. mITT modified intention-to-treat
Primary Endpoint
In the mITT population (n = 388), mean (SD) SPID norm,0–3h values in the ambroxol and placebo groups were −0.386 (0.259) and −0.366 (0.243), respectively (data not shown); the adjusted mean ± standard error (SE) SPIDnorm,0–3h difference between ambroxol and placebo was −0.020 (± 0.025; 95% CI −0.070 to 0.031). Although marked pain relief was seen, superiority of the treatment with ambroxol versus placebo was not demonstrated (p = 0.443) (Fig. 3).

Change from baseline in throat pain intensity 3 h after the first lozenge (mITT population). CI confidence interval, LS least square, mITT modified intention-to-treat, SD standard deviation
Secondary Efficacy Endpoints
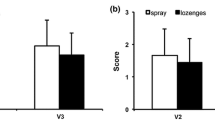
Similar results were obtained for sore throat pain relief over 24 h after the first lozenge administration. The mean (SD) SPIDnorm,0–24h was −0.506 (0.267) in the ambroxol group and −0.537 (0.279) in the placebo group, while the adjusted mean (SE) SPIDnorm,0–24h difference between ambroxol and placebo was −0.031 (0.028; 95% CI −0.024 to 0.085, p = 0.269). The time course of pain intensity change from baseline over 24 h following the first lozenge showed that the dynamics of pain relief were comparable between ambroxol and placebo. A rapid decrease in pain intensity was noted over the first 3 h, after which pain relief was maintained for the 24-h period (noting that patients were permitted to take additional lozenges between 3 and 24 h; Fig. 4). Overall, the median (Q1; Q3) total number of lozenges taken was 3.0 (2.0; 4.0) for ambroxol and 3.0 (2.0; 5.0) for placebo on day 1, 3.0 (2.0; 4.0) for both ambroxol and placebo on day 2, and 1.0 (0.0; 3.0) for both ambroxol and placebo on day 3. Comparable results between treatment groups were also found with regard to the patient overall assessment of efficacy at 3 and 24 h after the first lozenge. The odds ratio of patients being in a higher category (higher efficacy) when treated with ambroxol rather than placebo was 1.208 (95% CI 0.840–1.736; p = 0.308) and 0.915 (95% CI 0.636–1.315; p = 0.630) at 3 and 24 h, respectively. A higher proportion of patients in both treatment groups rated the treatment as “very good” or “excellent” after 24 h than after 3 h (Fig. 5a). No significant differences were noted between ambroxol and placebo in the final patient and investigator assessments of efficacy (Fig. 5b).

LS mean (SE) for the time course of PID from pre-dose baseline over the first 24 h (mITT population). LS least square, mITT modified intention-to-treat, PID pain intensity difference, SE standard error

a Patient assessments of efficacy at 3 and 24 h and b final patient and investigator assessments of efficacy (mITT population). mITT modified intention-to-treat population
Safety Outcomes
Overall, treatments were well tolerated during the study, and the proportion of patients experiencing treatment-emergent adverse events (TEAEs) was similar between groups (11.7%, ambroxol versus 9.3%, placebo). However, the incidence of TEAEs leading to treatment discontinuation was greater in the ambroxol arm, with four (2.0%) patients discontinuing versus only one (0.5%) receiving placebo. The most common TEAEs, defined as an event reported for more than one patient, were headache (n = 6, ambroxol, n = 3, placebo), dizziness (n = 5, ambroxol, n = 2, placebo), and accidental overdose (n = 2, ambroxol, n = 3, placebo). There were no serious AEs reported in the study. Lastly, patient and investigator assessments of tolerability were similar between ambroxol and placebo lozenges at the end of the study (Table 2). At the end of the study, more than 90% of patients and investigators rated the study medications as having “good”, “very good”, or “excellent” tolerability.
Discussion
Acute pharyngitis is a common condition encountered in clinical practice, and despite its self-limiting nature, most patients seek pain-relieving treatments [1, 2]. In this study, the efficacy of the novel ambroxol hard-boiled lozenge formulation was not found to be superior in relieving throat pain compared with placebo. Results were consistent for all the efficacy endpoints considered. Furthermore, the overall safety and tolerability profiles of the study drug and placebo were also comparable.
