
Abstract
Scalability is increasingly important for bioinformatics analysis services, since these must handle larger datasets, more jobs, and more users. The pipelines used to implement analyses must therefore scale with respect to the resources on a single compute node, the number of nodes on a cluster, and also to cost-performance. Here, we survey several scalable bioinformatics pipelines and compare their design and their use of underlying frameworks and infrastructures. We also discuss current trends for bioinformatics pipeline development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Bioinformatics analyses are increasingly provided as services that end users access through a web interface that has a powerful backend that executes the analyses. The services may be generic, such as those provided by research institutes such as EMBL-EBI (http://www.ebi.ac.uk/services), commercial companies such as Illumina (https://basespace.illumina.com/home/index), and research projects such as Galaxy (https://usegalaxy.org/). However, they can also be specialized and targeted, for example, to marine metagenomics as our marine metagenomics portal (https://mmp.sfb.uit.no/).
Scalability is increasingly important for these analysis services, since the cost of instruments such as next-generation sequencing machines is rapidly decreasing [1]. The reduced costs have made the machines more available which has caused an increase in dataset size, the number of datasets, and hence the number of users [2]. The backend executing the analyses must therefore scale up (vertically) with respect to the resources on a single compute node, since the resource usage of some analyses increases with dataset size. For example, short sequence read assemblers [3] may require TBs of memory for big datasets and tens of CPU cores [4]. The analysis must also scale out (horizontally) to take advantage of compute clusters and clouds. For example, the widely used BLAST [5] is computationally intensive but scales linearly with respect to the number of CPU cores. Finally, to efficiently support many users it is important that the analyses scale with respect to cost-performance [6].
The data analysis is typically implemented as a pipeline (workflow) with third-party tools that each processes input files and produces output files. The pipelines are often deep, with 10 or more tools [7]. The tools are usually implemented in a pipeline framework ranging from simple R scripts to full workbenches with large collections of tools (such as the Galaxy [8] or Apache Taverna [9]). A review of pipeline frameworks is in [10], but it does not focus on scalability. Here, we survey several scalable bioinformatics pipelines and compare their design and deployment. We describe how these scale to larger datasets or more users, how they use infrastructure systems for scalable data processing, and how they are deployed and maintained. Finally, we discuss current trends in large-scale bioinformatics analyses including containers, standardization, reproducible research, and large-scale analysis-as-a-service infrastructures.
2 Scalable Pipelines
We focus our review on scalable pipelines described in published papers. Many of the pipelines are configured and executed using a pipeline framework. It is difficult to differentiate between the scalability of a pipeline framework and the scalability of individual tools in a pipeline. If a pipeline tool does not scale efficiently, it may be necessary to replace it with a more scalable tool. However, an important factor for pipeline tool scalability is the infrastructure service used by the pipeline framework for data storage and job execution (Fig. 1). For example, a columnar storage system may improve I/O performance, but many analysis tools are implemented to read and write regular files and hence cannot directly benefit from columnar storage. We therefore structure our description of each pipeline as follows:

Scalable pipeline components. A pipeline consists of third-party tools, data parsers, and data transformations. The pipeline tools and their dependencies are specified using a workflow language or implemented as a program or script. A pipeline framework executes the pipeline tools on a cluster or cloud using a big data processing engine or a supercomputer job scheduler. The pipeline framework stores the data as files, objects, or matrices in a columnar storage. The execution environment allocates the resources needed for the pipeline, and a user interface provides access for end users to the pipeline
-
1.
We describe the compute, storage, and memory requirements of the pipeline tools. These influence the choice of the framework and infrastructure systems.
-
2.
We describe how the pipelines are used. A pipeline used interactively to process data submitted by end users has different requirements than a pipeline used to batch process data from a sequencing machine.
-
3.
We describe the pipeline framework used by the pipeline, how the pipeline tools are executed, how the pipeline data are stored, and the execution environment.
-
4.
