
Abstract
Purpose
An increasing body of evidence suggests that bone resorbing osteoclasts are important—but as yet underrated—cellular initiators of bone formation. Furthermore, macrophages also have shown stimulatory effects on the osteogenic differentiation of mesenchymal stromal cells (MSCs). Consequently, we here investigated whether human macrophage- and osteoclast-laden carrier materials can induce ectopic bone formation upon subcutaneous implantation in nude mice.
Methods
Human osteoclast precursors were isolated and differentiated toward macrophages. Subsequently, these macrophages were seeded onto two types of cell carrier materials (i.e., electrospun polymeric scaffolds and devitalized bovine bone granules) and differentiated for 14 days toward osteoclasts. DNA assay and fluorescent nuclei staining were performed. Osteoclast differentiation was assessed by a tartrate-resistant acid phosphatase (TRAP)-activity assay, TRAP, and immunocytochemical staining for β3 integrin. After 60 days of implantation into nude mice, specimens were retrieved, histologically processed, and stained with hematoxylin and eosin (HE) as well as for TRAP to study ectopic bone formation and osteoclast activity, respectively.
Results
Osteoclast precursors limitedly adhered to both material types. Osteoclast-laden samples showed increased intracellular gross TRAP-activity on both cell carrier types, TRAP staining on polymeric electrospun scaffolds, and positive β3 integrin staining on decellularized bovine bone granules compared to the macrophage-laden materials. We observed that only the positive control samples loaded with bone morphogenetic protein-2 (BMP-2) induced ectopic bone formation and TRAP signal.
Conclusion
We conclude that neither human macrophage- nor osteoclast-laden constructs are capable to induce ectopic bone formation under the current experimental set-up.
Lay summary
Interestingly, increasing amounts of evidence suggest that osteoclasts—the cells responsible for breaking down bone tissue—can trigger bone formation. Therefore, we here aimed to study whether blood-derived macrophages and osteoclasts can induce bone formation in vivo. Consequently, we generated human macrophage- and osteoclast-laden constructs using two types of scaffold materials and implanted them underneath the skin of nude mice. Although we confirmed the presence of macrophages and osteoclasts on the materials, we found no signs of bone formation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Impaired or delayed healing of bone injuries due to pre-existing co-morbidities (e.g., diabetes, osteoporosis) or lifestyle choices (e.g., smoking, alcohol abuse) and non-healing of critical-sized bone defects resulting from trauma or tumor resection are a major clinical challenge [1, 2]. In fact, bone is one of the most frequently transplanted tissues in Europe with more than 1 million transplantations occurring annually [3]. This is primarily related to the growth and aging of the world population and secondly due to an increase in cases of trauma, bone tumor resection, infection, and congenital bone malformations [1, 4]. Current treatment options for such injuries include implantation of auto-/allo-/xenografts, or synthetic bone substitute materials (alloplasts). However, all of these are associated with specific shortcomings; while autografts for example are only limitedly available, allo- or xenografts might induce an immunogenic response and lack osteogenic or -inductive properties [5,6,7].
In view of these limitations, bone tissue engineering (BTE) is a promising treatment strategy for this type of bone injuries by tackling shortcomings of (synthetic) bone substitute materials [8]. To this end, BTE combines 2 or 3 of the main elements of TE (i.e., biomaterials, differentiation factors, and cells) to generate constructs for improved bone healing compared to the sole use of biomaterials [8]. Generally, biomaterials are combined with a potent osteoinductive differentiation factor such as bone morphogenetic protein-2 (BMP-2) to trigger bone formation upon implantation [9]. However, clinical application of BMP-2 was reported to be associated with several potential life-threatening side effects, such as cervical swelling (reviewed by [9]), particularly when used off-label [10,11,12,13]. Alternatively, biomaterials loaded with autologous mesenchymal stromal cells (MSCs) were assumed to accelerate bone regeneration due to the active role those cells have in bone formation. However, the invasive harvesting procedures, time-consuming in vitro manipulation, and—most importantly—the inconsistent therapeutic efficacy have so far hampered widespread implementation of MSC-laden biomaterials in clinics [2, 14].
Consequently, to improve therapeutic efficacy and reduce complexity (i.e., cell harvesting), it might be worthwhile to explore other cell types for BTE approaches. As of recently, cells involved in the initiation of bone formation are increasingly considered for this purpose, instead of using cells which can directly form bone such as MSCs. Interestingly, during physiological bone remodeling, a tight coordination ensures that the exact amount of old or damaged bone resorbed by osteoclasts is replaced by new bone tissue deposited by osteoblasts, a process also known as osteoclast-osteoblast coupling [15]. Osteoclasts mediate this process either indirectly by releasing bone matrix-derived factors (e.g., transforming growth factor-β (TGF-β), insulin-like growth factor-1 (IGF-1)) following resorption, or directly via secretion of so-called clastokines (e.g., collagen triple repeat containing-1 (CTHCR1), complement component 3a (C3a), sphingosine-1-phosphate (S1P)) [15,16,17]. The release of these factors is critical for the subsequent attraction and differentiation of osteoprogenitor cells [15,16,17]. The particular importance of the direct contribution of osteoclast-osteoblast coupling was unveiled by preclinical and clinical studies on osteoclast-rich osteopetrosis, a bone disorder characterized by impaired osteoclast resorption activity [16, 18]. Interestingly, patients [19,20,21] and experimental animals [22,23,24,25,26,27,28,29,30,31,32] suffering from this condition show normal or even increased bone formation rates, suggesting that these dysfunctional osteoclasts are still able to couple osteoblastic activity. In contrast, patients [21] and experimental animals [33,34,35,36] with osteoclast-poor osteopetrosis demonstrate reduced bone formation, which suggests that coupling is impaired in the absence of osteoclasts. Furthermore, the ability of non-resorbing osteoclasts to secrete anabolic factors was confirmed by in vitro studies where medium collected from osteoclast cultures was reported to induce either migration [37] or osteogenic differentiation of osteoprogenitor cells [37,38,39,40,41,42]. Lastly, several preclinical animal studies suggest that osteoclasts are relevant for the initiation of bone formation at ectopic sites. For instance, implantation of osteoinductive biomaterials was previously reported to not only result in successful bone induction at ectopic sites, but also induce native osteoclast activity [43,44,45,46,47]. Similarly, osteoclasts were also observed when biomaterials were implanted in combination with either BMP-2 [48] or human MSCs [49]. More importantly, the dependence of ectopic bone formation on native osteoclast activity following implantation of cell-free [44] or MSC-loaded biomaterials [49] was demonstrated. This dependence was indicated following inhibition of osteoclast formation upon systemic administration of either liposomal clodronate [44] or monoclonal anti-RANKL antibody [49] to the experimental animals, which caused a significant reduction in ectopic bone formation. While these findings are generally interpreted as confirming the importance of developing “resorbable” bone replacement materials that can be remodeled in situ into new bone tissue upon implantation [50,51,52], we here hypothesize that osteoclasts can actually be considered cellular initiators of bone formation, which can be used (as initiators) instead of osteoblasts (as bone-forming cells) for cell-based construct preparation.