In contrast to the current study, the efficacy of white compressed lozenges containing ambroxol has been extensively demonstrated in previous studies with a similar design (randomized, placebo-controlled, double-blind, multicenter, with a maximum of 6 lozenges per day up to 3 consecutive days) [2, 12, 13]. In two clinical trials, Fischer et al. found that the effect of white compressed lozenges containing 20 mg ambroxol hydrochloride was “evident and consistently more favorable than placebo both in terms of efficacy and tolerability” [2]. The efficacy of 20 mg ambroxol white compressed lozenges was also evaluated in a pooled analysis of seven clinical trials conducted by de Mey et al. and was confirmed to be statistically superior to the control treatment [12]. That analysis showed that the observed pain relief effect of ambroxol white compressed lozenges across studies ranged between 38 and 52% of the maximum theoretically achievable effect versus 27–36% after placebo treatment. In contrast, comparable results were observed between treatment groups in the current study with the new formulation, with sore throat pain relief of 39% with ambroxol hard-boiled lozenges versus 37% with placebo over the first 3 h of treatment. The reasons that the efficacy of ambroxol and placebo hard-boiled lozenges could not be discriminated in this study are unclear. A small group of patients did not adhere to the washout phase (2.3%), and three in the ambroxol group used additional medications for the treatment of sore throat within 3 h following the first study lozenge intake. Additionally, over the study, the use of prohibited concomitant medications was observed in 8.5% (9.7% ambroxol; 7.2% placebo) of the patients. However, the overall impact of these study deviations on the efficacy results is questionable. Both active and placebo hard-boiled lozenges contained flavoring agents, such as menthol and eucalyptus oil, which are known to have local anesthetic and analgesic properties [10, 14]. Although any local beneficial effect that these excipients might have exhibited would be present in both ambroxol-treated and placebo-treated patients, this could potentially have diminished the pain relief effect attributable to the pharmacological action of ambroxol [6]. Moreover, the main difference between this study and earlier studies of ambroxol lozenges was the formulation used—previous studies tested a white compressed ambroxol lozenge, whereas here a new hard-boiled lozenge formulation was investigated. It could be hypothesized that hard-boiled lozenges have organoleptic characteristics that are less prominent in compressed lozenges with similar excipients, and that overall these account for a positive pain relief effect that the model used in this study did not allow us to evaluate. Despite the final outcome, the study design was considered appropriate and no limitation could be determined; an adequate number of patients were enrolled, and baseline characteristics were well-balanced between arms.
In conclusion, the present study failed to demonstrate the superiority of the new hard-boiled lozenges containing ambroxol for the treatment of sore throat pain. Compared with previously published placebo-controlled studies that have demonstrated significant sore throat pain reduction produced by ambroxol 20 mg white compressed lozenges, the factors contributing to this negative outcome remain unclear, and further investigations would be required to establish the causes of such a result.
References
Palm J, Fuchs K, Stammer H, Schumacher-Stimpfl A, Milde J. Efficacy and safety of a triple active sore throat lozenge in the treatment of patients with acute pharyngitis: results of a multi-centre, randomised, placebo-controlled, double-blind, parallel-group trial (DoriPha). Int J Clin Pract. 2018;72(12):17.
Fischer J, Pschorn U, Vix JM, Peil H, Aicher B, Muller A, de Mey C. Efficacy and tolerability of ambroxol hydrochloride lozenges in sore throat. Randomised, double-blind, placebo-controlled trials regarding the local anaesthetic properties. Arzneimittelforschung. 2002;52(4):256–63.
Russo M, Bloch M, de Looze F, Morris C, Shephard A. Flurbiprofen microgranules for relief of sore throat: a randomised, double-blind trial. Br J Gen Pract. 2013;63(607):e149–55.
McNally D, Shephard A, Field E. Randomised, double-blind, placebo-controlled study of a single dose of an amylmetacresol/2,4-dichlorobenzyl alcohol plus lidocaine lozenge or a hexylresorcinol lozenge for the treatment of acute sore throat due to upper respiratory tract infection. J Pharm Pharm Sci. 2012;15(2):281–94.
