
Abstract
In this work, the microstructure property relationship of D2 (X153CrMoV12; 1.2379) ledeburitic cold work steel processed by filament extrusion is investigated. Contrary to the conventional process, which involves a multi-step process of printing, debinding, and solid-state sintering, the specimens in this study were densified using Supersolidus Liquid-Phase Sintering (SLPS). SLPS occurs after thermal debinding in the interval between the solidus and liquidus temperatures. Optimized liquid-phase volume fraction was evaluated by means of thermodynamic calculations using the CALPHAD method and their experimental validation. The microstructure formation process during debinding, solid state, and SLPS sintering was investigated by X-ray diffraction and electron microscopy. Tomography studies confirm a relative density of 99.92% by volume during SLPS sintering at 1280 °C and provide a deep insight into local densification behavior during SLPS processing. In addition, surface roughness, as determined by confocal laser scanning microscopy, could be reduced. The reduction in porosity and surface roughness can be attributed to the presence of a liquid phase during SLPS. Using adapted heat treatment parameters determined by hardness-tempering curves, higher hardness values were achieved for SLPS-post-compacted specimens compared to conventionally processed specimens and the same material in the cast and heat-treated reference state.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Additive manufacturing (AM) offers great potential, especially for manufacturing complex-shaped tools with internal cavities (cooling channels, adding cooling lubrication) directly from geometric data. Although AM methods have a low build-up speed, time-consuming and costly production of preforms in tool manufacture can be avoided, so the term “rapid tooling” is often used in this context [1]. According to the review paper by Cheah et al., rapid tooling in AM can be subdivided into indirect (IRT = indirect rapid tooling) and direct methods (DRT = direct rapid tooling) [2]. The indirect processes aim to manufacture a mold using AM. In the case of DRT, the component is directly processed and can subsequently be used as a tool for material forming or cutting, for example. In particular, in the manufacture of tools that require optimal temperature control (e.g., press-hardening tools [4, 5]) or the addition of local lubricants (machining and forming tools [6,7,8]), AM makes it possible to introduce inner cavities, which is hardly achievable by subtractive or formative processes. The current state of knowledge on AM of tools is provided by the work of Hölker-Jäger [9] and Levy et al. [10].
For the tools that should feature a long service life, their material properties must be adapted to the tribomechanical and chemical stresses that are acting during operation. Depending on the processing temperature and the required material properties, carbon-martensitic hardenable tool steels are commonly used. Due to the high tendency to cold cracking in Powder Bed Fusion (PBF) and Direct Energy Deposition (DED) processing of carbon-martensitic hardening tool steels, these material classes can only be processed with special measures such as preheating [11,12,13,14,15] or with a correspondingly adapted alloy design [16]. As an alternative to carbon-martensitic hardening tool steels with a high tendency to cold cracking, the literature often refers to soft-martensitic (18Ni300) or precipitation-hardened grades (17-4PH, 15-5PH) being easy to process by AM. For many applications, however, these steels in the PBF-LB and heat-treated condition offer an insufficient hardness of 310 HV10 (18Ni300 [17]) to 500HV (15-5PH [18]). In the case of press-hardening tools or forming tools for cold work, in particular, carbon-martensitic hardenable steels with a higher hardness are mandatory. While the literature contains many works on the additive processing of maraging steels [17, 19,20,21,22,23,24] and carbon-martensitic hardenable hot- and high-speed tool steels [16, 25,26,27,28,29], only a few works on cold work steels, particularly on hard-phase-containing ledeburitic cold work steels, can be found in the literature. Botero et al. [30] investigated the processing of a Cr–Mo–V-alloyed ledeburitic cold work steel with a carbon content of 1.4 mass%, using Powder Bed Fusion/Electron Beam/Metal (PBF-EB/M). Here, the powder bed has to be preheated to approx. 850 °C with a defocused electron beam to counteract crack formation. The as-built microstructure consists of a metal matrix with finely dispersed Mo- and V-rich carbides with a hardness of 48 to 60 HRC. Further, hardening and tempering increase the hardness to 62–64 HRC. Hentschel et al. investigated the processing of 60CrMo18-5 cold work tool steel on X37CrMoV5-1 substrate using DED-L in Ref. [12], where preheating of 400 °C was mandatory for achieving dense microstructures free of defects. Sander et al. investigated PBF-LB/M processing of Fe85Cr4Mo8V2C1 tool steel using a building board form heating of 500 °C to produce low-defect specimens [31]. In the as-built condition, the specimens had a hardness of 67 HRC and a compressive strength of 3800 MPa with an elongation at fracture of 15%. The development of residual stress and the associated damage caused by crack formation and propagation during powder bed fusion of cold work steel was analyzed using synchrotron investigations and atom-probe studies by Platl et al. [32]. They determined that crack formation and propagation primarily originate from the network-like eutectic carbide structures. These findings were confirmed by the work of Lim et al. in [33], who demonstrate that crack propagation in D2 (X153CrMoV12; 1.2379) tool steel occurs along eutectic Cr-rich carbides of M7C3 type. Contrary to other work, they attribute crack formation to hot crack formation along the low-melting eutectics in the interdendritic areas, with the length of the cracks increasing with an increased number of layers, i.e., renewed heat input. Similar findings are reported by Yadav et al. in Ref. [34].
What all of the previously mentioned methods of rapid tooling have in common is that the microstructure and, thus, the defects are formed directly during the build-up process, thus limiting the PBF or DED processing of a wide range of different materials. To avoid defect formation during the build-up process, AM methods are available, in which the build-up process and the microstructure formation process for setting the material properties are decoupled from each other. One such method is the so-called Atomic Diffusion Additive Manufacturing (ADAM), which is a multi-step process of Fused Filament Fabrication (FFF) followed by sintering compaction. The work by Galati et al. [35] provides an overview of the ADAM process, the materials that can be processed, and the process sequence. The starting material is a filament consisting of approx. 60 vol.% of metal powder and a polymer binder mixture. During processing, the binder is melted in an extruder, and the liquid binder-metal powder mass is applied on a baseplate or further applied material. In a further process step, the binder is removed, and the metallic powder particles are sintered at a higher temperature so that the complex-shaped component retains its mechanical properties. Achievable properties of ADAM-processed maraging steel 17–4 PH [36,37,38], corrosion-resistant austenite AISI 316L [39, 40], or Cu-base materials [41] can be gathered from the literature. However, the mechanical properties of ADAM-processed components are inferior compared to the cast state of the same material; this is attributed to the relatively high residual porosity of approximately 3–6 vol.% [37]. The pores are internal cavities with a continuous connection to the sample surface and are essential for removing the binder constituents during the debinding process. Several works on ADAM-processed metals confirm that solid-state sintering following debinding provides insufficient sintering activity to fully reduce this open porosity [35,36,37,38, 42].
The need for research can be derived based on the current state of knowledge. On the one hand, this work will fundamentally describe the microstructure formation process of the ledeburitic cold work steel D2 (X153CrMoV12; 1.2379) during ADAM processing. The resulting material properties should be compared to the same materials in casting + heat treatment condition. On this basis, a statement can be made as to whether the ADAM process is, in principle, suitable for producing appropriate tools for cold work in the context of rapid tooling. On the other hand, there is a need to develop an adapted sintering strategy to reduce the residual porosity of ADAM-manufactured components. Supersolidus Liquid-Phase Sintering (SLPS) is considered as a solution, which is generally suitable for processing hard-phase-containing cold work tool steels and high-speed steels. Furthermore, the question arises as to what extent a SLPS sintering enables the reduction of the surface roughness of ADAM-processed samples. Thus, expensive post-processing machining processes can be reduced, especially for hard phase containing and thus hard materials like ledeburitic cold work tool steels or high-speed steels. Thereby, it should be investigated whether SLPS densification can promote complete compaction of filament extruded D2 cold work tool steel. In addition to designing optimal sinter windows using the CALPHAD method and its experimental validation, the aim is to describe the microstructure formation process during filament extrusion and SLPS using computer tomography, electron microscopy, and X-ray diffraction. Finally, the hardness of SLPS-densified material in the heat-treated condition is compared to the cast and conventional produced reference state.
2 Material and methods
2.1 Material
This work used filament D2 (X153CrMoV12; 1.2379) from Markforged. The filament has a diameter of d = 1.75 mm and is composed of gas-atomized steel powder of D2 (X153CrMoV12: 1.2379) steel and a polymeric binder system (e.g., paraffin wax and polyethylene). The steel powder content in the filament is approx. 60 vol.%. The chemical composition of the specimens was measured by optical emission spectrometry (OES) and is listed in Table 1 [35]. The D2 (X153CrMoV12; 1.2379) ledeburitic cold work tool steel with the chemical composition listed in Table 1 was taken as a reference state in as-cast and hot worked conditions.
2.2 Samples manufacture, printing, debinding, sintering, and heat treatment
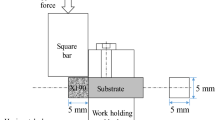
The metal X system by Markforged was used to process green compacts (in the as-built state). Figure 1 illustrates the schematic structure of the specimen and the deposition strategy. Concerning the later application (tools), cubic samples instead of cylindrical ones were manufactured to show how the material shrinkage on these edges behaved during the subsequent sinter compaction. Cubic specimens with a dimension of 12 × 12x12 mm were manufactured with an infill of 100% using a 45° filling strategy and a layer thickness of 0.125 mm in the z-direction. An outer contour with a width of four layers is applied to ensure low surface topography of the samples.