We describe how the pipeline tools scale out or up, how the pipeline framework supports multiple users or jobs, and whether the execution environment provides elasticity to adjust the resources allocated for the service.
-
5.
We discuss limitations and provide comparisons to other pipelines.
2.1 META-Pipe 1.0 Metagenomics Pipeline
Our META-pipe pipeline [11, 12] provides preprocessing, assembly, taxonomic classification, and functional analysis for metagenomics samples. It takes as input short reads from a next-generation sequencing instrument and outputs the organisms found in the metagenomics sample, predicted genes, and their corresponding functional annotations. The different pipeline tools have different resource requirements. Assembly requires a machine with at least 256 GB RAM, and it cannot run efficiently on distributed resources. Functional analysis requires many cores and has parts that are I/O intensive, but it can be run efficiently distributed on a cluster with thin nodes. Taxonomical classification has low resource requirements and can be run on a single node. A typical dataset is 650 MB in size and takes about 6 h to assemble on 12 cores and 20 h for functional annotation on 384 cores.
A Galaxy [13] interface provides META-pipe 1.0 to Norwegian academic and industry users (https://nels.bioinfo.no/). The pipeline is specified in a custom Perl-script-based framework [14]. It is executed on the Stallo supercomputer, which is a traditional HPC cluster with one job queue optimized for long-executing batch jobs. A shared global file system provides data storage. We manually install and maintain the pipeline tools and associated database versions on a shared file system on Stallo.
The job script submitted to the Stallo job scheduler describes the resources requested on a node (scale up) and the number of nodes requested for the job (scale out). Both Galaxy and the job scheduler allow multiple job submissions from multiple users at the same time, but whether the jobs run simultaneously depends on the load of the cluster. HPC clusters are typically run with a high utilization, so jobs are often queued for a long time and therefore jobs submitted at the same time may not run at the same time. HPC clusters are not designed for elastic resource provision, so it is difficult to efficiently scale the backend to support the resource requirement variations of multi-user workloads.
META-pipe 1.0 has several limitations as a scalable bioinformatics service. First, the use of a highly loaded supercomputer causes long wait times and limits elastic adjustment of resources for multi-user workloads. We manually deploy the service on Galaxy and Stallo, which makes updates time-consuming and prone to errors. Finally, our custom pipeline framework has no support for provenance data maintenance nor failure handling. For these reasons, we have re-implemented the backend in META-pipe 2.0 using Spark [15] so that it can take advantage of the same features as the pipelines described below do.
2.2 Genome Analysis Toolkit (GATK) Variant Calling Reference Pipeline
The GATK [16] best practices pipeline for germline SNP and indel discovery in whole-genome and whole-exome sequence (https://software.broadinstitute.org/gatk/best-practices/bp_3step.php?case=GermShortWGS) is often used as reference for scalable genomics data analysis pipelines. This pipeline provides preprocessing, variant calling, and callset refinement. (The latter usually is not included in benchmarking.) It takes as input short reads and outputs annotated variants. Some tools have high CPU utilization (BWA and HaplotypeCaller), but most steps are I/O bound. An Intel white paper [17] recommends using a server with 256 GB RAM and 36 cores for the pipeline, and they achieved the best resource utilization by running analysis jobs for multiple datasets at the same time and configuring the jobs to only use a subset of the resources. The pipeline is well suited for parallel execution as demonstrated by the MapReduce programming models used in [16] and the Halvade [18] Hadoop MapReduce implementation that analyzes a 86 GB (compressed) WGS dataset in less than 3 h on Amazon Elastic MapReduce (EMR) using 16 workers with a total of 512 cores.
The first three versions of GATK are implemented in Java and optimized for use on local compute infrastructures. Version 4 of GATK (at the time of writing in Beta) uses Spark to improve I/O performance and scalability (https://software.broadinstitute.org/gatk/blog?id=9644). It uses GenomicsDB (https://github.com/Intel-HLS/GenomicsDB) for efficiently storing, querying, and accessing (sparse matrix) variant data. GenomicsDB is built on top of Intel’s TileDB (http://istc-bigdata.org/tiledb/index.html) which is designed for scalable storage and processing of sparse matrices. To support tertiary (downstream) analysis of the data produced by GATK, the Hail framework (https://hail.is/) provides interactive analyses. It optimizes storage and access of variant data (sparse matrices) and provides built-in analysis functions. Hail is implemented using Spark and Parquet.
