
Abstract
In this research, the recycling of industrially collected and crushed nickel metal hydride battery waste, rich in valuable metals such as Ni and rare earth elements (REE), was investigated. The crushed waste was characterized based on elemental distribution per particle size class and density. Although issues with sieving, such as agglomeration of shredded separator fibers, were observed, a good separation of Fe and plastics could be achieved by using a 1-mm sieve size. It was observed that, as the waste battery particles were washed with water, some organic compounds were dissolved. Acid consumption of 14 mol H+ ions per 1 kg of battery sample (sieve fraction—1 mm) was determined to be sufficient to achieve the desired final pH of < 1. Selectivity of the leaching at higher equilibrium pH was also investigated by using dilute H2SO4. Pregnant leach solution rich in Ni (46 g/L) and REEs (La: 9 g/L, Ce: 7.5 g/L, Pr: 1.4 g/L, Sm: 0.29 g/L, Y: 0.17 g/L) was obtained and REE precipitation was investigated as a function of dilute Na2SO4 solution concentration (0.01–0.5 M) at a temperature of 50 °C. The best precipitation efficiency was achieved with a Na:REE ratio of 9.1, which resulted in a > 99% precipitation efficiency for the REEs.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Nickel metal hydride (NiMH) batteries are a battery class that has recently been overtaken by lithium-ion batteries (LIBs), nevertheless, these types of batteries still find extensive use in certain applications like hybrid electrical vehicles (HEV), consumer electronics, and tools [1, 2]. Despite this increased shift towards LIBs, NiMH batteries are still being used in appreciable numbers, although the current methods of recycling often neglect certain elements like rare earths [3]. As NiMH batteries contain significant quantities of vital metals that include rare earth elements (REE) and Ni, their recycling is still of great importance. This fact was recently highlighted in the 2017 European Union strategic resources overview, which classified both heavy and light REEs as extremely critical due to their high supply risk [4]. Mass-volume- wise, the NiMH batteries are still used and collected in significant quantities. For example, the European Commission reported in 2015 that the portable primary and secondary batteries market in Germany included moderate levels of LIBs (16%) or NiMH (6%) [5]. Moreover, additional data show that approximately 550 tons of NiMH batteries were collected in Germany during 2014 [6].
NiMH batteries are in principle hydrogen storage devices, where the charging cycle will result in the formation of metal hydride on the anode, whereas the discharging reaction involves loss of hydrogen from the anode and the reduction of NiO·OH on the cathode back to Ni(OH)2. NiMH batteries are composed of active powders that commonly contain large quantities of light REEs and various transition metals, especially Ni. These active powders are most often of AB5 type where A = La or, alternatively, mischmetal (La + Ce + Pr + Nd) and B = Ni or Ni + Co + Zn + Mn + Al [7]. As shown by Larsson et al., REEs may account for nearly 10 wt% of the NiMH battery cell, Ni nearly 50 wt%, Co 3.7 wt%, and Fe 30 wt% [8]. The cathode and anode are immersed in alkali electrolyte composed of aqueous NaOH and KOH solution [9] rather than the organic electrolytes found in LIBs [10]. The cathode and anode reside on nickel-plated steel current collector and the electrodes are separated by a non-conductive ion-permeable polymer membrane [11].
Several processes have been proposed for NiMH battery recycling and globally, a number has been implemented on an industrial scale like the Umicore ultra-high temperature (UHT) process [12]. Nonetheless, a more complete recycling of all the critical metal components within these batteries is often still lacking, e.g., the rare earth elements are lost in pyrometallurgical processes to the liquid iron silica slag where they are further diluted [13]. For example, SNAM and Inmetco undertake NiMH batteries recycling via pyrometallurgical means that result in REEs in the process of side streams [9], while the Nickelhütte smelting process generates slags that are subsequently used for road or highway construction [14]. In contrast, Honda is recycling NiMH batteries in collaboration with Japan Metals & Chemicals Co. Ltd by a method that allows REEs to be recovered by molten salt electrolysis.
In hydrometallurgical-based processes, a mechanical pre-treatment of the batteries is necessary in order to expose the active materials ready for the subsequent stages. This is can be a precarious process and different precautions such as discharging in aqueous solution [15] or cryogenic treatment [16] have been suggested. After mechanical treatment, separation of materials has been investigated by, e.g., magnetic separation and size separation [17, 18].