Schematic representation of a specimen generation, b sample, and c filling strategy with wall layer and 45° infill layer
Cubic-shaped specimens were built on a support structure (dimensions: length, width, height = 30 × 30 × 3 mm) made from the same material within 1 h and 57 min. The system’s printing parameters included a post-sintered layer height of 0.125 mm and the process options “sinter stability” and “use raft.” In addition, a solid fill as a fill pattern, a surrounding wall thickness of 1 mm (post-sintered), the material “D2 Tool Steel (v1),” and the use of the option "cloud slicing" were chosen.
Before sintering, the samples were debound in a two-stage process. In the first step, samples were washed in 3M™ Novec™ 7100 (methoxynonafluorobutane) solvent at 50 °C for 14 h, followed by air drying for 4 h. According to the manufacturer’s instructions, a weight loss of 4 mass% indicates a successful solvent debinding process. Therefore, the sample’s weight was measured (accuracy of precision balance = 0.01 g) prior to (green compacts) and after (brown compacts) solvent debinding. Solvent debinding, which was the same for all specimens, is followed by thermal debinding (second dependent step) and a sintering process to adjust the microstructure and the associated material properties.
The specimens’ nomenclature is given in Table 2. All samples were manufactured while varying the thermal debinding and sintering temperatures. In the conventional approach of the Margforged company, the samples (hereinafter referred to as S-conv) are fed into a sintering furnace (device Sinter-1) using a specific temperature–time control. The exact process parameters are a trade secret of the manufacturer and not traceable during the process because of the closed system. To determine the sintering temperatures in the used sintering furnace of company Markforged, indirect temperature measurement was carried out using Bullers rings (TempTAB 600 JSI), which enable precise temperature measurement (accuracy 1 °C) in the temperature range of 1100–1300 °C. After sintering, the temperature was determined indirectly by measuring the sintering shrinkage of the ceramic ring diameter with a µm micrometer (accuracy 1/100 mm). With the knowledge of the ring diameter of d = 25.79 mm (measurement position close to furnace wall) and d = 25.46 mm (measurement position close to the middle of the furnace), a sintering temperature of 1169 °C to 1186 °C (average temperature = 1177.5 ± 12 °C) was maintained. The sintering temperature corresponds to approximately 85% of the calculated liquidus temperature (Tliq = 1388 °C). The total processing time for the conventionally manufactured samples is 27.5 h.
Besides the conventional route, thermal debinding and SLPS sintering were performed in a vacuum tube furnace (Heraeus ROF7/75). During SLPS compaction, sinter compaction took place in the temperature range between solidus and liquidus temperatures. A liquid phase is specifically created, with the volume content of the liquid phases increasing from the solidus temperature (0 vol.%) to the liquidus temperature (100 vol.%). With the help of thermodynamic calculations, the liquid-phase volume content was calculated as a function of the sintering temperature in thermodynamic equilibrium (see Sect. 2.3). The calculations were used to determine sintering temperatures at which a liquid-phase volume content of 6.5 vol.% (1260 °C), 28.5% by volume (1280 °C), and 35 vol.% should be present. In addition, a sintering temperature below the solidus temperature (i.e., solid-state sintering without liquid-phase volume content) of 1240 °C was considered. The aim was to check whether the carbon from the binder system during post-processing led to the carburization of the steel powder and, thus, to a reduction in the solidus temperature. Before SLPS, the solvent-debonded specimens were heated to 450 °C (10 °C/min) and further to 650 °C (3.3 °C/min) in a vacuum atmosphere to remove the long-chain polymeric binder. Subsequently, the samples were heated to the appropriate sintering temperatures at a heating rate of 10 °C/min and sintered for 30 min in a vacuum atmosphere. The samples were then cooled to room temperature at a cooling rate of 10 °C/min. The numerical values used in the terminology of the SLPS samples in Table 2 correspond to the sintering temperature selected in each case.
The material under consideration is a secondary-hardening cold work tool steel, which is hardened and tempered to its maximum secondary hardness to set its functional properties. The sintering and austenitizing temperatures influence the formation of tempering carbides and, thus the secondary hardness maximum. Hardness-tempering diagrams were recorded for all states considered to determine optimal tempering parameters. All samples were austenitized at 1050 °C (30 min) in an inert gas furnace (argon atmosphere), oil quenched, and then double tempered for 2 h in a temperature regime of RT to 600 °C (air cooled). The hardness of the heat-treated specimens was measured according to DIN EN ISO 6507 using a hardness tester (ATM Carat 930), and a load of 98.07 N. Average hardness and the standard mean were calculated from ten measurements. The hardness tests were carried out at the center of the surface of the metallographically prepared specimens.
2.3 Thermodynamic calculation and post-processing
To calculate the solidus and liquidus temperatures and the formed liquid phase as a function of the SLPS temperature, ThermoCalc software (version 2020.2.72656) was used. The calculations were carried out using the TCFE9 thermodynamic database at atmospheric pressure and a system size of 1 mol, considering the nominal chemical composition listed in Table 1 (mean values were taken into account). In the calculations, the following phases were considered: “liquid (LIQUID)”, “γ-Fe (FCC_A1#1)”, “MC (FCC_A1#2)”, “M7C3 (M7C3)” and “M23C6 (M23C6)” [43].
2.4 Microscopy and metallography
Metallographic specimens of every specimen were prepared to investigate the microstructures. In the first step, the cubic specimens were divided in half using an automatic micro-cutting machine with a cBN cutoff wheel. Afterward, the samples were hot-mounted using an electrically conductive phenolic resin. The metallographic preparation consisted of grinding with SiC abrasive paper with a decreasing grit size (80 → 320 → 500 → 800 → 1000 mesh) and polishing with diamond suspension (grit: 6 µm → 3 µm → 1 µm). In addition, metallographically prepared surfaces were contrasted with V2A etchant (HCI, H2O, HNO3) or polished with OPS. The microstructure of the specimens in different conditions was examined at low magnifications of 50 × to 100 × using a Leica DM2000 optical light microscope (OM) in bright-field mode to determine porosity and pore distribution. Microstructures were examined at a higher magnification using a Tescan Vega 3 SBH scanning electron microscope (SEM). Images were taken in SE mode at a working distance of 10 to 15 mm and an accelerating voltage of 18 to 20 keV. The beam intensity varied in the range of 8 to 10.
2.5 Phase analysis
X-ray diffraction measurements were performed at beamline BL9 of the DELTA synchrotron radiation source (Dortmund, Germany) [44]. The incident photon energy was set to 27 keV, i.e., a wavelength of 0.4592 Å. The beam size at the sample position was 0.7 × 0.05 mm2 (h x v), and the sample was aligned so that the angle of incidence was 1°. 2D diffraction images were measured using a MAR345 image plate detector. A diffraction image of a CeO2 reference sample was utilized to calibrate the setup, i.e., detector–sample distance, detector tilt, and beam center, applying the Fit2D software package [45]. Based on the calibration, the 2D images of all samples were integrated to obtain the corresponding diffraction patterns on a scattering-angle (2Θ) scale (alternatively: on a wave-vector transfer (q) scale). To perform a quantitative and qualitative evaluation of the measurements, results were analyzed with Maud software and Crystallography Open Database (COD) using the Rietveld method. Furthermore, EDX and EBSD analyses were performed using the Tescan Vega 3 SBH scanning electron microscope and the Bruker xFlash 5030 system at an acceleration voltage of 20 keV and a working distance of 15 mm. To investigate the binder system, measurements were carried out on a dynamic differential scanning calorimeter (DSC, DSC 214 Nevio, Netsch). The reference crucible was made of Al, and a sample mass of 20.3 mg of the polymer was used. N2 gas was used as an atmosphere with a flow rate of 40 ml/min to 60 ml/min. The temperature range during the measurement was set between 25 °C and 210 °C, with a heating and cooling rate of 10 °C/min.
2.6 Surface roughness
A KEYENCE VK-X250 confocal laser scanning microscope was used to determine the surface roughness of the sintered sample surfaces. To that end, images with a 10 × magnification of a 3 × 3 image area were recorded in four areas with an image size of 1900 × 1300 µm. The images were analyzed with the MultiFileAnlayzer software, version 1.3.0.116, from Keyence Corp. Surface correction for plane tilt and wave removal were applied. The surface roughness measurement was carried out on the upper surface of the cube sample and a lateral surface (“side-face”) to analyze the different surface structures created by the build-up process. The measurement positions are labeled in Fig. 1b.
2.7 Determination of density
Computer tomography (CT) images were collected using the EasyTom 160.X. device and xact software from RX Solutions to measure the density of the sample. For the CT measurements, smaller samples with the dimensions “length × width × thickness = 10 × 5 × 1 mm” were removed from the cubic samples by cutting with a cBN cutting disk. The CT scans were performed with an accelerating voltage of 160 keV and a sample flow between 40 and 42 µm. The voxel size varied between 1 and 3 µm per pixel, and the scan time was approximately 3 h. The CT images generated were stacked with ImageJ software and evaluated using quantitative image analysis. The CT measurement allows 3D defect imaging, whereby pores with a size of 3 voxels are recognized as defects. Considering the voxel size, only defects larger than 1.6—3.2 µm can be detected. Smaller pores were detected using quantitative image analysis. To that end, micrographs were taken by scanning electron microscopy at a magnification of 1000x. The porosity was determined using ImageJ software and evaluated using quantitative image analysis by image binarization.
3 Results and discussion
In the following presentation and discussion of the results, the debinding process will be described first in Sect. 3.1. Based on a basic description of the brown compacts that form after debinding, the microstructure formation process during solid-phase sintering and SLPS sintering on the mesoscale will be described in Sect. 3.2. Using thermodynamic calculations, suitable SLPS temperatures and optimal liquid-phase volume contents should first be derived. By means of sintering tests, these calculation results are validated experimentally, and an optimal SLPS sintering window for filament-extruded D2 (X153CrMoV12; 1.2379) steel is derived. The influence of the sintering temperature on the geometric stability and the surface roughness that develops during sintering will be discussed in Sect. 3.3. Microstructure formation during SLPS sintering will be explained in Sect. 3.4. Finally, suitable heat treatment parameters will be determined by hardness-tempering diagrams, and the hardness values achieved will be compared to reference samples in the as-cast condition in Sect. 3.5.
3.1 Description of the microstructure and the change in sample properties during sample debinding
Microstructural investigations using optical light microscopy were performed to understand the development of the microstructure from the filler starting material (Fig. 2a) through the filament-extruded specimens (Fig. 2b) to the solvent debond sample (Fig. 2c). The filament starting material is characterized by spherical metal particles (light areas) with a diameter of 0.9 to 26 µm, homogeneously distributed in the binder system (dark areas) with a packing density of about 61 ± 0.8 vol.%. At higher magnification (Fig. 1a), the steel particles have high sphericity and show a hypoeutectic microstructure consisting primarily of solidified metal cells surrounded by a Cr-rich eutectic M7C3. Cube-shaped samples (green compacts) were manufactured from this filament, with an edge length of 11.98 ± 0.03 mm and a weight of approximately. 7.97 ± 0.01 g.