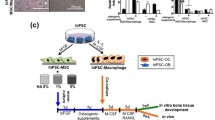
Therefore, we here aim to investigate whether constructs loaded with osteoclasts are also able to induce ectopic bone formation (Fig. 1). Due to their clinical relevance, we here used primary human osteoclast precursors which were isolated and differentiated toward macrophages. Those cells were then seeded on different types of materials (i.e., Bio-Oss granules and electrospun polymeric scaffolds) to assess whether different types of organic and inorganic biomaterials can be rendered osteoinductive by loading osteoclasts. Those macrophages were subsequently stimulated to form osteoclasts according to a previously established protocol [53]. After 14 days of osteoclast differentiation, osteoclast formation was confirmed by tartrate-resistant acid phosphatase (TRAP)-activity assay, TRAP, and immunocytochemical staining for integrin β3, and these osteoclast-laden constructs were implanted subcutaneously in nude mice. Macrophage-laden constructs were implanted as additional experimental group in addition to BMP-2-loaded and cell-free scaffolds as positive and negative controls, respectively. After 60 days of implantation, constructs were retrieved and analyzed for ectopic bone formation and presence of osteoclasts following histological processing and staining.

Experimental overview of the performed study. Monocytes were isolated from apheresis-derived PBMCs by adhesion and differentiated toward macrophages for 4 days. Then, macrophages were detached and seeded onto FBS-soaked electrospun PLGA/PCL scaffolds and Bio-Oss granules for 2.5 h. Cell loading was evaluated by DNA assay and DAPI staining. Remaining samples were further differentiated toward macrophages or osteoclasts for 14 days. Osteoclast differentiation on materials was assessed by TRAP-activity assay, TRAP, and immunocytochemical staining. Macrophage- and osteoclast-laden materials were implanted subcutaneously in nude mice for 60 days. Finally, bone induction and osteoclastic activity were evaluated by histological staining. Abbreviations: Mφ, macrophage; MC, monocytes; MCSF, macrophage colony stimulating factor; PLGA/PCL, poly(lactic-co-glycolic acid)/polycaprolactone; DAPI, 4′,6-diamidino-2-phenylindole; RANKL, receptor activator of nuclear factor κB ligand; OC, osteoclast; TRAP, tartrate-resistant acid phosphatase; BMP-2, bone morphogenetic protein-2; HE, hematoxylin/eosin; PBMCs, peripheral blood-derived mononuclear cells; FBS, fetal bovine serum. This illustration was created with BioRender.com
Materials and Methods
For composition of used buffers and media, see Supplementary Table 1.
Peripheral Blood-Derived Mononuclear Cell Isolation and Macrophage Differentiation
A commercially available apheresis product (Sanquin) enriched in the mononuclear cell fraction was obtained from human whole blood of one donor. The apheresis product was diluted in dilution buffer. Next, 30 ml of the diluted product was distributed over 50-ml tubes and 12–14 ml lymphoprep (Axis-Shield Diagnostics) pipetted underneath. Cells were spun at room temperature (RT) for 25 min at 800 g with acceleration set to 1 and deceleration to 1. The peripheral blood mononuclear cell (PBMC) interphase was collected into new 50-ml tubes. Cells were diluted with dilution buffer and spun for 10 min at 650 g at RT with maximum brake. Following several washing steps at 500 g for 5 min at 4 °C, cells were pooled until finally only one tube was left, and cells were resuspended in medium or proliferation medium (PM). Cells were counted and frozen until further use. Cells were thawed and seeded in PM at a concentration of ~ 0.5 × 106 PBMCs per cm2 into T175 cell culture flasks (Greiner bio-one). After 1-h incubation at 37 °C and 5% CO2, cells were washed twice with PBS and macrophage differentiation medium was added. Cells were differentiated toward macrophages for 4 days.
Preparation of Cell-Laden Constructs
Electrospun poly(lactic-co-glycolic acid)/polycaprolactone (PLGA/PCL) meshes were prepared as previously described [54, 55]. The electrospinning solution was prepared by dissolving PLGA (Purasorb® PDLG 8531, Corbion, The Netherlands) and PCL (Sigma-Aldrich) in a weight ratio of 3:1 at a concentration of 0.12 g/ml overnight using a magnetic stirrer. 2,2,2-Trifluoroethanol (TFE, Sigma-Aldrich) served as organic solvent. Three-dimensional (3D) scaffolds were generated using the so-called wet electrospinning technique in a commercially available electrospinning set-up (E-sprayer™ ES-2000S, Fuence). The processing parameters for stable formation of electrospun fibers were chosen based on an earlier publication [55]. In brief, the polymer solution was fed into a glass syringe and delivered at a feeding rate of 50 µl/min to an 18G nozzle. To generate a stable polymer jet, a voltage of 25 kV was applied at the tip. A grounded bath filled with 99.5% ethanol located 15 cm under the nozzle was used to collect the resulting fiber meshes. The size of those was controlled by stopping the electrospinning process every 15 min for fiber mesh collection. Henceforth, the wet electrospun material was washed thoroughly several times in Milli-Q water and freeze-dried (VirTis BenchTop Pro with Omnitronics Freeze Dryer, SP Scientific) for 3 days. Scaffolds obtained using these parameters were previously reported to display an uncompressed structure with an average fiber diameter of 1.98 ± 0.51 µm and a porosity of 99% [55]. Disk-shaped scaffolds with a diameter of 4 mm were punched out from these PLGA/PCL meshes using a disposable biopsy punch (Kai Industries). Next, scaffolds were sterilized for 3 h in 70% ethanol and washed 4 times with PBS. PLGA/PCL scaffolds and Bio-Oss granules (0.25–1 mm size, Geistlich) were soaked for 3 days in FBS at 37 °C and 5% CO2. Scaffolds and granules were placed into ultralow attachment 24-well plates (Corning). To load cells onto scaffolds and granules, macrophages were first enzymatically detached for 15 min with 0.25% Trypsin–EDTA (Sigma-Aldrich) at 37 °C and 5% CO2 followed by mechanical detachment using cell scrapers, respectively. Cells were counted and macrophages were seeded at a concentration of 0.5 × 106 cells in 20 µl onto each scaffold type and incubated for 2.5 h at 37 °C and 5% CO2 to allow initial cell attachment. Scaffolds and granules loaded with 20 µl PM served as cell-free negative controls. Additionally, two-dimensional (2D) control cultures to confirm osteoclastogenic potential of the donor were prepared by seeding macrophages at a concentration of 2.5 × 104 per cm2. Afterwards, cell-laden constructs were transferred in 48-well cell suspension plates (Greiner bio-one) and 500 µl of RANKL-free control medium, osteoclast differentiation medium, or PM was added. Similarly, medium was changed to 200 µl macrophage or osteoclast differentiation medium for the 2D control. Cells were cultured for 14 days with two full medium changes per week. Positive controls were prepared by incubating scaffolds and granules with BMP-2 (R&D Systems, 100 µg/ml) for 24 h at RT. Thereafter, materials were air-dried for several hours and incubated at 4 °C until implantation.