Tools in the GATK can be run manually through the command line, specified in the workflow definition language (WDL) and run in Cromwell, or use written in Scala and run on Queue (https://software.broadinstitute.org/gatk/documentation/pipelines). GATK provides multiple approaches to parallelize tasks: multi-threading and scatter–gather. Users enable multi-threading mode by specifying command-line flags and use Queue or Cromwell to run GATK tools using a scatter–gather approach. It is also possible to combine these approaches (https://software.broadinstitute.org/gatk/documentation/article.php?id=1988).
2.3 ADAM Variant Calling Pipeline
ADAM [6] is a genomics pipeline that is built on top of the Apache Spark big data processing engine [15], Avro (https://avro.apache.org/) data serialization system, and Parquet (https://parquet.apache.org/) columnar storage system to improve the performance and reduce the cost of variant calling. It takes as input next-generation sequencing (NGS) short reads and outputs sites in the input genome where an individual differs from the reference genome. ADAM provides tools to sort reads, remove duplicates, do local realignment, and do base quality score recalibration. The pipeline includes both compute and I/O-intensive tasks. A typical dataset is 234 GB (gzip compressed) and takes about 74 min to run on 128 Amazon EC2 r3.2xlarge (4 cores, 30.5 GB RAM, 80 GB SSD) instances with 1024 cores in total.
ADAM focuses on backend processing, and hence, user-facing applications need to be implemented as, for example, Scala or Python scripts. ADAM uses Spark to scale out parallel processing. The data are stored in Parquet, a columnar data storage using Avro serialized file formats that reduce I/O load by providing in-memory data access for the Spark pipeline implementation. The pipeline is implemented as a Spark program.
Spark is widely supported on commercial clouds such as Amazon EC2, Microsoft Azure HDInsight, and increasingly in smaller academic clouds. It can therefore exploit the scale and elasticity of these clouds. There are also Spark job schedulers that can run multiple jobs simultaneously.
ADAM improves on MapReduce-based pipeline frameworks by using Spark. Spark solves some of the limitations of the MapReduce programming model and runtime system. It provides a more flexible programming model than just the map-sort-reduce in MapReduce, better I/O performance by better use of in-memory data structures between pipeline stages and data streaming, and reduced job startup time for small jobs. Spark is therefore becoming the de facto standard for big data processing, and pipelines implemented in Spark can take advantage of Spark libraries such as GraphX [19] for graph processing and MLlib [20] for machine learning.
ADAM has two main limitations. First, it implemented as part of research projects that may not have the long-term support and developer efforts required to achieve the quality and trust required for production services. Second, it requires re-implementing the pipeline tools to run in Spark and access data in Parquet, which is often not possible for analysis services with multiple pipelines with tens of tools each.
2.4 GESALL Variant Calling Pipeline
GESALL [21] is a genomic analysis platform for unmodified analysis tools that use the POSIX file system interface. An example pipeline implemented with GESALL is their implementation of the GATK variant calling reference pipeline that was used as an example in the ADAM paper [6]. GESALL is evaluated on fewer but more powerful nodes (15, each with 24 cores, 64 GB RAM, and 3 TB disk) than the ADAM pipeline. A 243 GB compressed dataset takes about 1.5 h to analyze.
GESALL pipelines are implemented and run as MapReduce programs on resources allocated by YARN (https://hadoop.apache.org/). The pipeline can run unmodified analysis tools by wrapping these using their genome data parallel toolkit. The tools access their data using the standard file system interface, but GESALL optimizes data access patterns and enables correct distributed execution. It stores data in HDFS (https://hadoop.apache.org/) and provides a layer on top of HDFS that optimizes storage of genomics data type, including custom partitioning and block placement.