Before acidic leaching of the mechanically treated spent NiMH battery materials, it is beneficial to wash them. Washing is very important as in this manner in-situ precipitation of insoluble REE double sulfates can be avoided by removing K and Na [19]. Several studies report that the spent NiMH raw material was washed prior leaching experiments [20,21,22], however, the reports are often only tangential and do not consider the implications of generating a new wastewater stream [23]. Innocenzi et al. reported in detail the removal of K by washing, but were not able to analyze the presence of Na, nor was removal of possible hydrophilic binder reported. Wang et al. reported removal of “hydrophilic binder,” but did not observe any adverse effects [24].
A recent review by Innocenzi et al. [18] represents leaching of NiMH battery waste as simple neutral reactions, Eq. (1):
where M = Ni, Co, Cd, Zn, Mn. However, this can be considered as an oversimplification, as it is impossible for the leaching to proceed in this manner, due to the fact that reactive metal alloys are present in the raw material and therefore redox reactions must be involved in the dissolution reaction. In reality, dissolution proceeds through charge transfer, as shown by Meshram et al. in the suggested total leaching reaction, Eq. (2). Here, the leaching of the electrode powder is simplified assuming that the cathode material is rather of the form of LaNi5 than mischmetal [25]:
Also Pietrelli et al. [11] suggested a leaching reaction (Eq. 3), where O2 acts as an oxidant in acidic medium for the elemental Ni, however, reaction (2) can be suggested to represent the NiMH waste dissolution more accurately.
At T = 30 °C, reaction (2) has ΔG = − 2664.629 kcal, ΔH = − 3147.371 kcal, however, reaction (3) has ΔG = − 540.790 kcal, ΔH = − 862.114 kcal [26]. The metal alloy present in the raw material acts as a reductant and is able to reduce hydrogen in acidic solutions, which has been observed as vigorous gas generation during leaching [27]. The reactions in leaching are strongly exothermic, as shown in Eq. (2). In addition to metallic alloys on anode, cathode can contain Ni(OH)2, depending on the end-of-life charge state and of the degree of exposure to air, which can cause further heat generation upon neutralization according to Eq. (4):
Based on observed gas generation and temperature increase during leaching [19], as well as the Gibbs energies of reactions (2) and (3), the principal reaction in acidic leaching can be suggested to be the reduction of hydrogen ions (Eq. 3), which in turn generates heat. This indicates that during the recycling process design, temperature control needs to be addressed either by appropriate heat exchangers (cooling) in the reactor or by the control of the feed material—in the current study the reaction is controlled by acid addition.
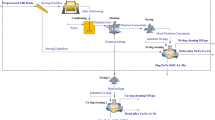
The present work focuses on the effect of mechanical processing of industrially crushed NiMH battery waste and the aspects related to hydrometallurgical treatment, i.e., leaching without prior thermal treatment. Contemporary reports in prior literature that cadmium can end up into these waste streams along consumer battery recycling systems [28]. Metallic Cd has relatively low boiling point of 766.8 °C [29], necessitating the removal of Cd from gases generated in the high temperature processes. The mechanical processing aspects such as optimal sieve size for size separation and gravity separation of different elements were investigated in order to observe if they could offer advantageous separation of metals. The raw material was water washed in order to minimize the presence of alkali elements that can cause REE losses due to in-situ precipitation during leaching [19]. The raw material was subsequently treated with sulfuric acid solutions to ascertain the consumption of acid. These metallic active electrode materials can react violently with acids, producing hydrogen gas [25]. Leaching by controlled acid addition was investigated in order to investigate if selective extraction could be achieved over certain metals, such as Al, and whether H2(g) could be controlled. Furthermore, the REE recovery through double salt precipitation, directly from pregnant leach solution was demonstrated, accompanied with analysis of Na and K. Different, low and high stoichiometric ratios of sodium per REE were investigated in order to ascertain the optimal range for REE precipitation. The context and the focus of the work performed are shown in Fig. 1.

Experimental
Raw Material
The NiMH batteries were initially hand-sorted at the facility and no discharging process was performed prior to crushing. This sorted battery waste was then crushed by a 2-stage process and the ensuing material was provided as-is by the operator. The raw material received was characterized by particle size distribution and density analysis. Particle size distribution of the crushed spent batteries was determined by sieving using 200-mm-diameter test sieves (Retsch) in a vibrating sieving machine (Retsch AS 30). The density of the particles was determined with a gas pycnometer (ULTRAPYC1200, Quantachrome, USA). All chemical analyses of aqueous solutions were performed using either flame atomic absorption spectroscopy (FAAS, Varian AA240) or inductively coupled plasma-optical emission spectroscopy (ICP-OES, PerkinElmer 7100 DV, USA). Solid samples of the different size and density fractions were dissolved in aqua regia prior to chemical composition analyses by ICP-OES and AAS. The PerkinElmer 7100 DV is unable to distinguish Ce from Nd due to spectral interference, therefore results of Ce tend to be overestimated while Nd results are absent. Further size fractions were sieved for the hydrometallurgical experiments with the same sieves and from these fractions, samples were obtained by using a rotary riffle splitter. A scanning electron microscope (SEM, A Zeiss (LEO) 1450VP) attached with Oxford Instruments INCA analyzer was utilized to observe the fibers obtained from the batteries after the crushing and sieving process. X-ray diffraction (XRD, Malvern Panalytical X’Pert Pro MPD Powder with CoKα radiation source operated at 40 kV and 40 mA, equipped with PIXcel1D detector and iron beta filter without monochromator) was utilized for the crystallographic characterization of the raw material.