Optical microscope images (magnification 200 ×, unetched state) of a filament, b as-built state (green compact), c debindered state (brown compact) to illustrate the powder packing and the powder distribution
The DSC analysis of the filament in Fig. 3 showed that the binder system consists of a low-melting polymer (paraffin wax, melting point: 58–70 °C) and a higher melting polymer (polyethylene, melting point: 120–140 °C).

DSC analysis of the examined filament
The microstructure of the green compacts (Fig. 2b) corresponds to the microstructure of the filament starting material (Fig. 2a). The spherical metal particles (diameter from 0.6 to 24 µm; packing density = 62 ± 0.5 vol.%.) continue to be homogeneously distributed in the binder system after printing. The slight increase in the packing density of the steel particles can be attributed to the printing process, where the filament is conveyed through a heated nozzle at a temperature of 220 °C, leading to a remelting (melting points: polyethylene: ~ 120–140 °C; paraffin wax: ~ 58–70 °C) and steaming off of the binder material. The complete remelting of the binder material creates a flowable mass consisting of liquid polymer binder and solid steel powder particles. After depositing the plasticized extrudate, the binder system solidifies again and forms a solid compound of steel particles embedded in a solid binder system. However, due to the layer-by-layer deposition strategy at 45°, pores appear in the printed green compacts at regular intervals (every 250 mm in width, every 50 µm in height) between the deposited extrudate strands (Fig. 4a). This suggests an insufficient plasticization of the binder system, leading to insufficient material closure of the interfaces between the extrudate lines, as illustrated in Fig. 4c.

Optical microscope images of a green compact and b brown compact, microstructural images at 200 × magnification in the unetched state; c schematic representation of the deposited extrudate lines and the tethering errors that occur
During solvent debinding in methoxynonafluorobutane, the short-chain primary binder (paraffin wax) is dissolved. A porous structure of backbone binder (polyethylene) and metal particles, as shown in Fig. 4b, with an open porosity remains. An increased porosity in the brown compact due to the dislodged primary binder cannot be detected, as shown by comparing Figs. 2c and 4b. The metallographic preparation steps of grinding and polishing partially dissolve out metal particles and backbone binder, so that the resulting pores are not visible due to solvent debinding. Removal of the primary binder by solvent did not affect the geometric dimensions of the specimens. The brown compacts remained dimensionally stable, having an edge length (w x d x h) of approx. 11.97 ± 0.01 × 11.98 ± 0.03 × 11.89 ± 0.02 mm (same as the green compacts). By removing the primary binder, a mass loss of about 4.15 ± 0.14 mass% could be measured. As a result, the brown compacts weighed only 7.63 ± 0.02 g, corresponding to the manufacturer’s specification (mass loss > 4 mass%) and optimum debinding.
During thermal debinding, the long-chain binder decomposed by exceeding a temperature of 400 °C, as shown in the work of Wagner et al. [46], the open porosity of the brown compact support the gaseous escape of the binder [47,48,49,50]. Integrating thermal debinding (as described in Sect. 2.2) into the sintering process is necessary. After the backbone binder is burned out, the sintering and diffusion processes ensure that the specimen geometry retains its shape [48]. When burning out the remaining backbone binder, two model developments are available to describe the porosity evolution [48,49,50]. A hypothetical model case is that during the first phase—low debinding rate—the polymer images initially escape at the sample surface [48,49,50]. A porous outer layer is formed in this process, while the inner structure is filled with a liquid polymer core [47, 49, 50]. As the debinding time increases, the inner polymer binder evaporates and escapes through the exposed outer cavities. For this to occur, the polymer binder’s escape requires an open porosity and sufficient time. The second approach assumes that liquid polymer is uniformly distributed in the porous structure of the sample due to the influence of capillary effects [49, 50]. In both model assumptions, as the debinding rate increases, on the one hand, the liquid polymer is transported to the outer surfaces by pressure-induced flows, decomposes, and escapes [49, 50]. On the other hand, the liquid polymer inside the specimens is partially decomposed. The polymer vapor is driven by convection and diffusion to the outer surfaces, where it can escape from the structure [49, 50]. Temperature–time cycles for thermal debinding were selected in such a way that only as much binder evaporates as can be released into the environment at the same time via the open porosity. Preliminary tests show that when the sample is heated directly to the sintering temperature, bloating of the specimen occurs (Fig. 5). This “bloating” can be traced back to a quick transition of the solid–liquid binder into the gas phase so that an internal pressure has built up, which leads to an intense deformation of the sample.

Inflated and “bloated” sample during direct heating to SLPS temperature: a side plan view of the SLPS-sintered sample and b structural cross-section of an SLPS-sintered sample separated in the middle; the porosity corresponds to approx. 15–25 vol.% and varies greatly locally
3.2 Description of densification of filament extrudated materials during solid-state and SLPS sintering on the mesoscale
After thermal debinding, specimens designated as “S-conv” were solid-state sintered following the manufacturer’s instructions, as mentioned in Sect. 2.2. Because the sintering furnace used (Sinter-1) is a closed system, the temperature–time curves cannot be measured and are, therefore, not detailed here. Even if the acting temperatures are not precisely known (a sintering temperature of 0.8⋅TSOL = ~ 1000 to 1050 °C is assumed [51]), densification can be described by way of the three known sintering stages. In the first stage (initial step), particle rearrangement occurs, and sinter necks are formed primarily by surface diffusion (driving force = reduction of surface energy). In the second stage, the sinter necks enlarge, and new grain boundaries form. In addition, open porosity will close as the sintering time increases, so isolated pores are present. In the third sintering stage, these isolated pores are closed by preferential vacancy diffusion, and the microstructure begins to coarsen due to Ostwald ripening [51]. Besides solid-state sintering, SLPS was performed to improve the densification of the filament extrudated specimens and to reduce surface roughness at the same time. This SLPS focuses on achieving fully dense specimens with improved material and component properties. SLPS sintering can be carried out using pre-alloyed powders, with a target liquid-phase volume content adjusted by sintering between the solidus and liquidus temperatures. According to the studies by German et al. [52,53,54], a liquid-phase volume fraction of about 30 vol.% is the target. Improved sinter activity during SLPS can be attributed to capillary forces, improved particle rearrangement processes, and separation of the pre-alloyed powder particles along grain boundaries due to the liquid phase formation. To gain a deeper understanding of the melting behavior, the solidification interval, and the solidus and liquidus temperatures, phase quantity diagrams were calculated using ThermoCalc software and the TCFE9 database. By means of these equilibrium diagrams, the liquid-phase volume fraction between the solidus and liquidus temperatures could be calculated (Fig. 6a, b).