Bouroubi A, Donazzolo Y, Donath F, Eccles R, Russo M, Harambillet N, Gautier S, Montagne A. Pain relief of sore throat with a new anti-inflammatory throat lozenge, ibuprofen 25 mg: a randomised, double-blind, placebo-controlled, international phase III study. Int J Clin Pract. 2017;71(9):4.
de Mey C, Peil H, Kolsch S, Bubeck J, Vix JM. Efficacy and safety of ambroxol lozenges in the treatment of acute uncomplicated sore throat. EBM-based clinical documentation. Arzneimittelforschung. 2008;58(11):557–68.
Ferrer F. Sprays and lozenges for sore throats. S Afr Family Pract. 2012;54(2):120–2. https://doi.org/10.1080/20786204.2012.10874190.
HMA. Mucoangin 20 mg lozenges—Summary of Product Characteristics. https://mri.cts-mrp.eu/Human/Downloads/BE_H_0104_001_FinalPI.pdf. 2016.
EMA. Ambroxol and bromhexine containing medicinal products—revised assessment report. https://www.ema.europa.eu/en/documents/referral/ambroxol-bromhexine-article-31-referral-prac-assessment-report_en.pdf. Accessed 15 Sept 2015.
Chenot JF, Weber P, Friede T. Efficacy of Ambroxol lozenges for pharyngitis: a meta-analysis. BMC Family Pract. 2014;15(1):45. https://doi.org/10.1186/1471-2296-15-45.
Malerba M, Ragnoli B. Ambroxol in the 21st century: pharmacological and clinical update. Expert Opin Drug Metab Toxicol. 2008;4(8):1119–29.
de Mey C, Koelsch S, Richter E, Pohlmann T, Sousa R. Efficacy and safety of ambroxol lozenges in the treatment of acute uncomplicated sore throat—a pooled analysis. Drug Res. 2016;66(7):384–92.
Schutz A, Gund HJ, Pschorn U, Aicher B, Peil H, Muller A, de Mey C, Gillissen A. Local anaesthetic properties of ambroxol hydrochloride lozenges in view of sore throat. Clinical proof of concept. Arzneimittelforschung. 2002;52(3):194–9.
Tsuchiya H. Anesthetic agents of plant origin: a review of phytochemicals with anesthetic activity. Molecules. 2017;22(8):1369.
Acknowledgements
The authors wish to thank all the participants of the study.
Funding
This study and the Rapid Service Fee were funded by Sanofi-Aventis Group.
Medical Writing Assistance
Writing support was provided by Martina Klinger-Sikora of inScience Communications, Springer Healthcare Ltd, UK, and funded by Sanofi-Aventis Group.
Authorship
All named authors meet the Internal Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
Rita Sousa provided medical input during the design phase of the study. Deepak Ramjee Lakha served as investigator during the study. Sandrine Brette oversaw the statistical analysis of the study. Simon Hitier provided statistical input during the design phase of the study.
Disclosures
Simon Hitier is an employee of Sanofi-Aventis Group. Rita Sousa is an employee of Sanofi-Aventis Group. Sandrine Brette is employed by Aixial, contracted by Sanofi. Deepak Ramjee Lakha received funds from Sanofi-Aventis Group.
Compliance with Ethics Guidelines
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines as defined by the International Conference on Harmonization, and approved by the institutional review boards and independent ethics committees for all participating centers (additional information can be found in the Supplementary Appendix). This study is registered on ClinicalTrials.gov (NCT03583658), and written informed consent was obtained from all patients prior to study enrollment.
Data Availability
Qualified researchers may request access to patient-level data and related study documents including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and data set specifications. Patient-level data will be anonymized and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data-sharing criteria, eligible studies, and process for requesting access can be found at: https://www.clinicalstudydatarequest.com
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.9907487.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any non-commercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sousa, R., Lakha, D.R., Brette, S. et al. A Randomized, Double-Blind, Placebo-Controlled Study to Assess the Efficacy and Safety of Ambroxol Hard-Boiled Lozenges in Patients with Acute Pharyngitis. Pulm Ther 5, 201–211 (2019). https://doi.org/10.1007/s41030-019-00100-w
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41030-019-00100-w