Water Leaching
Based on mechanical and characterization experiments, the < 1-mm sieve size fraction was used for the hydrometallurgical experiments with the collected underflow divided by a rotating sample divider. De-ionized water was used in the water leaching experiments. The water leachate was analyzed for its metals content and the water leaching conditions are shown in Table 1. The water leaching time (5 or 30 min) contains variation as the filtration was a time-consuming process due to small particle size.
The washed samples were allowed to settle, after which some of the initial water was decanted through the filter due to the fine particles which may easily make the filter medium impermeable to solution. Wash water was then acidified with sulfuric acid by mixing 10 mL of 4 M H2SO4 with 200 mL of wash water. The resulting precipitate was filtered, and while on filter, washed with DI water several times. The precipitate was analyzed with Thermoscientific Flash Smart MVC CHNS/O Elemental Analyser controlled by EagerSmart Software. Oxygen flow rate = 250 mL/min, cycle time = 720 s, sample delay = 12 s, and oxygen injection = 4 s. Furnace temperature was 950 °C. Samples between 2 and 3 mg in weight were sealed in tin prior to combustion and Cystine was used as the standard reference material.
Extraction of Metals
Leaching experiments were performed on the raw materials by using sulfuric acid (H2SO4; Emsure ISO 95–97%, VWR Chemicals) and the dissolution process was performed as a stepwise addition of acid under constant stirring by either magnetic stirrer or mechanical agitation (VWR VOS16). Two experiments were performed: firstly a leaching experiment with stepwise acid addition 1E, to measure the acid consumption and secondly, leaching experiment 2E with stepwise acid addition to investigate whether preferential dissolution of certain metals occurs under acid-starved conditions (Table 2).
Sample 1E comprised of washed sample 2W, and 2E was washed sample 3W. In 1E, a short equilibration time (ca. 15 min) was utilized between the acid additions, whereas in 2E, there was 1-h equilibration time between steps. In both leaching experiments, the 1-L glass leaching reactor was immersed in a heated water bath (Haake DC 10) and a water condenser was installed in order to avoid solution evaporation during leaching. Mechanical agitation (400 rpm, reactor d = 14 cm) was employed throughout the experiments. Between acid additions, the pH of the solution was measured by a pH Meter (Easy Seven Mettler-Toledo), equipped with an InLab Expert Pro Electrode (Mettler-Toledo) and the solution redox potential was monitored by a Radiometer Analytical REF201 Red Rod reference electrode (Hach) and platinum strip. Due to the heterogeneity of the raw material samples, the leach residues were also analyzed after the experiments were complete.
Bulk Recovery of Rare Earths
Rare earths were recovered as double salt precipitates by sodium sulfate addition. Four samples obtained from the pregnant leach solution (PLS, 1E) were used, in each experiment 80 mL of PLS was added into a small water-jacketed reactor. REE double sulfate solubility decreases as a function of increasing temperature, so a higher T will result in higher supersaturation and precipitation yield of REEs. Nonetheless, in terms of future process scalability, it is desirable to limit the temperature to such a level that it does not necessitate a higher temperature than that used in leaching—therefore a T = 50 °C was chosen. Under magnetic stirring, 20 mL of sodium sulfate (Na2SO4; Anhydrous ACS Reag. ISO) solution was added with varying, pre-determined Na2SO4 quantities, totaling solution volume of 100 mL. The amount of Na2SO4 was calculated in such way that each of these 100 mL solutions had different, well-defined Na2SO4 molarity at the beginning of the experiment (0.01, 0.05, 0.1, and 0.5 M, respectively). Each precipitation experiment lasted an hour, after which, the solution samples were retrieved, diluted, and analyzed.