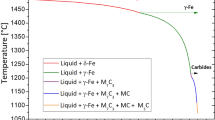
a Phase quantity diagram of D2 (X153CrMoV12; 1.2379) steel to represent the phases formed at a given temperature in the thermodynamic equilibrium; b phase quantity diagram with SLPS temperatures and the corresponding liquid-phase fractions plotted
The phase quantity diagram of D2 (X153CrMoV12; 1.2379) steel (Fig. 6a) was calculated with the chemical composition listed in Table 1. According to Fig. 6, austenite and Cr-rich M7C3 carbides are present below the solidus temperature of TSOL = 1257°C. Concerning the thermodynamic calculations, eutectic Cr-rich M7C3 represents the low-melting phase. Due to the high volume fraction of the eutectic Cr-rich M7C3, a liquid-phase volume content of approx. 24 vol.% forms at a temperature of 1263 °C. At a temperature of 1263 °C, the slope of the forming liquid-phase volume fraction decreases as a function of temperature. This behavior can be traced back to the fact that the liquid phase is formed solely by the metal cells (austenite phase) at higher temperatures. A fully liquid phase is present when the liquidus temperature at TLIQ = 1388 °C is exceeded. To analyze the influence of the formed liquid-phase volume content on the densification of the microstructure, three different SLPS temperatures were derived from the phase quantity diagrams in Fig. 6b, taking into account the temperatures and liquid-phase volume contents selected in other works for LSPS compaction of similar material systems [55, 56]. Here, suitable SLPS temperatures should be selected with regard to stable process control. On the one hand, temperature ranges should be selected in which there is only a small change in the liquid-phase volume content forming as a function of the sintering temperature so as to counteract adverse effects concerning temperature fluctuations in the sintering furnaces. On the other hand, a sufficiently high liquid-phase volume content of 20 to 40 vol.% (ideally 30 vol.%) should be present [52]. According to the calculations, both conditions are present when sintering temperatures are selected in the range of 1263 °C (approx. 20 vol.% liquid phase) to 1312 °C (40 vol.%). Liquid-phase volume contents that are too high are avoided for geometric stability and the drop in mechanical properties due to coarse grain formation. For the subsequent experimental validation of the calculations, the SLPS temperatures were derived from the calculated solidus temperature, at which a liquid-phase volume content of 0 vol.% (T1 = 1240 °C, meaning solid-state sintering at high temperature), 6.5 vol.% (T2 = 1260 °C), 28.5 vol.% (T3 = 1280 °C) and 35 vol.% (T4 = 1300 °C) was present. Figure 7 shows the relationship between the selected sintering temperature, the liquid-phase content that forms during SLPS sintering, the porosity, and the microstructure. The density values as a function of sintering temperature, determined using quantitative image analysis on at least three microstructural cross-sections, with the respective standard deviation, are listed in Table 3.

Microstructural images of the sintered samples a S-conv, b S-1240, c L-1260, d L-1280, and e L-1300. All microstructural images at 50 × magnification, 1 µm polished and unetched. The micrographs examined correspond to the sample extraction level of the specimens taken according to Fig. 8a
The prepared and examined micrographs correspond to the sample extraction layer of the samples taken for CT measurement in Fig. 8a. The sintered samples S-conv and S-1240 have pores with similar morphology to that of the green (Fig. 4a) and brown compacts (Fig. 4b). The pores have a triangular or diamond-shaped morphology and are arranged symmetrically. Pores with a diamond-shaped morphology are located in three rows with a vertical distance of approx. 100 µm and a horizontal distance of approx. 400 µm to each other. In contrast, pores with a triangular shape are evenly distributed with a vertical and horizontal spacing of approx. 250 µm.

Representation of pore distribution using created CT images. a Schematic representation of sample extraction for CT analysis from the printed cube samples. 3D CT volume image of samples: b S-conv, c S-1240, d L-1260, e L-1280, and f L-1300
Concerning the measured density values in Table 3, it is noticeable that the values determined using µCT measurements are at a higher density level than those determined using quantitative image analysis. This behavior is due to the inability to detect defects with a size of less than 1.6–3.2 µm, as described in [57]. With the help of quantitative image analysis, pores with smaller sizes could also be recorded so that the density is correspondingly lower. Compared to quantitative image analysis (measurement based on a cross-section of the structure), the µCT measurement allows the determination of the density in the sample volume. Despite the lower measurement resolution by µCT, especially for detecting small-sized pores, this data appears more representative and will be considered in the discussion below.
In the 3D volume representation of the experimental specimens S-conv (Fig. 8b) and S-1240 (Fig. 8c) obtained by µCT-investigations, the geometry of the voids (dark areas) addressed in Fig. 7a and b is clearly visible. The samples were taken in such a way that the volumes close to the edge (wall layer) and the volume in the sample (infill) can be examined simultaneously (Fig. 8a). In the region at the right edge, the insufficient connection of the extrudate strands in the wall layer is apparent. In the region on the left, the alternating 45° aligned gaps between the extrudate lines of the infill layer are evident. These different morphologies result from the build-up strategy of infill and the wall layer used (Fig. 1c). The row of diamond-shaped pores is formed during the build-up of the four-fold wall layer, as the extrudate is deposited with each successive layer covering the previous extrudate line of the wall layer. The triangular morphologies result from the 45° alternating deposition of the infill layer (Fig. 1c). This can be seen from the horizontal and vertical offset of the pores. The sinter compaction of both samples (S-conv; S-1240) took place below the solidus temperature and, thus, in the solid state. The porous microstructure confirms that sintering activity during solid-state sintering is not sufficiently high to eliminate the larger cavities [54]. Therefore, the specimens S-conv (density: 98.49 ± 0.05 vol.%) and S-1240 (density: 98.54 ± 0.04 vol.%) sintered in the solid state have equal high porosity of approx. 1.5 vol.%. Eliminating these cavities by adapting the material application strategy during the printing process is unsuitable since these cavities are essential as channels for the escape of the binder in the first and second debinding stages, as described above. In the case of a closed cavity due to a higher infill, internal pressures from the gas formation during thermal debinding would result, possibly causing fragmentation of the component. In contrast, the SLPS-sintered samples L-1260 (6.5 vol.% liquid phase), L-1280 (28.5 vol.% liquid phase), and L-1300 (35 vol.% liquid phase) are free from triangular or diamond-shaped pores. The µCT investigations in Fig. 8d to f confirm that by increasing the sintering temperature or the liquid-phase volume fraction formed, the pores created by the generative structure are compressed, and the porosity decreases. Sample L-1260 has a porosity of 0.23 ± 0.03 vol.%, whereas the microstructure of samples with an increased liquid-phase content L-1280 (0.08 ± 0.05 vol.%) and L-1300 (0.09 ± 0.07 vol.%) has a lower porosity in each case (Table 3). Even if the porosity of the samples sintered at 1280 and 1300 °C is at the same level (density values are within the measurement uncertainty), one might get the impression that higher SLPS sintering temperatures lead to poorer sinter compaction behavior. An explanation is provided by the work of German et al., who mentioned in Ref. [57] that higher SLPS-sinter temperatures promote Ostwald ripening of the metal cells, which is accompanied by bigger particle–particle distances and, thus, a lower capillary force. Because of the reduced capillary forces, the degree of sinter compaction decreases, associated with increased porosity. It can be concluded that optimum densification of the microstructure occurs at an SLPS temperature of 1280 °C and a volume fraction of the liquid phase of 28.5 vol.%. An increase in SLPS temperature to 1300 °C and a higher liquid-phase volume fraction of 35 vol.% show no significant improvement in density.
3.3 Influence of the sintering technique on the surface roughness of filament extruded parts
Additively manufactured components must often be post-treated by machining processes to achieve the required surface quality. Depending on the component’s geometry, this post-processing can be a complex and, therefore, time-consuming and costly process [58]. The aim is to achieve the lowest possible surface roughness during additive manufacturing. Therefore, the influence of the sintering temperature on the surface roughness of filament extruded components is described below. Figure 9 shows the images of the top and the lateral surfaces (“side-face”), taken by confocal laser scanning microscopy. The sample surface is marked by vertical (surface structure infill) and the lateral surface by horizontal lines (surface structure outfill). In the case of specimens S-conv and S-1240, a high surface roughness of values of 5.08 ± 0.62 µm and 4.12 ± 0.48 µm, respectively, was measured on the top surface (Fig. 9a and c). For the lateral surfaces, a roughness value of 11.45 ± 0.72 µm (S-conv) and 10.37 ± 0.65 µm (S-1240) was determined. Thus, solid-state sintering does not strongly affect the roughness values of the surfaces. If the specimens were SLPS sintered at a temperature of 1260 °C (sample S-1260, liquid-phase content: 6.5 vol.%), the roughness of the top surface is reduced to 3.62 ± 0.65 µm and that of the lateral surface to 9.45 ± 0.27 µm (Fig. 9e and f). As the liquid-phase volume fraction is further increased to 28.5 vol.% and 30 vol.%, the extrudate lines of the infill layer and the wall layer are no longer visible in Fig. 9g and h (L-1280) and Fig. 9i and k (L-1300). With increasing temperature, the liquid-phase volume fraction increases inside the sample and on its surface. Thus, the solid surface is wetted by the liquid phase. Due to cohesive forces, liquids always strive to form a low surface tension [63]. The surface tension measures the energy required to increase the surface area. The liquid phase has a lower surface tension than the solid surface components. As a result of the liquid phase striving to adopt the most energetically favorable surface tension, the solid components are assumed to be uniformly wetted with the liquid phase [63]. This effect is evident in the images of the top and lateral surfaces of samples L-1280 (Fig. 9g and h) and L-1300 (Fig. 9i and k).