Assessment of Cell Loading Efficiency
Additional cell-laden constructs were prepared for DNA quantification and nuclear staining. Samples for the DNA assay were transferred to an empty ultralow attachment 24-well after 2.5-h incubation and rinsed twice with PBS, before being transferred in a new 1.5-ml tube. Next, 1 ml demineralized water was added per tube. In addition to loading macrophages onto scaffolds or granules, 20 µl-containing 0.5 × 106 cells were pipetted into new 1.5-ml tubes and 1 ml demineralized water was added. These samples served as reference for assessment of loading efficiency onto the cell carrier materials. Tubes were stored at − 20 °C. After two freeze/thaw cycles at − 20 °C/RT, the lysates were centrifuged for 1 min at 4000 g and the cellular DNA content was quantified using the QuantiFluor® dsDNA System Kit (Promega Corporation) as described previously [56]. Furthermore, samples were prepared for fluorescent nuclear staining. After rinsing samples with PBS and fixation using 3.7% formaldehyde (Boom), Bio-Oss granules were washed with PBS and incubated for 10 min with 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich, 0.4 µg/ml) diluted in PBS. After washing, granules were transferred onto microscopy slides (Marienfeld) and mounted [53]. Similarly, PLGA/PCL samples were stained with DAPI after histological processing as described below. Samples were imaged using the ImagerZ.2 with the Axiocam 705 microscope camera and ZEN (blue edition) version 3.3 software (all from Zeiss).
Osteoclast Characterization
Gross TRAP-Activity Assay
Cells and constructs were washed once with PBS, lysed using 300 µl of 50 mM sodium acetate lysis buffer, and stored at − 70 °C. After two repetitive freeze/thaw cycles at − 70 °C/RT, intracellular TRAP-activity of cell-loaded construct and 2D control lysates was determined using a previously described assay [53].
TRAP Staining
To visualize osteoclasts on the different cell carrier materials, samples were stained for TRAP. Bio-Oss granules were stained using a previously described protocol [53]. PLGA/PCL samples were dehydrated and infiltrated with Surgipath Paraplast Plus paraffine using the TP1020 tissue processor (all from Leica). Following paraffine embedding, 5-µm sections were cut using the RM2165 microtome (Leica). Sections were melted to microscopy slides (Thermo Fisher Scientific or Epredia), de-paraffinized in xylene, and rehydrated in a descending alcohol series. Next, samples were incubated for 2 h at 37 °C in a 0.2 M Tris (Serva)/magnesium chloride (Sigma-Aldrich) buffer with pH 9. After rinsing with demineralized water, sections were incubated up to 2 h in acid phosphatase medium at 37 °C. The composition of this buffer can be found in Supplementary Table 2. Finally, samples were rinsed in tap water, dehydrated, and mounted with DPX mountant (Sigma-Aldrich) before image acquisition.
Immunocytochemistry
Besides TRAP staining, integrin β3 was used as a marker to confirm osteoclast formation on Bio-Oss granules by means of immunocytochemical staining. Staining was performed as described earlier, with small modifications [53]. Cells were incubated with the primary mouse anti-human CD61 antibody (Biorbyt, 1:200) overnight at 4 °C, followed by incubation with a secondary biotin-conjugated donkey anti-mouse antibody (Jackson, 1:500) diluted in 2% normal donkey serum (NDS, GeneTex) for 1 h at RT. After rinsing with PBS supplemented with 0.05% Tween-20 (Sigma-Aldrich), granules were incubated for 45 min with ABC solution (VECTOR Laboratories) protected from light. Following another washing step, granules were treated for 10 min with 3,3′-diaminobenzidine (DAB, Sigma-Aldrich). Samples were rinsed in demineralized water and incubated for 5 min in 0.5% copper sulfate in 0.9% sodium chloride (Merck) before staining with DAPI and mounting.
Animal Experiment
Ethical approval for the animal experiment was obtained by the French Department of Research under the license number 12/07/16-7B. Nine male 4-week-old BALB/cAnNRj-foxn1nu/nu mice were obtained from Janvier. After 1 week of acclimatization, surgery was performed on 5-week-old mice with an approximate weight of 28–30 g. Animals were anaesthetized using vetflurane (isoflurane) inhalation. Mice were placed in dorsal position. Six subcutaneous pockets per animal—3 pockets on each site of the vertebral column—were created using blunt dissection in the back of the animal and constructs were inserted. For the cell-free condition, 6 constructs were included, while for the BMP-2-, macrophage-, and osteoclast-loaded conditions, 7 constructs were implanted (n = 6–7). Wounds were closed using clamps. Animals were kept at a nycthemeral cycle with food and water being available at libitum. After 60 days, animals were euthanized via cervical dislocation and samples collected. Samples were fixed for 26 h in 3.7% neutral-buffered formaldehyde, and stored in 70% ethanol until histological processing.