Like Spark, MapReduce is widely used in both commercial and academic clouds and GESALL can therefore use the horizontal scalability, elasticity, and multi-job support features of these infrastructures. The unmodified tools executed by a GESALL pipeline may also be multi-threaded. A challenge is therefore to find the right mix of MapReduce tasks and per-tool multi-threading.
2.5 Toil: TCGA RNA-Seq Reference Pipeline
Toil is a workflow software to run scientific workflows on a large scale in cloud or high-performance computing (HPC) environments [22]. It is designed for large-scale analysis pipelines such as The Cancer Genome Atlas (TCGA) [23] best practices pipeline for calculating gene- and isoform-level expression values from RNA-seq data. The memory-intensive STAR [24] aligner requires 40 GB of memory. As with other pipelines, the job has a mix of I/O- and CPU-intensive tasks. In [22], the pipeline runs on a cluster of AWS c3.8xlarge (32 cores, 60 GB RAM, 640 GB SSD storage) nodes. Using about 32.000 cores, they processed a 108 TB with 19,952 samples in 4 days.
Toil can execute workflows written in both the Common Workflow Language (CWL, http://www.commonwl.org/), the Workflow Definition Language (WDL, https://github.com/broadinstitute/wdl), or Python. Toil is written in Python, so it is possible to interface it from any Python application using the Toil Application Programming Interface (API). Toil can be used to implement any type of data analysis pipeline, but it is optimized for I/O-bound NGS pipelines. Toil uses file caching and data streaming, and it schedules work on the same portions of a dataset to the same compute node. Toil can run workflows on commercial cloud platforms, such as AWS, and private cloud platforms, such as OpenStack (https://www.openstack.org/), and it can execute individual pipeline jobs on Spark. Users interface with Toil through a command-line tool that orchestrates and deploys a data analysis pipeline. Toil uses different storage solutions depending on platform: S3 buckets on AWS, the local file system on a desktop computer, network file systems on a high-performance cluster, and so on.
3 Current Trends
In addition to the scalability considerations discussed above, we see several other trends in pipelines developed for scalable bioinformatics services.
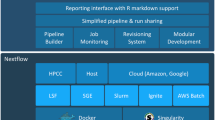
Containers are increasingly used to address the challenges of sharing bioinformatics tools and enabling reproducible analyses in projects such as BioContainers (http://biocontainers.pro/). A bioinformatics pipeline is often deep, with more than 15 tools [7]. Each tool typically has many dependencies on libraries and especially reference databases. In addition, some tools are seldom updated. Pipelines therefore often require a large effort to install, configure, and run bioinformatics tools. Software containerization packages an application and its dependencies in an isolated execution environment. One popular implementation of software container is Docker [25]. With Docker, developers can build a container from a configuration file (Dockerfile) that includes machine and human-readable instructions to install the necessary dependencies and the tool itself. Both the Dockerfile and the resulting container can be moved between machines without installing additional software, and the container can be rerun later with the exact same libraries. Containers can be orchestrated for parallel execution using, for example, Kubernetes (https://kubernetes.io/) or Docker Swarm (https://github.com/docker/swarm), and there are now multiple pipelining tools that use Docker or provide Docker container support including Nextflow [26], Toil [22], Pachyderm (http://www.pachyderm.io/), Luigi (https://github.com/spotify/luigi) [27], Rabix/bunny [28], and our own walrus system (http://github.com/fjukstad/walrus).
There are several efforts to standardize pipeline specifications to make it easier to port pipelines across frameworks and execution environments (including Toil described above). For example, the Common Workflow Language (CWL) is an effort supported by many of the developers of the most popular pipeline frameworks. CWL is a standard for describing data analysis pipelines. Developers can describe a data analysis pipeline in YAML or JSON files that contain a clear description of tools, input parameters, input and output data, and how the tools are connected. There are multiple systems that implement the CWL standard, including Galaxy [13], Toil, Arvados (https://arvados.org/), and AWE [29], making it possible to write a single description of a pipeline and run it in the most suitable pipeline execution environment. It is an open challenge to implement support for the standardized pipeline descriptions on execution environments such as Spark.