Representation of the surfaces of a S-conv, c S-1240, e L-1260, g L-1280, i L-1300 and the lateral surfaces of b S-conv, d S-1240, f L-1260, h L-1280, k L-1300, recorded by laser scanning microscope
In the figures, extrudate lines are no longer visible, and the roughness values for the top and lateral surfaces of the samples with increased liquid-phase volume content (L-1280: 28.5 vol%; and L-1300: 30 vol%) are lower than those of the other sintered samples; this relationship is summarized in Fig. 10. The Sa roughness values of the top and lateral surfaces are about 2.73 ± 0.53 µm and 4.71 ± 0.32 µm for sample L-1280, and approx. 2.62 ± 024 µm and 4.64 ± 1.13 µm for sample L-1300. Here, the roughness of the lateral surface is about 1.7 to 1.8 times higher than the roughness of the top surface. In summary, SLPS sintering allows both the elimination of internal porosity and the smoothening of the surface of the components, so less post-processing by, e.g., machining is required to adjust the required surface finish. The SLPS-sintered samples have a surface roughness similar to those of products manufactured by investment casting [63]. Comparable to the density values of the SLPS compacted microstructure, the surface analysis also shows minimum roughness values at an SLPS temperature of 1280 °C (liquid-phase volume content: 28.5 vol.%). No significant reduction in roughness is observed at an SLPS temperature of 1300 °C.

Measurement of roughness values Sa in µm of the top and lateral surfaces of the respective test specimens
To classify these results concerning the state of knowledge, reference should be made to the work of Tay et al. [59] and Shrestha et al. [60], which measured the surface roughness of a PBF-processed material (AISI316L) with Ra = 18–21 µm or for MIM-processed materials made of AISi316L with Ra = 401–58 nm. In addition to the influence of the manufacturing process on the surface roughness, the starting powder significantly influences the surface roughness that develops. Shrestha et al. used a starting powder for PBF processing of 9–45 µm; the MIM feedstock in work by Tay et al. possesses a powder size of less than 5 µm (d50 = 2.4 µm). In this study, the size of the starting particles was less than 20 µm. Thereby, the achieved roughness of the solid-state sintered samples is approximately half, and the SLPS-stinted samples with Sa = 5 µm have a surface roughness that corresponds to only a quarter of the PBF-produced samples in Ref. [60]. In particular, SLPS compaction can effectively reduce the roughness, and thus the necessary amount of post-machining compared to the solid-state sintered or PBF-processed samples.
3.4 Microstructure formation during SLPS
In the following, microstructure formation on the microscale of the filament extruded specimens, post-compacted by solid-state and SLPS sintering, will be discussed. In Fig. 11b, the microstructure of the solid-sintered sample according to the manufacturer's specifications (sample S-conv) is shown. According to the XRD investigations at Delta, TU Dortmund (Fig. 12), the microstructure consists of a bcc-Fe phase, fcc-phase (retained austenite) and orthorhombic Cr-rich carbides of the M7C3 type. The fine microstructure of sample S-conv can be traced back to the gas-atomized starting powder used (Fig. 11a) and the solid-state sintering process associated with short-range diffusion processes. The gas-atomized powder contains small metal cells with a cell size lower than 5 µm, surrounded by eutectic carbides of the M7C3 type. During the solid-state sintering process, the metal cells undergo a slight Ostwald ripening, and the morphology of the Cr-rich carbides is transformed from network-like to spherical, with a diameter of 1 to 4 µm. The effective driving force for the spheroidization of the eutectic carbides with previous herringbone morphology is the reduction of the interfacial energy between orthorhombic M7C3 and fcc metal matrix, considering the strain energy and the associated misfit stresses [61]. However, the carbides counteract the pronounced Ostwald ripening of the metal cells during solid-state sintering so that a fine microstructure is retained. Sample S-1240 (densified with no liquid phase, Fig. 11c) has a microstructure similar to sample S-conv (spherical M7C3 carbides finely distributed in the metal matrix). Quantitative and qualitative diffraction analysis according to the measurements at Beamline 9 was performed with the Maud software to measure the retained austenite volume content of the sintered specimens more precisely. The results are summarized in Table 4.

SEM microstructural images of the test specimens: a powder particle (10k × magnification), b S-conv (5k × magnification), c S-1240 (5k × magnification), d L-1260 (1k × magnification), e L-1280 (1k × magnification), and f L-1300 (1k × magnification)

Intensities of the XRD measurement on Beamline 9 (Delta, TU Dortmund) at states S-conv (red), S-1240 (green), L-1260 (purple), L-1280 (turquoise), and L-1300 (pink). The diagram shows the reflections of austenite (fcc), ferrite (bcc), M7C3, M23C6, and MC phases determined in the microstructure for the respective test specimens
Thus, both solid-sintered samples have the same microstructure. Korobahee et al. [62] investigated the influence of the austenitizing temperature on the formation of the microstructure of D2 (X153CrMoV12; 1.2379) steel using Magnetic Hysteresis and Barkhausen Noise Parameters. They discovered that as the austenitizing temperature increases, the retained austenite volume fraction increases from 15.7 vol.% for austenitizing at 1000 °C to 42 vol.% at 1130 °C. As a reason for the increased retained austenite volume content, Kahrobaee et al. [62] mentioned the increasing dissolution of the M7C3-type carbides, which was confirmed by means of equilibrium calculations in Fig. 6. In particular, the elements Cr, Mo, C, and V accumulate in the austenite phase with increasing austenitization or sintering temperature and stabilize it. With the stabilization of the austenitic phase, proeutectoid carbide precipitation and the phase formation of pearlite and bainite are shifted toward longer times. At the same time, the martensite start temperature and martensite finish temperature drop. If the martensite finish temperature falls below the temperature of the quenching medium (here, room temperature), untransformed austenite, so-called retained austenite (RA), remains in the microstructure. Contrary to the work of Kahrobaee et al. [62], which mentions a retained austenite volume content of 50% to 60% by volume after quenching from 1130 °C to 1200 °C for D2 (X153CrMoV12; 1.2379) steel, the samples S-conv and S-1240 have no RA volume content. This fact can be attributed to the slow furnace cooling in the vacuum atmosphere. With respect to the time–temperature transformation behavior of D2 (X153CrMoV12; 1.2379) steel (see TTT diagram in Pillai and Karthikeyan [63]), proeutectoid carbide formation and cooling through the pearlite and bainite phases took place. The microstructure of the samples S-conv and S-1240, thus, consists of the phases ferrite (bcc) and Cr-rich carbides of type M7C3, which can be confirmed using the EBSD phase map in Fig. 13b and the results of the XRD investigations in Table 4. EBSD mappings were performed to investigate the phase distribution.

Crystal orientation maps of the samples a) S-conv and c) L-1280, highlighted by inverse pole figure (IPF). Phase maps of the samples b) S-conv and d) L-1280: ferrite (blue), austenite (yellow), and M7C3 (red). Representative for all other SLPS-sintered specimens (L-1260 to L-1300)
In contrast to the solid-state sintered specimens, the microstructure of the SLPS-sintered specimens consists of austenitic metal cells surrounded by a seam of eutectic Cr-rich carbides of M7C3 type with a herringbone-like morphology (Fig. 11d–f and Fig. 13d). In Fig. 13d, the EBSD mapping of specimen L-1280 is shown as a representative for all other SLPS-sintered specimens (L-1260 to L-1300). EBSD mappings were performed to investigate the phase distribution. With rising SLPS temperature or formed liquid-phase volume content, the average metal cell size and the width of the Cr carbide seams increase, as shown in Table 5. EDS mappings were performed to investigate the distribution of the elements. In Fig. 14, the EDS mapping of specimen L-1280 is shown as a representative for all other SLPS-sintered specimens (L-1260 to L-1300). In particular, large amounts of the elements Cr, Mo V, and C are located in the eutectic carbides. By means of EDS point measurements at three different positions, the chemical composition of the mixed carbides was measured to Fe34Cr26Mo2V4C34.

EDS mapping of sample L-1280 showing the element distribution on the microscale, representative for all other SLPS-sintered specimens (L-1260 to L-1300)
In the following, the microstructure formation process of the SLPS-sintered specimens will be explained based on the microstructure of the starting material used, the gas-atomized powder. In this regard, reference is made to Fig. 15, which shows a schematic of the flow of the SLPS process. When the solidus temperature of 1256 °C is exceeded, the low-melting phase (Cr-rich M7C3) within the powder particles melts, separating the powder particles into smaller fragments [52, 53, 64].