Histology
All Bio-Oss and 9 PLGA/PCL implants which showed mineral deposition upon X-ray radiography were decalcified in 10% EDTA (Merck) for up to 10 days until no mineral was detected anymore. Next, decalcified samples and PLGA/PCL implants without signs of mineral deposition were dehydrated and infiltrated with Surgipath Paraplast Plus paraffine using the TP1020 tissue processor. Following paraffine embedding, 5-µm sections were cut using the RM2165 microtome. Sections were melted to microscopy slides, de-paraffinized, and rehydrated before staining.
For hematoxylin and eosin (HE) staining, sections were first incubated for 8 min in hematoxylin (Merck), followed by rinsing in running tap water for 10 min. Following dehydration up to 96% ethanol, sections were stained for 2 min with eosin (Merck). After complete dehydration, sections were incubated in xylene and mounted with DPX. To check for the presence of osteoclasts, sections were stained for TRAP as described afore. Finally, sections were scanned using the Pannoramic 1000 digitalization system at 20 × magnification, and images from scans were prepared using the CaseViewer version 2.3 software (both 3DHISTECH).
Statistical Analysis
Statistical analysis of data was performed with SPSS Statistics version 25 (IBM). Data were analyzed using independent t-tests and one-way ANOVA followed by Dunnett’s post hoc test with the loading control set as control group. p-values < 0.05 were considered statistically significant. Data are presented as mean ± standard deviation (SD).
Results
Cell Attachment to Both Cell Carrier Materials Is Limited
After seeding macrophages onto electrospun PLGA/PCL discs and Bio-Oss granules, samples were incubated for 2.5 h to allow for initial cell attachment. To qualitatively confirm the presence of cells by fluorescent nuclear staining and quantitatively by performing a DNA assay, samples were collected after this incubation period (Fig. 2).

Cell attachment on different cell carrier materials after 2.5 h of static loading. Electrospun PLGA/PCL scaffolds and Bio-Oss granules were stained with DAPI (n = 1–2) (a). Asterisk (*) indicates PLGA/PCL scaffold. Images were taken with 20 × and 10 × magnification, respectively. Scale bars indicate 100 µm. DNA content was assessed for the loading control consisting of 0.5 × 106 macrophages directly pipetted into 1 ml demineralized water and the same number of macrophages loaded onto electrospun PLGA/PCL scaffolds and Bio-Oss granules (n = 3–4) (b). Data were analyzed using one-way ANOVA with a Dunnett’s post hoc test with the loading control set as control. The asterisk symbol “***” indicates statistical significance at p < 0.001 level. Loading efficiency is expressed relative to the loading control for both material types (n = 3) (c). Abbreviations: PLGA/PCL, poly(lactic-co-glycolic acid)/polycaprolactone; DAPI, 4′,6-diamidino-2-phenylindole
DAPI staining revealed that multiple nuclei were present on both material types (Fig. 2a). While nuclei on PLGA/PCL were observed predominantly on one side of the material, the distribution of nuclei showed to be heterogeneous on Bio-Oss granules, which themselves showed strong autofluorescence.
The quantitative DNA assay indicated that the DNA content of both cell-loaded electrospun PLGA/PCL scaffolds (129 ± 21 ng/ml) and Bio-Oss samples (24 ± 14 ng/ml) was low compared to the loading control corresponding to the initial total cell number used at cell seeding (395 ± 67 ng/ml, p < 0.001, Fig. 2b). Based on these data, the loading efficiency for electrospun PLGA/PCL scaffolds (33 ± 5%) and Bio-Oss granules (6 ± 4%) relative to the loading control was calculated (Fig. 2c).
Osteoclasts Form on Both Types of Cell Carrier Materials
Following loading of macrophages onto electrospun PLGA/PCL discs and Bio-Oss granules, cell-laden carrier materials were transferred to new 48-well cell suspension plates and differentiated in macrophage or osteoclast differentiation medium for 14 days. On the day of implantation, samples were collected to confirm osteoclast differentiation on the different cell carrier materials (Fig. 3).

Osteoclast characterization on different cell carrier materials after 14 days of in vitro culture. Intracellular gross TRAP-activity was assessed for cells differentiated on tissue culture-treated polystyrene (2D control) and cell carrier materials (n = 3–4) (a). Data were analyzed using an independent t-test. The asterisk symbols “**” and “***” indicate statistical significance at p < 0.01 and p < 0.001 level. In addition, TRAP staining was performed to visualize differentiated cells on the cell carrier materials (n = 1) (b). Asterisk (*) indicates PLGA/PCL scaffold. Images were taken with 10 × magnification. Scale bars indicate 100 µm. Black arrow heads indicate the presence of TRAP-positive cells. Bio-Oss granules were also stained for integrin β3 and DAPI (n = 1) (c). Integrin β3 signal appears as dark brown precipitate. Images were taken with 10 × magnification with scale bars indicating 100 µm. Magnified image of an integrin β3-positive, multinuclear osteoclast on a Bio-Oss granule (d). Scale bar indicates 100 µm. White arrow head indicates an integrin β3-positive, mononuclear cell. Abbreviations: PLGA/PCL, poly(lactic-co-glycolic acid)/polycaprolactone; TRAP, tartrate-resistant acid phosphatase; 4-NP, 4-nitrophenol; 2D, two-dimensional; RANKL, receptor activator of nuclear factor κB ligand; DAPI, 4′,6-diamidino-2-phenylindole
A quantitative assay to determine intracellular gross TRAP-activity was performed with lysates of cells differentiated on tissue culture-treated polystyrene which served as 2D control and on the two types of cell carrier materials (Fig. 3a). Lysed cells cultured with RANKL showed increased gross TRAP-activity for the 2D control (3.81 ± 0.19 mM vs. 0.32 ± 0.06 4-NP/h, p < 0.001), electrospun PLGA/PCL discs (1.76 ± 0.28 vs. 0.17 ± 0.07 mM 4-NP/h, p < 0.01), and Bio-Oss granules (2.89 ± 0.45 vs. 0.19 ± 0.02 mM 4-NP/h, p < 0.01) compared to the respective RANKL-free control cultures.