The needs and challenges for reproducible analyses [30] require a standardized way to specify and document pipelines and all their dependencies, in addition to maintaining all provenance information of pipeline executions [31]. Specifications such as CWL can be used to standardize the specification, and for example, Spark has built-in data lineage recording. However, there is not yet an analysis standard that describes the minimum information required to recreate bioinformatics analyses [32].
Finally, there are several large infrastructures and platforms that provide scalable bioinformatics services. The European ELIXIR (https://www.elixir-europe.org/) distributed infrastructure for life science data resources, analysis tools, compute resources, interoperability standards, and training. The META-pipe pipelines described above are developed as part of the ELIXIR project. Another example is the Illumina BaseSpace Sequence Hub (https://basespace.illumina.com/home/index), which is a cloud-based genomics analysis and storage platform provided by the producer of the currently most popular sequencing machines. Other commercial cloud platforms for bioinformatics analyses are DNAnexus (https://www.dnanexus.com/), Agave (https://agaveapi.co), and SevenBridges (https://www.sevenbridges.com/platform/). We believe the efforts required to maintain and provide the resources needed for future bioinformatics analysis services will further consolidate such services in larger infrastructures and platforms.
4 Summary and Discussion
We have provided a survey of scalable bioinformatics pipelines. We compared their design and use of underlying infrastructures. We observe several trends (Table 1). First, there are few papers that describe the design, implementation, and evaluation of scalable pipeline frameworks and pipelines, especially compared to the number of papers describing bioinformatics tools. Of those papers, most focus on a specific type of analysis (variant calling) using mostly the same tools. This suggests that there is a need to address the scalability and cost-effectiveness of other types of bioinformatics analysis.
Most papers focus on the scalability of a single job. Only our META-pipe paper evaluates the scalability of the pipeline with respect to multiple users and simultaneous jobs. With analyses provided increasingly as a service, we believe multi-user job optimizations will become increasingly important. Staggered execution of multiple pipeline jobs can also improve resource utilization as shown in [17, 27].
It is becoming common to standardize pipeline descriptions and use existing pipeline frameworks rather than implementing custom job execution scripts. An open challenge is how to optimize the execution of pipelines specified in, for example, CWL. Frameworks such as GESALL provide genomic dataset optimized storage which can be difficult to utilize from a generic pipeline specification. ADAM uses an alternative approach where the pipeline is a Spark program like for data analyses in many other domains.
In addition to standardizing pipeline description, there is a move to standardize and enable completely reproducible execution environments through software containers such as Docker. Although not yet widely adopted, containerized bioinformatics tools simplify deployment, sharing, and reusing of tools between research groups. We believe that standardizing the execution environment, together with standardizing the pipeline descriptions, is a key feature for reproducible research in bioinformatics.
Most pipelines save data in a traditional file system since most analysis tools are implemented to read and write files in POSIX file systems. GESALL provides a layer that enables using HDFS for data storage by wrapping tools and providing optimized genomic data-specific mapping between POSIX and HDFS. ADAM uses a different, data-oriented approach, with a layered architecture for data storage and analysis that exploits recent advancement in big data analysis systems. Like ADAM, GATK4 is also built on top of Spark and a columnar data storage system. ADAM requires re-implementing the analysis tools, which may be practical for the most commonly used tools and pipelines such as the GATK reference pipelines, but is often considered impractical for the many other tools and hence pipelines.
Pipeline frameworks such as GESALL and ADAM use MapReduce and Spark to execute pipeline jobs on clouds or dedicated clusters, and Toil supports job execution on HPC clusters with a job scheduler, which is important since most bioinformatics analysis pipelines are not implemented in MapReduce or Spark. HPC job schedulers are also provided on commercial and private clouds, so also these pipelines can take advantage of the elasticity provided by these infrastructures.