Schematic description of the mechanisms involved in SLPS sintering and the associated liquid-phase formation, powder fragmentation, powder rearrangement, and densification of the material
Gaps can be closed by particle rearrangement processes and melt infiltration due to capillary forces. As sintering progresses, several smaller metal cells can sinter together to form larger metal cells or grow through Ostwald ripening. During subsequent cooling from the sintering heat, the low-melting phase solidifies along the metal cell interface and forms Cr-rich carbide seams along the metal cells. In the process, the seam width increases toward a larger metal cell size diameter ranging from 1.7 ± 0.5 µm (cell size = 63 ± 10 µm; sample L-1260) to 3.7 ± 0.4 µm (cell size = 86 ± 8 µm; sample L-1300). Due to the reduced specific surface of the metal cells and a constant M7C3 volume fraction, the eutectic carbide seams to increase as a consequence of raising the SLPS temperature. Differences in the microstructure of the SLPS-sintered specimens can be seen in the phase composition shown in the diffractograms in Fig. 12. The retained austenite volume content was determined using X-ray diffraction and the Rietveld method and is listed in Table 4.
The RA volume content of the phase increases from sample L-1260 (6.5 ± 0.1 vol.%) through sample L-1280 (35.17 ± 0.0 vol.%) to sample L-1300 (38.73 ± 0.9 vol.%). The high RA content can be attributed to the increase in the solution state by the carbide dissolution, as described above. In contrast to the solid-state sintered samples, however, no pearlitic–bainitic–austenitic structure forms due to slow cooling; rather, a martensitic–bainitic–austenitic microstructure is formed. This finding can be verified indirectly via the measured hardness of the SLPS-sintered samples. For example, the hardness of sample L-1260 is 633 ± 8 HV10. The SLPS-sintered samples L-1280 and L-1300 have a lower hardness of 506 ± 3 HV10 and 538 ± 4 HV10, due to the higher content of soft retained austenite. Even if the SLPS-sintered samples were subjected to slow sample cooling in the vacuum furnace, the high RA volume content is based on the high solution state. This result correlates with the results of Biggnozi et al., which investigated the effect of the austenitization temperature on the formed RA volume fraction of the ledeburitic cold work tool steel X190CrMoV20-4-1 [65]. They were able to show that RA volume fraction increases from 4.3 vol.% (less than 1100 °C), over 29.6 vol.%(1100–1120 °C) to 55.6 vol.% (1120 °C) by increasing austenitization temperature. They attributed the increased retained austenite volume fraction to the more pronounced dissolution of the Cr-rich carbides in the direction of a higher austenitization temperature. This behavior results in a higher solution state of the metal matrix, especially of the elements Cr and C, lowering the Ms and Mf temperatures. According to the thermodynamic calculations in Fig. 6, the austenite phase in the SLPS-sintered samples has significantly higher Cr, Mo, V, and C contents than in the solid-phase sintered samples (Table 6). EDS point analyses (average of three measurements) within the metal cells in sample L-1280 confirm that the Cr (12.13 ± 0.34 mass.%), Mo (1.69 ± 0.53 mass.%), and V (0.62 ± 0.17 mass.%) contents dissolved in the austenite phase during sintering are retained. In particular, the elements Cr, Mo, and V prevent a subsequent C diffusion and, thus, the formation of a bainitic–pearlitic microstructure. Consequently, the bainite and pearlite phase fields are shifted in the direction of longer cooling times in the TTT diagram by Pillai and Karthikeyan [63].
3.5 Hardness-tempering behavior
The material group under consideration has to feature a high hardness so that wear can be counteracted during operation. Even if the wear properties (system properties) play an important role, the following section only focuses on the hardness because the complex tribomechanical relationships cannot be described in the necessary depth in a short section. Further work deals with the basic description of the wear properties, taking into account the microstructures manufactured in consideration of the manufacturing process and the hat treatment state. In the case of ledeburitic cold work steels, hardness is adjusted by an appropriate heat treatment consisting of quenching and tempering in the regime of secondary hardness. Since the location of the secondary hardness maximum depends mainly on the solution state after hardening (matrix potential), the hardness-tempering behavior will be examined below, and an optimal heat treatment specification will be derived. The hardness values of the sintered samples show that the material has insufficient properties for direct use as a material for tools after solid state (hardness ~ 247–289 HV10) or SLPS sintering (hardness ~ 506–633 HV10). The material under consideration is a ledeburitic cold work steel whose properties are adjusted by quenching and subsequent multiple tempering in the area of the secondary hardness maximum. The desired microstructure consists of tempered martensite with secondary carbides, finely distributed in the metal matrix and thus increasing the hardness and strength due to precipitation hardening. The selection of optimal austenitizing temperatures depends on the C content dissolved in the austenite phase during austenitization, which should be in the range of 0.5 to 0.6 mass%. Sufficiently high C contents in the phase austenite are set by selecting sufficiently high austenitizing temperatures. With increasing austenitization temperatures, the stability of the Cr-rich M7C3 decreases, and the austenitic phase is enriched by the elements Cr, Mo, V, and C due to carbide dissolution (see the thermodynamic calculation in Fig. 6). According to the manufacturer's specification [66], hardening occurs at TAUS = 1040 °C, associated with a dissolved C content of 0.552 mass% in the austenite phase. In the following discussion, the samples S-conv (conventional processing route) and L-1280 (sample with the best combination of high density, low surface roughness, and fine microstructure) and cast (cast and hot worked reference material) will be considered. All materials were oil quenched from 1050 °C, and the hardness-tempering diagram was evaluated by double-tempered samples in the temperature range from room temperature to 600 °C using Vickers hardness testing. According to the hardness and tempering diagrams (Fig. 16), maximum secondary hardness occurs at a tempering temperature of 525 °C. Table 7 compares the hardness values of the samples in the different states. Sample S-conv has the lowest secondary hardness at 530 ± 12 HV10. In comparison, the highest secondary hardness of 682 ± 6 HV10 (58–60 HRC) was determined for sample L-1280, concurring with hardness values from other studies on material D2 (X153CrMoV12; 1.2379) [67,68,69].