Furthermore, electrospun PLGA/PCL discs and Bio-Oss granules were fixed after 14 days of differentiation and stained for TRAP (Fig. 3b). While strong signal was observed for RANKL-supplemented electrospun PLGA/PCL scaffolds and reduced signal in the respective RANKL-free controls, hardly, any TRAP signal was observed for RANKL-supplemented and RANKL-free control Bio-Oss granules.
An additional immunocytochemical staining for the osteoclast marker integrin β3 on Bio-Oss granules revealed a clear signal in RANKL-supplemented granules and lack thereof in the RANKL-negative control (Fig. 3c). Moreover, besides mononuclear, integrin β3-positive cells, also multinuclear osteoclasts were observed on the granules (Fig. 3d).
Only BMP-2-Loaded Carrier Materials Induce Ectopic Bone Formation
Cell-free, BMP-2-, macrophage-, and osteoclast-laden electrospun PLGA/PCL discs and Bio-Oss granules were implanted subcutaneously in nude mice and retrieved after 60 days to investigate their capacity to induce ectopic bone formation.
Using HE staining, we observed that ectopic bone formation had occurred only for both types of BMP-2-laden carrier materials (Fig. 4). All other groups showed only soft tissue formation surrounding the granules or within the electrospun PLGA/PCL discs. A higher amount of new bone and bone marrow formation was observed for BMP-2-laden Bio-Oss granules compared to BMP-2-laden PLGA/PCL discs.

HE staining of different cell carrier materials after 60 days of ectopic implantation. Electrospun PLGA/PCL scaffolds and Bio-Oss granules without cells (cell-free), loaded with BMP-2, human macrophages, or osteoclasts were subcutaneously implanted into nude mice (n = 6–7). HE staining revealed ectopic bone formation only occurred in the BMP-2-loaded positive controls. Images were prepared at 10 × magnification from scans obtained with the Pannoramic 1000 digitalization system at 20 × magnification using CaseViewer version 2.3. Scale bars indicate 100 µm. Asterisks (*) indicate Bio-Oss granules. Abbreviations: BMP-2, bone morphogenetic protein-2; M, macrophages; OC, osteoclasts; PLGA/PCL, poly(lactic-co-glycolic acid)/polycaprolactone; HE, hematoxylin/eosin
TRAP staining was performed as an indicator of osteoclast presence within the implanted cell carrier materials (Fig. 5). Only the BMP-2-laden positive controls for both types of carrier materials revealed a strong TRAP signal. All other groups hardly showed any TRAP signal, except for the PLGA/PCL scaffolds, which showed some weak TRAP staining in the vicinity of the scaffold.

TRAP staining of different materials after 60 days of ectopic implantation. Electrospun PLGA/PCL scaffolds and Bio-Oss without cells (cell-free), loaded with BMP-2, human macrophages, or osteoclasts, were subcutaneously implanted into nude mice (n = 6–7). TRAP staining revealed strong signal in BMP-2-loaded positive controls. Images were prepared at 20 × magnification from scans obtained with the Pannoramic 1000 digitalization system at 20 × magnification using CaseViewer version 2.3. Scale bars indicate 50 µm. Black arrow heads indicate TRAP-positive cells and asterisks (*) indicate Bio-Oss granules. Abbreviations: BMP-2, bone morphogenetic protein-2; M, macrophages; OC, osteoclasts; PLGA/PCL, poly(lactic-co-glycolic acid)/polycaprolactone; TRAP, tartrate-resistant acid phosphatase
Discussion
Based on previous work demonstrating the crucial role osteoclasts play in ectopic bone formation upon implantation of cell-free [44] or MSC-laden biomaterials [49], we here aimed to evaluate whether carrier materials directly loaded with human osteoclasts have osteoinductive capacity.
Cell loading on both types of carrier material appeared to be far from efficient despite soaking PLGA/PCL scaffolds and Bio-Oss granules in FBS for 3 days before loading to increase cell attachment. Following fluorescent DAPI staining, we observed the presence of multiple nuclei on the different cell carrier materials, indicative for the presence of attached cells. While nuclei were observed on one side of the sectioned electrospun PLGA/PCL discs, likely the side onto which the cells were loaded, nuclei were heterogeneously distributed on and between Bio-Oss granules. We additionally determined the cell loading efficiency for both cell carrier materials using a DNA assay based on reference values consisting of the same number of cells directly pipetted in demineralized water. This revealed a relatively low cell loading efficiency, particularly for Bio-Oss granules. Although there is no consensus on the optimal cell density for cell-based construct preparation, it is conceivable to speculate that a higher number of osteoclast precursors adhering to the material likely results in increased osteoclast fusion. Strategies to achieve increased cell loading efficiency include for example the use of fibrin glue to retain all cells with the cell carrier material [57,58,59], or seeding of a higher number of osteoclast precursors.
To achieve successful osteoclast formation, we developed an optimized differentiation protocol for 2D osteoclast cultures [53] which was then adapted for osteoclast differentiation on the different cell carrier materials. We demonstrated a clear increase in intracellular gross TRAP-activity for both material types for RANKL-supplemented cultures compared to the negative control despite low cell numbers present on the cell carrier materials. However, those values were substantially lower compared to the 2D differentiation control cultured in a 48-well plate with much lower cell seeding density (i.e., 2.5 × 104 cells per well). These quantitative data were supplemented with visual confirmation of osteoclast differentiation following staining. While we confirmed presence of TRAP-positive cells on electrospun PLGA/PCL scaffolds visually by TRAP staining, hardly any TRAP signal was observed on Bio-Oss granules. While TRAP staining was hardly observed on Bio-Oss granules, we were able to confirm osteoclast differentiation by means of an immunocytochemical staining for the osteoclast marker integrin β3. Besides few multinuclear osteoclasts, also many mononuclear integrin β3-positive cells were present on the granules. Next to those mononuclear cells, also single nuclei without an integrin β3-positive cell body reflect the heterogeneity of osteoclast cultures as not all cells—also in 2D culture—will fuse to form osteoclasts. Thus, in vitro as well as in vivo experiments ideally require the inclusion of an additional undifferentiated macrophage control as presented in this study.