In addition to enabling and evaluating horizontal scalability, the cost of an analysis and the choice of virtual machine flavors are becoming increasingly important for efficient execution of bioinformatics analysis, since pipelines are increasingly deployed and evaluated on commercial clouds [6, 21, 22]. However, even on dedicated clusters it is important to understand how to scale a pipeline up and out on the available resources to improve the utilization of the resources. However, with the exception of [17], none of the reviewed papers have evaluated multiple pipeline job executions from the cluster provider’s point of view.
We believe deployment, provenance data recording, and standardized pipeline descriptions are necessary to provide easy-to-maintain and reproducible bioinformatics pipelines in infrastructures such as ELIXIR or platforms such as BaseSpace. These three areas are typically not addressed in the reviewed papers, suggesting that more research is required to address these areas in the context of scalable bioinformatics pipelines.
Summarized, we have described many scalability problems and their solutions in the reviewed papers. These include: scaling up nodes to run tools with large memory requirements (META-pipe), scale out for parallel execution (all reviewed pipelines), use of optimized data structures and storage systems to improve I/O performance (GATK 4.0, ADAM, GESALL), and the choice of machine flavor to optimize either the execution or cost (GATK, ADAM, GESALL). Although many of the pipelines have the same scalability issues, such as I/O performance for variant calling, the infrastructure system and optimizations differ depending on overall design choices (e.g., the use of unmodified vs modified analysis tools) and the software stack (e.g., Spark vs HPC schedulers and file systems). We therefore believe there is no right solution or platform that solves all scalability problems, and that more research is needed to scale up and cost-optimize the many types of bioinformatics data analyses. The increasing use of standardized layers, standards, and interfaces to implement these analyses should allow reusing the developed solutions across pipelines and pipeline frameworks.
References
Sboner A, Mu XJ, Greenbaum D et al (2011) The real cost of sequencing: higher than you think! Genome Biol 12:125. doi:10.1186/gb-2011-12-8-125
Schuster SC (2008) Next-generation sequencing transforms today’s biology. Nat Methods 5:16–18. doi:10.1038/nmeth1156
Vollmers J, Wiegand S, Kaster A-K (2017) Comparing and evaluating metagenome assembly tools from a microbiologist’s perspective—not only size matters! PLoS ONE 12:e0169662. doi:10.1371/journal.pone.0169662
Couger MB, Pipes L, Squina F et al (2014) Enabling large-scale next-generation sequence assembly with Blacklight. Concurr Comput Pract Exp 26:2157–2166. doi:10.1002/cpe.3231
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410. doi:10.1016/S0022-2836(05)80360-2
Nothaft FA, Massie M, Danford T et al (2015) Rethinking data-intensive science using scalable analytics systems. In: Proceedings of 2015 ACM SIGMOD international conference on management of data. ACM, New York, pp 631–646
Diao Y, Abhishek R, Bloom T (2015) Building highly-optimized, low-latency pipelines for genomic data analysis. In: Proceedings of the 7th biennial Conference on Innovative Data Systems Research (CIDR 2015), The New York Genome Center
Blankenberg D, Von Kuster G, Bouvier E et al (2014) Dissemination of scientific software with Galaxy ToolShed. Genome Biol 15:403. doi:10.1186/gb4161
Wolstencroft K, Haines R, Fellows D et al (2013) The Taverna workflow suite: designing and executing workflows of Web Services on the desktop, web or in the cloud. Nucleic Acids Res 41:W557–W561. doi:10.1093/nar/gkt328
Leipzig J (2016) A review of bioinformatic pipeline frameworks. Br Bioinform. doi:10.1093/bib/bbw020