Hardness–temperature diagram for specimens L-1280 (black), S-conv (red), and Cast (blue)
Compared to the hardness values presented here, Markforged lists higher hardness values of 55 HRC in its product data sheet [66], corresponding to a Vickers hardness of 595 HV10. It should be noted that Markforged’s classic heat treatment involves hardening at 1040 °C, followed by a lower tempering temperature of 200 °C. According to the hardness–temperature diagram in Fig. 16, a hardness of approx. 608 ± 16 HV10 was determined for the S-conv condition at a tempering temperature of 200 °C, which is in close agreement with the data given by Markforged.
4 Conclusion
This work investigated the filament extrusion processing of D2 (X153CrMoV12; 1.2379) tool steel and a novel post-compaction by SLPS sintering. To the authors’ knowledge, it was shown for the first time that additive manufacturing of ledeburitic cold work tool steel D2 using FFF (ADAM technology) with subsequently adapted SLPS compaction and heat treatment leads to dense microstructures, with properties (hardness) being at a similar level as that same material in cast and heat-treated condition. In the considered manufacturing route, the shape of the component (green body) is processed using FFF, whereby the microstructure is adjusted in a subsequent sintering and heat treatment process. Decoupling the microstructure formation process from the "printing" process offers the advantage that materials can also be processed that cannot be processed by PBF or DED without additional measures (preheating, alloy adjustment). The following key findings could be derived from this work.
Before SLPS, sufficient thermal debinding at 400 °C is necessary to remove the long-chain binder to avoid swelling of the samples.
The optimal SLPS sintering temperature was determined to be 1280 °C, whereby specimens with a high relative density of 99.11 ± 0.21 vol.% could be achieved. At lower sintering temperatures, an insufficient liquid-phase volume content is formed, leading to samples with higher porosity. Sintering temperatures exceeding 1280 °C cause increased Ostwald ripening of the metal cells, which is associated with reduced sinter compaction due to decreasing capillary forces. Due to the lowered capillary forces, sinter compaction is counteracted, which can result in a higher porosity.
SLPS compaction enables the reduction of surface roughness, whereby the driving force can be attributed to the reduction of the surface tension of the formed liquid phase on the specimens’ surfaces.
The lateral roughness of the samples is approximately 1.7–1.8 times greater than the surface roughness of the top layer. These differences in surface roughness should be considered in the pre-processing step (meaningful placement of the structure to be built on the construction platform) to reduce a high effort for post-processing by machining, for example.
The microstructure sintered in solid-state features spherical Cr-rich M7C3, finely embedded in a metal matrix consisting of pearlite and bainite. After SLPS sintering, the carbides form a network-shaped morphology, and the metal matrix consists of retained austenite and martensite. Thereby, the retained austenite volume fraction in the microstructure increases in the direction of a higher SLPS temperature. The higher retained austenite volume fraction was attributed to increased carbide dissolution and a corresponding higher solution state in the metal matrix. The higher solution state reduces the martensite start and martensite finish temperature, so that non-transformed austenite remains in the metal matrix.
The hardness of the SLPS-sintered material (675–680 HV10) exceeds the hardness of the cast reference material (~ 600–620 HV10) after quenching and tempering. The described method, therefore, offers high potential for industrial purposes in the context of rapid tooling to additively manufacture corresponding tools from the ledeburitic cold work steel D2.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Leal R, Barreiros FM, Alves L et al (2017) Additive manufacturing tooling for the automotive industry. Int J Adv Manuf Technol 92:1671–1676. https://doi.org/10.1007/s00170-017-0239-8
Cheah C, Chua C, Lee C et al (2005) Rapid prototyping and tooling techniques: a review of applications for rapid investment casting. Int J Adv Manuf Technol 25:308–320. https://doi.org/10.1007/s00170-003-1840-6
Fedorov K, Fayazbakhsh K, Ravindran C (2022) Surface roughness and dimensional tolerances in A319 alloy samples produced by rapid investment casting process based on fused filament fabrication. Int J Adv Manuf Technol 119:4423–4437. https://doi.org/10.1007/s00170-021-08644-5
Chantzis D, Liu X, Politis DJ et al (2020) Review on additive manufacturing of tooling for hot stamping. Int J Adv Manuf Technol 109(1–2):87–107. https://doi.org/10.1007/s00170-020-05622-1
Palmieri ME, Tricarico L (2022) Influence of Conformal Cooling Channel Parameters on Hot Stamping Tool and Press-Hardening Process. KEM 926:645–654. https://doi.org/10.4028/p-5kczh8
Lakner T, Bergs T, Schraknepper D (2020) A Novel Test Bench to Investigate the Effects of the Tool Rotation on Cutting Fluid Jets to Improve the Tool Design via Additive Manufacturing. Procedia CIRP 91:9–14. https://doi.org/10.1016/j.procir.2020.02.144
Larsson J, Karlsson P, Ekengren J, Pejryd L (2021) Enhanced Cooling Design in Wire Drawing Tooling Using Additive Manufacturing. In: Meboldt M, Klahn C (eds) Industrializing Additive Manufacturing. Springer International Publishing, Cham, pp 426–436
Traxel KD, Bandyopadhyay A (2019) First Demonstration of Additive Manufacturing of Cutting Tools using Directed Energy Deposition System: Stellite™-Based Cutting Tools. Addit Manuf 25:460–468. https://doi.org/10.1016/j.addma.2018.11.019
R. Hölker-Jäger, A.E. Tekkaya (2017) 17: Additive manufacture of tools and dies for metal forming, Editor(s): Milan Brandt, In Woodhead Publishing Series in Electronic and Optical Materials, Laser Additive Manufacturing, Woodhead Publishing https://doi.org/10.1016/B978-0-08-100433-3.00017-8
Levy GN, Schindel R, Kruth JP (2003) Rapid Manufacturing And Rapid Tooling With Layer Manufacturing (LM) Technologies, State Of The Art And Future Perspectives. CIRP Ann 52(2):589–609. https://doi.org/10.1016/S0007-8506(07)60206-6
Boes J, Röttger A, Mutke C et al (2018) Microstructure and mechanical properties of X65MoCrWV3-2 cold-work tool steel produced by selective laser melting. Addit Manuf 23:170–180. https://doi.org/10.1016/j.addma.2018.08.005
Hentschel O, Scheitler C, Fedorov, et al (2017) Experimental investigations of processing the high carbon cold-work tool steel 1.2358 by laser metal deposition for the additive manufacturing of cold forging tools. J Laser Applicat 29(2):22307. https://doi.org/10.2351/1.4983247
Krell J, Röttger A, Geenen K, Theisen W (2018) General investigations on processing tool steel X40CrMoV5-1 with selective laser melting. J Mater Process Technol 255:679–688. https://doi.org/10.1016/j.jmatprotec.2018.01.012
Mertens R, Vrancken B, Holmstock N et al (2016) Influence of Powder Bed Preheating on Microstructure and Mechanical Properties of H13 Tool Steel SLM Parts. Phys Procedia 83:882–890. https://doi.org/10.1016/j.phpro.2016.08.092
Wu L, Das S, Gridin W et al (2021) Hot Work Tool Steel Processed by Laser Powder Bed Fusion: A Review on Most Relevant Influencing Factors. Adv Eng Mater 23(7):2100049. https://doi.org/10.1002/adem.202100049
Röttger A, Boes J, Großwendt F, Weber S (2023) Description of a new concept for the development of adapted hot-work tool steels for laser-powder bed fusion. Addit Manuf. https://doi.org/10.1016/j.addma.2022.103292
Jägle EA, Sheng Z, Kürnsteiner P et al (2016) Comparison of Maraging Steel Micro- and Nanostructure Produced Conventionally and by Laser Additive Manufacturing. Materials (Basel, Switzerland) 10:1
Roberts D, Zhang Y, Charit I, Zhang J (2018) A comparative study of microstructure and high-temperature mechanical properties of 15–5 PH stainless steel processed via additive manufacturing and traditional manufacturing. Prog Addit Manuf 3(3):183–190. https://doi.org/10.1007/s40964-018-0051-5
Ben-Artzy A, Reichardt A, Borgonia J-P et al (2021) Compositionally graded SS316 to C300 Maraging steel using additive manufacturing. Mater Des. https://doi.org/10.1016/j.matdes.2021.109500
Bourell D, Kruth JP, Leu M et al (2017) Materials for additive manufacturing. CIRP Ann 66(2):659–681. https://doi.org/10.1016/j.cirp.2017.05.009
Guo L, Zhang L, Andersson J, Ojo O (2022) Additive manufacturing of 18% nickel maraging steels: Defect, structure and mechanical properties: A review. J Mater Sci Technol 120:227–252. https://doi.org/10.1016/j.jmst.2021.10.056
Meneghetti G, Rigon D, Cozzi D et al (2017) Influence of build orientation on static and axial fatigue properties of maraging steel specimens produced by additive manufacturing. Proced Struct Integrity 7:149–157. https://doi.org/10.1016/j.prostr.2017.11.072
Meneghetti G, Rigon D, Gennari C (2019) An analysis of defects influence on axial fatigue strength of maraging steel specimens produced by additive manufacturing. Int J Fatigue 118:54–64. https://doi.org/10.1016/j.ijfatigue.2018.08.034
Shakerin S, Hadadzadeh A, Amirkhiz BS et al (2019) Additive manufacturing of maraging steel-H13 bimetals using laser powder bed fusion technique. Addit Manuf. https://doi.org/10.1016/j.addma.2019.100797
Boes J, Röttger A, Mutke C et al (2023) Microstructure and Properties of a Novel Carbon-Martensitic Hot Work Tool Steel Processed by Laser Additive Manufacturing without Preheating. Steel Res Int. https://doi.org/10.1002/srin.202200439
Ghaffari M, Vahedi Nemani A, Nasiri A (2022) Microstructural evolution and mechanical performance after precipitation hardening of PH 13–8Mo martensitic stainless steel fabricated by wire arc additive manufacturing. Materialia. https://doi.org/10.1016/j.mtla.2022.101507
Großwendt F, Röttger A, Strauch A et al (2021) Additive manufacturing of a carbon-martensitic hot-work tool steel using a powder mixture: microstructure, post-processing, mechanical properties. Mater Sci Eng, A. https://doi.org/10.1016/j.msea.2021.142038