Herein, however, we demonstrated that using the current experimental set-up, both human macrophage- and osteoclast-laden constructs did not induce in vivo ectopic bone formation. It remains unclear whether the low loading efficiency, the limited number of formed osteoclasts, or construct transportation to another facility for several hours before implantation might be the cause for the lack of osteoinductive capacity observed in this work. The latter cause cannot be excluded, as samples to confirm osteoclast formation were collected before, and not after transport. Furthermore, even if cells survived transport and implantation, it was previously observed that for example human MSCs disappeared a few weeks after ectopic implantation [49]. However, despite clearance from the implantation site, those cells were still able to initiate an osteoinductive host response, likely via paracrine mechanisms [49]. Another explanation for our finding might simply be that neither human macrophages nor human osteoclasts have the ability to induce bone formation. To our knowledge, there is only one earlier study available which describes seeding and osteoclastic priming of human monocytes on bone-derived MSC (BMSC)-produced, engineered hypertrophic cartilage (HC) in an attempt to increase its endochondral ossification in vivo [60]. Such an approach is of particular interest as monocytes can easily be isolated from patient blood. This would allow for the cell isolation and cell-based construct preparation to occur on the day the patient undergoes surgery [61]. Furthermore, these minimally manipulated cells might trigger bone tissue regeneration in situ [61]. However, following subcutaneous implantation of living and devitalized HC with or without osteoclastic primed monocytes in nude mice, the authors of this study noted that the addition of these monocytes on either one did not enhance ectopic bone formation [60]. Moreover, besides several studies reporting on anabolic effects of osteoclast-derived clastokines [37, 40] or extracellular vesicles [62, 63] on osteoprogenitor cells, there is also a body of in vitro evidence that demonstrated a negative effect of osteoclast-derived factors on osteogenic cells. Furthermore, there are studies available reporting that osteoclasts are not only capable of releasing stimulatory, but also inhibitory signals (e.g., sclerostin [64], vesicular miRNAs [65,66,67]) acting on osteoprogenitor or osteogenic cells.
To clarify whether the absence of ectopic bone formation is due to low cell and/or osteoclast numbers, or due to lack of osteoinductive efficacy of implanted cells, further experiments are required. To this end, another study using 2D polymer coverslips (e.g., Thermanox™) as carrier material instead of electrospun scaffolds or devitalized bone granules might be performed. Such coverslips can be seeded and cultured with human osteoclast precursors at concentrations typically used for 2D osteoclast differentiation and therefore avoid any of the earlier issues associated with cell seeding and osteoclast differentiation on carrier materials. Furthermore, this also simplifies analysis of cell adhesion and osteoclast formation by quantitative assays and staining. Macrophage- and osteoclast-laden coverslips can then be implanted subcutaneously in nude mice and, after several weeks of implantation, retrieved and histologically processed to observe whether ectopic bone formation was induced.
Conclusion
We here aimed to study whether human macrophages and osteoclasts have osteoinductive capacity in vivo. Consequently, we generated human macrophage- and osteoclast-laden constructs using two types of cell carrier materials and implanted them subcutaneously in nude mice. Although we confirmed osteoclast formation on the scaffold materials after in vitro culture, we found no signs of ectopic bone formation.
Data Availability
The research data associated with this paper are available upon request from the corresponding author.
References
Gómez-Barrena E, Rosset P, Müller I, Giordano R, Bunu C, Layrolle P, et al. Bone regeneration: stem cell therapies and clinical studies in orthopaedics and traumatology. J Cell Mol Med. 2011;15(6):1266–86.
Jakob M, Saxer F, Scotti C, Schreiner S, Studer P, Scherberich A, et al. Perspective on the evolution of cell-based bone tissue engineering strategies. Eur Surg Res. 2012;49(1):1–7.
CORDIS. Final report summary - REBORNE (regenerating bone defects using new biomedical engineering approaches). n.d. Retrieved 4 February 2022 from https://cordis.europa.eu/project/rcn/92715/reporting/en.
Fernández RF, Bucchi C, Navarro P, Beltrán V, Borie E. Bone grafts utilized in dentistry: an analysis of patients’ preferences. BMC Med Ethics. 2015;16(1):71.
Fernandez de Grado G, Keller L, Idoux-Gillet Y, Wagner Q, Musset AM, Benkirane-Jessel N, et al. Bone substitutes: a review of their characteristics, clinical use, and perspectives for large bone defects management. J Tissue Eng. 2018;9:2041731418776819.
Klijn RJ, Meijer GJ, Bronkhorst EM, Jansen JA. A meta-analysis of histomorphometric results and graft healing time of various biomaterials compared to autologous bone used as sinus floor augmentation material in humans. Tissue Eng Part B Rev. 2010;16(5):493–507.
Rosset P, Deschaseaux F, Layrolle P. Cell therapy for bone repair. Orthop Traumatol Surg Res. 2014;100(1 Suppl):S107–12.
Perez JR, Kouroupis D, Li DJ, Best TM, Kaplan L, Correa D. Tissue engineering and cell-based therapies for fractures and bone defects. Front Bioeng Biotechnol. 2018;6:105.
James AW, LaChaud G, Shen J, Asatrian G, Nguyen V, Zhang X, et al. A review of the clinical side effects of bone morphogenetic protein-2. Tissue Eng Part B Rev. 2016;22(4):284–97.
Carragee EJ, Hurwitz EL, Weiner BK. A critical review of recombinant human bone morphogenetic protein-2 trials in spinal surgery: emerging safety concerns and lessons learned. Spine J. 2011;11(6):471–91.
Poeran J, Opperer M, Rasul R, Mazumdar M, Girardi FP, Hughes AP, et al. Change in off-label use of bone morphogenetic protein in spine surgery and associations with adverse outcome. Global Spine J. 2016;6(7):650–9.
Poon B, Kha T, Tran S, Dass CR. Bone morphogenetic protein-2 and bone therapy: successes and pitfalls. J Pharm Pharmacol. 2016;68(2):139–47.
Ramly EP, Alfonso AR, Kantar RS, Wang MM, Siso JRD, Ibrahim A, et al. Safety and efficacy of recombinant human bone morphogenetic protein-2 (rhBMP-2) in craniofacial surgery. Plast Reconstr Surg Glob Open. 2019;7(8):e2347.
Grayson WL, Bunnell BA, Martin E, Frazier T, Hung BP, Gimble JM. Stromal cells and stem cells in clinical bone regeneration. Nat Rev Endocrinol. 2015;11(3):140–50.
Sims NA, Martin TJ. Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. Bonekey Rep. 2014;3:481.
Henriksen K, Karsdal MA, Martin TJ. Osteoclast-derived coupling factors in bone remodeling. Calcif Tissue Int. 2014;94(1):88–97.