Robertsen EM, Kahlke T, Raknes IA et al (2016) META-pipe—pipeline annotation, analysis and visualization of marine metagenomic sequence data. ArXiv160404103 Cs
Robertsen EM, Denise H, Mitchell A et al (2017) ELIXIR pilot action: marine metagenomics—towards a domain specific set of sustainable services. F1000Research 6:70. doi:10.12688/f1000research.10443.1
Afgan E, Baker D, van den Beek M et al (2016) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res 44:W3–W10. doi:10.1093/nar/gkw343
Pedersen E, Raknes IA, Ernstsen M, Bongo LA (2015) Integrating data-intensive computing systems with biological data analysis frameworks. In: 2015 23rd Euromicro international conference on parallel, distributed and network-based processing (PDP). IEEE Computer Society, Los Alamitos, pp 733–740
Zaharia M, Franklin MJ, Ghodsi A et al (2016) Apache Spark: a unified engine for big data processing. Commun ACM 59:56–65. doi:10.1145/2934664
McKenna A, Hanna M, Banks E et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi:10.1101/gr.107524.110
Prabhakaran A, Shifaw B, Naik M et al (2015) Infrastructure for GATK* best practices pipeline deployment. Intel, Santa Clara
Decap D, Reumers J, Herzeel C et al (2017) Halvade-RNA: parallel variant calling from transcriptomic data using MapReduce. PLoS ONE 12:e0174575. doi:10.1371/journal.pone.0174575
Gonzalez JE, Xin RS, Dave A et al (2014) GraphX: graph processing in a distributed dataflow framework. In: Proceedings of 11th USENIX conference on operating systems design and implementation. USENIX Association, Berkeley, pp 599–613
Meng X, Bradley J, Yavuz B et al (2016) MLlib: machine learning in Apache Spark. J Mach Learn Res 17:1235–1241
Roy A, Diao Y, Evani U et al (2017) Massively parallel processing of whole genome sequence data: an in-depth performance study. In: Proceedings of 2017 ACM international conference on management of data. ACM, New York, pp 187–202
Vivian J, Rao AA, Nothaft FA et al (2017) Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 35:314–316. doi:10.1038/nbt.3772
The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA et al (2013) The cancer genome atlas pan-cancer analysis project. Nat Genet 45:1113–1120. doi:10.1038/ng.2764
Dobin A, Davis CA, Schlesinger F et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi:10.1093/bioinformatics/bts635
Merkel D (2014) Docker: lightweight linux containers for consistent development and deployment. Linux J 239:2
Di Tommaso P, Chatzou M, Floden EW et al (2017) Nextflow enables reproducible computational workflows. Nat Biotechnol 35:316–319. doi:10.1038/nbt.3820
Schulz WL, Durant T, Siddon AJ, Torres R (2016) Use of application containers and workflows for genomic data analysis. J Pathol Inform 7:53. doi:10.4103/2153-3539.197197
Kaushik G, Ivkovic S, Simonovic J et al (2016) Rabix: an open-source workflow executor supporting recomputability and interoperability of workflow descriptions. Pac Symp Biocomput Pac Symp Biocomput 22:154–165
Gerlach W, Tang W, Keegan K et al (2014) Skyport: container-based execution environment management for multi-cloud scientific workflows. In: Proceedings of 5th international workshop on data-intensive computing in the clouds. IEEE Press, Piscataway, pp 25–32
Peng RD (2011) Reproducible research in computational science. Science 334:1226–1227. doi:10.1126/science.1213847
Huntemann M, Ivanova NN, Mavromatis K et al (2016) The standard operating procedure of the DOE-JGI Metagenome Annotation Pipeline (MAP v.4). Stand Genomic Sci 11:17. doi:10.1186/s40793-016-0138-x
ten Hoopen P, Finn RD, Bongo LA et al (2017) The metagenomics data life-cycle: standards and best practices. GigaScience. doi:10.1093/gigascience/gix047
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fjukstad, B., Bongo, L.A. A Review of Scalable Bioinformatics Pipelines. Data Sci. Eng. 2, 245–251 (2017). https://doi.org/10.1007/s41019-017-0047-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41019-017-0047-z