Iakovakis E, Avcu E, Roy MJ et al (2022) Wear resistance of an additively manufactured high-carbon martensitic stainless steel. Sci Rep 12(1):12554. https://doi.org/10.1038/s41598-022-15621-9
Zhang M, Chen C, Qin L et al (2018) Laser additive manufacturing of M2 high-speed steel. Mater Sci Technol 34(1):69–78. https://doi.org/10.1080/02670836.2017.1355584
Botero C, Ramsperger M, Selte A et al (2020) Additive Manufacturing of a Cold-Work Tool Steel using Electron Beam Melting. Steel Res Int 91(5):1900448. https://doi.org/10.1002/srin.201900448
Sander J, Hufenbach J, Giebeler L et al (2016) Microstructure and properties of FeCrMoVC tool steel produced by selective laser melting. Mater Des 89:335–341. https://doi.org/10.1016/j.matdes.2015.09.148
Platl J, Bodner S, Hofer C et al (2022) Cracking mechanism in a laser powder bed fused cold-work tool steel: the role of residual stresses, microstructure and local elemental concentrations. Acta Mater. https://doi.org/10.1016/j.actamat.2021.117570
Lim S-H, Ryou K, Jang K et al (2021) Hot cracking behavior of additively manufactured D2 steel. Mater Charact. https://doi.org/10.1016/j.matchar.2021.111217[36]
Yadav S, Dileep K, Jinoop AN et al (2022) Laser Directed Energy Deposition of High-Carbon High-Chromium D2 Tool Steel Structures: Processing, Heat Treatment and Material Behavior. J of Materi Eng and Perform. https://doi.org/10.1007/s11665-022-07470-4
Galati M, Minetola P (2019) Analysis of Density, Roughness, and Accuracy of the Atomic Diffusion Additive Manufacturing (ADAM) Process for Metal Parts. Materials (Basel, Switzerland) 12:24. https://doi.org/10.3390/ma12244122
Bouaziz MA, Djouda JM, Kauffmann J, Hild F (2020) Microscale mechanical characterization of 17–4PH stainless steel fabricated by Atomic Diffusion Additive Manufacturing (ADAM). Proced Struct Integrity 28:1039–1046. https://doi.org/10.1016/j.prostr.2020.11.119
Henry TC, Morales MA, Cole DP et al (2021) Mechanical behavior of 17–4 PH stainless steel processed by atomic diffusion additive manufacturing. Int J Adv Manuf Technol 114(7–8):2103–2114. https://doi.org/10.1007/s00170-021-06785-1
Jagtap BM, Kakandikar GM, Jawade SA (2022) Mechanical Behavior of Inconel 625 and 17–4 PH Stainless Steel Processed by Atomic Diffusion Additive Manufacturing. In: Dave, H.K., Dixit, U.S., Nedelcu, D. (eds.) Recent Advances in Manufacturing Processes and Systems. Lecture Notes in Mechanical Engineering, pp. 583–594. Springer Nature Singapore, Singapore
Jiang D, Ning F (2021) Additive Manufacturing of 316L Stainless Steel by a Printing-Debinding-Sintering Method: Effects of Microstructure on Fatigue Property. J Manufact Sci Eng 143:9. https://doi.org/10.1115/1.4050190
Sadaf M, Bragaglia M, Nanni F (2021) A simple route for additive manufacturing of 316L stainless steel via Fused Filament Fabrication. J Manuf Process 67:141–150. https://doi.org/10.1016/j.jmapro.2021.04.055
Roccetti Campagnoli M, Galati M, Saboori A (2021) On the processability of copper components via powder-based additive manufacturing processes: Potentials, challenges and feasible solutions. J Manuf Process 72:320–337. https://doi.org/10.1016/j.jmapro.2021.10.038
Kingery WD (1959) Densification during Sintering in the Presence of a Liquid Phase I. Theory. J Appl Phys 30(3):301–306. https://doi.org/10.1063/1.1735155
Andersson J-O, Helander T, Höglund L et al (2002) Thermo-Calc & DICTRA, computational tools for materials science. Calphad 26(2):273–312. https://doi.org/10.1016/S0364-5916(02)00037-8
Krywka C, Sternemann C, Paulus M et al (2007) The small-angle and wide-angle X-ray scattering set-up at beamline BL9 of DELTA. J Synchrotron Radiat 14(Pt 3):244–251. https://doi.org/10.1107/S0909049507009727
Rodi DJ, Mandava S, Gore DB et al (2007) Detection of functional ligand-binding events using synchrotron x-ray scattering. J Biomol Screen 12(7):994–998. https://doi.org/10.1177/1087057107306104
Wagner MA, Hadian A, Sebastian T et al (2022) Fused filament fabrication of stainless steel structures - from binder development to sintered properties. Addit Manuf. https://doi.org/10.1016/j.addma.2021.102472
Calvert P, Cima M (1990) Theoretical Models for Binder Burnout. J Am Ceram Soc 73(3):575–579. https://doi.org/10.1111/j.1151-2916.1990.tb06555.x
Donald F (2012) Heaney, In Woodhead Publishing Series in Metals and Surface Engineering, Handbook of Metal Injection Molding, Woodhead Publishing Pages i-iii, ISBN 9780857090669, https://doi.org/10.1533/9780857096234.frontmatter
Shengjie Y, Lam YC, Yu SCM, Tam KC (2001) Two-dimensional simulation of mass transport in polymer removal from a powder injection molding compact by thermal debinding. J Mater Res 16(8):2436–2451. https://doi.org/10.1557/JMR.2001.0334
Shengjie Y, Lam YC, Yu SCM, Tam KC (2002) Thermal debinding modeling of mass transport and deformation in powder-injection molding compact. Metall and Materi Trans B 33(3):477–488. https://doi.org/10.1007/s11663-002-0058-6
German RM, Suri P, Park SJ (2009) Review: liquid phase sintering. J Mater Sci 44(1):1–39. https://doi.org/10.1007/s10853-008-3008-0
German RM (1991) A Quantitative Theory for Supersolidus Liquid Phase Sintering. Powder Metall 34(2):101–107. https://doi.org/10.1179/pom.1991.34.2.101
German RM (1997) Supersolidus liquid-phase sintering of prealloyed powders. Metall and Mat Trans A 28(7):1553–1567. https://doi.org/10.1007/s11661-997-0217-0
Röttger A, Weber S, Theisen W (2011) Supersolidus liquid-phase sintering of ultrahigh-boron high-carbon steels for wear-protection applications. Mater Sci Eng, A. https://doi.org/10.1016/j.msea.2011.10.118
Farayibi PK, Blüm M, Weber S (2020) Densification of a high chromium cold work tool steel powder in different atmospheres by SLPS: Microstructure, heat treatment and micromechanical properties. Mater Sci Eng A. https://doi.org/10.1016/j.msea.2020.139053
Farayibi PK, Hankel J, F. van gen Hassend, et al (2023) Tribological characteristics of sintered martensitic stainless steels by nano-scratch and nanoindentation tests. Wear. https://doi.org/10.1016/j.wear.2022.204547
Lu QY, Nguyen NV, Hum AJW et al (2020) Identification and evaluation of defects in selective laser melted 316L stainless steel parts via in-situ monitoring and micro computed tomography. Addit Manuf. https://doi.org/10.1016/j.addma.2020.101287
Xing P, Kong L, Fuh JYH, Wang H (2021) A Review of Post-Processing Technologies in Additive Manufacturing. J Manufact Mater Process 5(2):38. https://doi.org/10.3390/jmmp5020038
Tay BY, Liu L, Loh NH et al (2005) Surface roughness of microstructured component fabricated by μMIM. Mater Sci Eng A 396(12):311–319. https://doi.org/10.1016/j.msea.2005.01.033
Shrestha R, Simsiriwong J, Shamsaei N (2019) Fatigue behavior of additive manufactured 316L stainless steel parts: Effects of layer orientation and surface roughness. Addit Manuf 28:23–38. https://doi.org/10.1016/j.addma.2019.04.011
Li Z, Jia P, Liu Y, Qi H (2019) Carbide Precipitation, Dissolution, and Coarsening in G18CrMo2–6 Steel. Metals 9(9):916. https://doi.org/10.3390/met9090916
Kahrobaee S, Kashefi M (2015) Assessment of Retained Austenite in AISI D2 Tool Steel Using Magnetic Hysteresis and Barkhausen Noise Parameters. J of Materi Eng and Perform 24(3):1192–1198. https://doi.org/10.1007/s11665-014-1337-5
Pillai N, Karthikeyan R (2018) Prediction of ttt curves of cold working tool steels using support vector machine model. IOP Conf Ser Mater Sci Eng 346:12067. https://doi.org/10.1088/1757-899X/346/1/012067
Liu Y, Tandon R, German RM (1995) Modeling of supersolidus liquid phase sintering: II. Densification. Metall and Mat Trans A 26(9):2423–2430. https://doi.org/10.1007/BF02671256
Bignozzi MC, Calcinelli L, Carati M et al (2020) Effect of Heat Treatment Conditions on Retained Austenite and Corrosion Resistance of the X190CrVMo20-4-1 Stainless Steel. Met Mater Int 26:1318–1328. https://doi.org/10.1007/s12540-019-00384-2
Markforged: D2 Tool Steel. Material Datasheet. https://static.markforged.com/downloads/D2-tool-steel.pdf
Al-Qawabah S, Mostafa A, Al-Rawajfeh A, Al-Qawabeha U (2020) Effect of heat treatment on the grain size, microhardness and corrosion behavior of the cold-working tool steels AISI D2 and AISI O1. Mater Tehnol 54(6):785–790. https://doi.org/10.17222/mit.2020.035
Bourithis L, Papadimitriou GD, Sideris J (2006) Comparison of wear properties of tool steels AISI D2 and O1 with the same hardness. Tribol Int 39(6):479–489. https://doi.org/10.1016/j.triboint.2005.03.005
Kara F, Karabatak M, Ayyıldız M, Nas E (2020) Effect of machinability, microstructure and hardness of deep cryogenic treatment in hard turning of AISI D2 steel with ceramic cutting. J Market Res 9(1):969–983. https://doi.org/10.1016/j.jmrt.2019.11.037
Acknowledgements
The authors thank Dr. Andrea Thöne of Bruker Nano GmbH (Am Studio 2D, 12489 Berlin, Germany) and Dr.-Ing. Michael Kroh of Vorwerk Elektrowerke GmbH and Co. KG (Blombacher Bach 3, 42287 Wuppertal, Germany) for their support. We also want to thank, in particular, for granting measurement time at the Center for Synchrotron Radiation at the TU Dortmund on beamline 9 to carry out the diffraction experiments. We thank Dr. Christian Sternemann of TU Dortmund (Maria-Goeppert-Mayer-Str. 2, 44221 Dortmund, Germany) for his support during the diffraction experiments.
Funding
Open Access funding enabled and organized by Projekt DEAL. The present work was developed and funded as part of the Federal Ministry for Economic Affairs and Climate Action (Bundesministerium für Wirtschaft und Klimaschutz—BMWK) joint project “Aif ZIM: AddTOOL-Additive manufacturing of complex-formed tools for cold machining of wear-resistant materials” under grant number KK5066807KX2.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Röttger, A., Wieczorek, L., Schmidtseifer, N. et al. Additive manufacturing of carbon-martensitic hardening ledeburitic cold work tool steels using Fused Filament Fabrication and subsequent Supersolidus Liquid-Phase Sintering. Prog Addit Manuf (2024). https://doi.org/10.1007/s40964-024-00650-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40964-024-00650-9