Teti A. Mechanisms of osteoclast-dependent bone formation. Bonekey Rep. 2013;2:449.
Karsdal MA, Martin TJ, Bollerslev J, Christiansen C, Henriksen K. Are nonresorbing osteoclasts sources of bone anabolic activity? J Bone Miner Res. 2007;22(4):487–94.
Alatalo SL, Ivaska KK, Waguespack SG, Econs MJ, Väänänen HK, Halleen JM. Osteoclast-derived serum tartrate-resistant acid phosphatase 5b in Albers-Schonberg disease (type II autosomal dominant osteopetrosis). Clin Chem. 2004;50(5):883–90.
Bollerslev J, Steiniche T, Melsen F, Mosekilde L. Structural and histomorphometric studies of iliac crest trabecular and cortical bone in autosomal dominant osteopetrosis: a study of two radiological types. Bone. 1989;10(1):19–24.
Del Fattore A, Peruzzi B, Rucci N, Recchia I, Cappariello A, Longo M, et al. Clinical, genetic, and cellular analysis of 49 osteopetrotic patients: implications for diagnosis and treatment. J Med Genet. 2006;43(4):315–25.
Horne WC, Neff L, Chatterjee D, Lomri A, Levy JB, Baron R. Osteoclasts express high levels of pp60c-src in association with intracellular membranes. J Cell Biol. 1992;119(4):1003–13.
Kornak U, Kasper D, Bösl MR, Kaiser E, Schweizer M, Schulz A, et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104(2):205–15.
Li CY, Jepsen KJ, Majeska RJ, Zhang J, Ni R, Gelb BD, et al. Mice lacking cathepsin K maintain bone remodeling but develop bone fragility despite high bone mass. J Bone Miner Res. 2006;21(6):865–75.
Li YP, Chen W, Liang Y, Li E, Stashenko P. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet. 1999;23(4):447–51.
Lotinun S, Kiviranta R, Matsubara T, Alzate JA, Neff L, Lüth A, et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest. 2013;123(2):666–81.
Marzia M, Sims NA, Voit S, Migliaccio S, Taranta A, Bernardini S, et al. Decreased c-Src expression enhances osteoblast differentiation and bone formation. J Cell Biol. 2000;151(2):311–20.
Pennypacker B, Shea M, Liu Q, Masarachia P, Saftig P, Rodan S, et al. Bone density, strength, and formation in adult cathepsin K (-/-) mice. Bone. 2009;44(2):199–207.
Rzeszutek K, Sarraf F, Davies JE. Proton pump inhibitors control osteoclastic resorption of calcium phosphate implants and stimulate increased local reparative bone growth. J Craniofac Surg. 2003;14(3):301–7.
Schaller S, Henriksen K, Sveigaard C, Heegaard AM, Hélix N, Stahlhut M, et al. The chloride channel inhibitor NS3736 [corrected] prevents bone resorption in ovariectomized rats without changing bone formation. J Bone Miner Res. 2004;19(7):1144–53.
Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64(4):693–702.
Visentin L, Dodds RA, Valente M, Misiano P, Bradbeer JN, Oneta S, et al. A selective inhibitor of the osteoclastic V-H(+)-ATPase prevents bone loss in both thyroparathyroidectomized and ovariectomized rats. J Clin Invest. 2000;106(2):309–18.
Dai XM, Zong XH, Akhter MP, Stanley ER. Osteoclast deficiency results in disorganized matrix, reduced mineralization, and abnormal osteoblast behavior in developing bone. J Bone Miner Res. 2004;19(9):1441–51.
McClung MR, Lewiecki EM, Cohen SB, Bolognese MA, Woodson GC, Moffett AH, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354(8):821–31.
Sakagami N, Amizuka N, Li M, Takeuchi K, Hoshino M, Nakamura M, et al. Reduced osteoblastic population and defective mineralization in osteopetrotic (op/op) mice. Micron. 2005;36(7–8):688–95.
Wang ZQ, Ovitt C, Grigoriadis AE, Möhle-Steinlein U, Rüther U, Wagner EF. Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992;360(6406):741–5.
Kreja L, Brenner RE, Tautzenberger A, Liedert A, Friemert B, Ehrnthaller C, et al. Non-resorbing osteoclasts induce migration and osteogenic differentiation of mesenchymal stem cells. J Cell Biochem. 2010;109(2):347–55.
Henriksen K, Andreassen KV, Thudium CS, Gudmann KN, Moscatelli I, Crüger-Hansen CE, et al. A specific subtype of osteoclasts secretes factors inducing nodule formation by osteoblasts. Bone. 2012;51(3):353–61.
Karsdal MA, Neutzsky-Wulff AV, Dziegiel MH, Christiansen C, Henriksen K. Osteoclasts secrete non-bone derived signals that induce bone formation. Biochem Biophys Res Commun. 2008;366(2):483–8.
Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci U S A. 2008;105(52):20764–9.
Stessuk T, Husch J, Hermens IAT, Hofmann S, van den Beucken JJJP. Osteogenic differentiation driven by osteoclasts and macrophages. J Immunol Regen Med. 2021;12:100044.
Zhang Y, Chen SE, Shao J, van den Beucken J. Combinatorial surface roughness effects on osteoclastogenesis and osteogenesis. ACS Appl Mater Interfaces. 2018;10(43):36652–63.
Colnot C, Romero DM, Huang S, Helms JA. Mechanisms of action of demineralized bone matrix in the repair of cortical bone defects. Clin Orthop Relat Res. 2005;435:69–78.
Davison NL, Gamblin AL, Layrolle P, Yuan H, de Bruijn JD, Barrère-de Groot F. Liposomal clodronate inhibition of osteoclastogenesis and osteoinduction by submicrostructured beta-tricalcium phosphate. Biomaterials. 2014;35(19):5088–97.
Davison NL, Luo X, Schoenmaker T, Everts V, Yuan H, Barrère-de Groot F, et al. Submicron-scale surface architecture of tricalcium phosphate directs osteogenesis in vitro and in vivo. Eur Cell Mater. 2014;27:281–97 (discussion 96-7).
Davison NL, Su J, Yuan H, van den Beucken JJ, de Bruijn JD, Barrère-de Groot F. Influence of surface microstructure and chemistry on osteoinduction and osteoclastogenesis by biphasic calcium phosphate discs. Eur Cell Mater. 2015;29:314–29.
Kondo N, Ogose A, Tokunaga K, Umezu H, Arai K, Kudo N, et al. Osteoinduction with highly purified beta-tricalcium phosphate in dog dorsal muscles and the proliferation of osteoclasts before heterotopic bone formation. Biomaterials. 2006;27(25):4419–27.
Irie K, Alpaslan C, Takahashi K, Kondo Y, Izumi N, Sakakura Y, et al. Osteoclast differentiation in ectopic bone formation induced by recombinant human bone morphogenetic protein 2 (rhBMP-2). J Bone Miner Metab. 2003;21(6):363–9.
Gamblin AL, Brennan MA, Renaud A, Yagita H, Lézot F, Heymann D, et al. Bone tissue formation with human mesenchymal stem cells and biphasic calcium phosphate ceramics: the local implication of osteoclasts and macrophages. Biomaterials. 2014;35(36):9660–7.
Detsch R, Boccaccine AR. The role of osteoclasts in bone tissue engineering. J Tissue Eng Regen Med. 2015;9(10):1133–49.
Kylmaoja E, Nakamura M, Tuukkanen J. Osteoclasts and remodeling based bone formation. Curr Stem Cell Res Ther. 2016;11(8):626–33.
Zhang Z, Egana JT, Reckhenrich AK, Schenk TL, Lohmeyer JA, Schantz JT, et al. Cell-based resorption assays for bone graft substitutes. Acta Biomater. 2012;8(1):13–9.
Husch JFA, Stessuk T, den Breejen C, van den Boom M, Leeuwenburgh SCG, van den Beucken J. A practical procedure for the in vitro generation of human osteoclasts and their characterization. Tissue Eng Part C Methods. 2021;27(7):421–32.
Tang H, Husch JFA, Zhang Y, Jansen JA, Yang F, van den Beucken J. Coculture with monocytes/macrophages modulates osteogenic differentiation of adipose-derived mesenchymal stromal cells on poly(lactic-co-glycolic) acid/polycaprolactone scaffolds. J Tissue Eng Regen Med. 2019;13(5):785–98.
Yang W, Yang F, Wang Y, Both SK, Jansen JA. In vivo bone generation via the endochondral pathway on three-dimensional electrospun fibers. Acta Biomater. 2013;9(1):4505–12.
Hayrapetyan A, Bongio M, Leeuwenburgh SC, Jansen JA, van den Beucken JJ. Effect of nano-HA/collagen composite hydrogels on osteogenic behavior of mesenchymal stromal cells. Stem Cell Rev Rep. 2016;12(3):352–64.
Mehrkens A, Saxer F, Güven S, Hoffmann W, Müller AM, Jakob M, et al. Intraoperative engineering of osteogenic grafts combining freshly harvested, human adipose-derived cells and physiological doses of bone morphogenetic protein-2. Eur Cell Mater. 2012;24:308–19.
Müller AM, Mehrkens A, Schäfer DJ, Jaquiery C, Güven S, Lehmicke M, et al. Towards an intraoperative engineering of osteogenic and vasculogenic grafts from the stromal vascular fraction of human adipose tissue. Eur Cell Mater. 2010;19:127–35.
Todorov A, Kreutz M, Haumer A, Scotti C, Barbero A, Bourgine PE, et al. Fat-derived stromal vascular fraction cells enhance the bone-forming capacity of devitalized engineered hypertrophic cartilage matrix. Stem Cells Transl Med. 2016;5(12):1684–94.
Todorov A, Scotti C, Barbero A, Scherberich A, Papadimitropoulos A, Martin I. Monocytes seeded on engineered hypertrophic cartilage do not enhance endochondral ossification capacity. Tissue Eng Part A. 2017;23(13–14):708–15.
Krasilnikova OA, Baranovskii DS, Yakimova AO, Arguchinskaya N, Kisel A, Sosin D, et al. Intraoperative creation of tissue-engineered grafts with minimally manipulated cells: new concept of bone tissue engineering in situ. Bioengineering. 2022;9(11):704.
Ikebuchi Y, Aoki S, Honma M, Hayashi M, Sugamori Y, Khan M, et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature. 2018;561(7722):195–200.
Liang M, Yin X, Zhang S, Ai H, Luo F, Xu J, et al. Osteoclast-derived small extracellular vesicles induce osteogenic differentiation via inhibiting ARHGAP1. Mol Ther Nucleic Acids. 2021;23:1191–203.
Kusu N, Laurikkala J, Imanishi M, Usui H, Konishi M, Miyake A, et al. Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J Biol Chem. 2003;278(26):24113–7.
Li D, Liu J, Guo B, Liang C, Dang L, Lu C, et al. Osteoclast-derived exosomal miR-214-3p inhibits osteoblastic bone formation. Nat Commun. 2016;7:10872.
Sun W, Zhao C, Li Y, Wang L, Nie G, Peng J, et al. Osteoclast-derived microRNA-containing exosomes selectively inhibit osteoblast activity. Cell Discov. 2016;2:16015.
Yang JX, Xie P, Li YS, Wen T, Yang XC. Osteoclast-derived miR-23a-5p-containing exosomes inhibit osteogenic differentiation by regulating Runx2. Cell Signal. 2020;70:109504.
Acknowledgements
We would like to thank Ben Joosten from the Department of Cell Biology, Radboudumc, for providing the apheresis product. We would like to acknowledge the Department of Pathology for providing the service of their histological scanning facility.
Funding
This study received financial support from the Netherlands Organization for Health Research and Development (ZonMw) GameChanger Project “ioCOMPONENTS” (No. 40-41400-98-16031).
Author information
Authors and Affiliations
Contributions
J.H.: conceptualization, investigation, methodology, and writing—original draft; L.C., N.C., N.D.: methodology, S.L.: writing—review and editing, J.B.: conceptualization, funding acquisition, supervision, and writing—review and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical Approval
Ethical approval for the animal experiment was obtained by the French Department of Research under the license number 12/07/16-7B.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Husch, J.F.A., Coquelin, L., Chevallier, N. et al. Human Macrophage- and Osteoclast-Based Constructs Do Not Induce Ectopic Bone Formation. Regen. Eng. Transl. Med. 10, 220–231 (2024). https://doi.org/10.1007/s40883-023-00315-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40883-023-00315-z