Results and Discussion
NiMH Battery Waste Characterization
The analysis of chemical elements reporting to different particle size classes is presented in Fig. 2. Particle size separation is a commonly applied method used (i) to enrich the fraction with respect to a desired metal as well as (ii) to separate desired metals from impurity metals such as Fe. In this work, La was taken as the indicator element for the behavior of other REEs present in the active electrode powder. In hydrometallurgical processes, dissolved Fe is an impurity which is difficult to precipitate with good filtration properties without high pressure and temperature, and is generally discarded as a plain hydroxide or jarosite. Therefore, to minimize Fe entering to the hydrometallurgical process, the selection of the optimum particle size was targeted at ensuring an acceptable separation efficiency for Fe-rich and REE-rich fractions. In Fig. 2, it is clearly seen that REEs concentrate in particle sizes < 1500 µm, indicating that the fine anode active materials are mostly separated from the larger electrode particles, such as the steel current collectors and are concentrated to the underflow of the 1000 µm sieve. However, Ni is found in significant quantities in particle sizes above 1500 µm (ca. 20 wt% and 5 wt% per 3000–6000 µm and > 6000 µm, respectively), which might be related to the other sources than the REE-containing anode materials. For example, Ni is also present in cathode materials such as Ni(OH)2 and NiO·OH. The “others” category, which contains plastics and metal traces, has a higher contribution at larger particle sizes as seen in Fig. 2a, b where different particles have been imaged. It must also be noted that La is part of the alloyed active materials that also contain Ce, Pr, Nd, Ni, Co, Al, Mn, and Zn, all of which would most likely be similarly distributed to La in the sieving—as emphasized by the way Al and Co are distributed between the different size fractions.

a The mass fractions of NiMH battery waste particle size classes and b the chemical composition of different particle size classes. (Color figure online)
Cumulative distribution of different elements was also determined (Fig. 3) as a combination of data from Fig. 2a, b. The figure shows that both, a perfect separation of Fe from La and high yield, cannot be achieved for the NiMH waste crushed by the state-of-the-art methods. For example, the largest difference in distribution based on particle sizes is found between Fe and La. Accordingly, a separation of Fe vs. La is possible with the 250 µm sieve, although 28% of La would still get lost to the Fe-rich fraction. On the other hand, sieving with the 1000 µm sieve results in 92% extraction for La. By choosing a sieve size of 1000 µm, the initial mass ratio of Fe/La = 177 for the whole raw material is reduced to 25.35 in the − 1000 µm size fraction. For the subsequent leaching experiments, sieving was performed at a sieve size of 1000 µm (with a 92%:14% of La:Fe extraction and enrichment of La in relation to Fe by factor of 6.99) using the underflow as the feed—i.e., pre-concentrated—material. Enrichment of La was calculated by using Eq. (5):
where \(m_{x_{\text{uflow}}}\) indicates the mass of the investigated element found in underflow of sieve size 8000 µm and \(m_{y_{\text{uflow}}}\) at sieve size of 1000 µm.

Cumulative particle size distribution for the crushed NiMH battery waste. (Color figure online)
The density distribution of elements, measured by pycnometry and ICP-OES, is shown in Fig. 4. No advantageous regions of separation were observed, even though it could be possible to separate a fraction relatively rich in Fe and plastics from the finer electrode powders. However, according to previous studies, magnetic separation may be a more viable separation method for the enrichment of Fe-containing powders [32].

Distribution of elements based on effective density of each size fraction. Size is defined as the average particle size on sieve. (Color figure online)
An interesting observation during the sieving procedure was that agglomerates of small fibrillar particles were observed. These fiber agglomerates are most likely parts of the broken polymeric separators. The fibers were able to pass even the smallest sieve size of 75 µm and it appears that the shaking motion of the sieving apparatus caused them to agglomerate. Furthermore, in the presence of a permanent magnet, the fibers appeared to have a positive magnetic susceptibility. Subsequently, some fibers were hand-picked for investigation by SEM (Fig. 5) and although energy dispersive X-ray spectroscopy (EDS) analysis was not possible due to the electrical charge buildup in the samples, the distinct shapes of the ferromagnetic steel current collectors can still be discerned from the images. Furthermore, it is clear that particles—most likely either anode or cathode active materials—are also distributed across the surface of the current collector material. Larger particles were also found tangled in the fibers as shown by Fig. 5, whereas it can be seen that the current collector pieces are only millimeters in size.

SEM pictogram presenting hand-picked fiber agglomerates
With prolonged sieving, it was found that the fibers agglomerate such that they were unable to pass through the perforations anymore. This characteristic could be exploited for the separation of the fibers from the rest of the raw materials, by the application of an additional vibration without the intent to sieve them into coarse fractions. However, without further treatment of the fibers, the extraction of valuable metals would most likely be reduced due to contamination of the agglomerated current collector-rich phase with active anode and cathode materials (REE, Ni). To the best of the authors’ knowledge, such observations have not been reported earlier and suggest that separation based on sieving may suffer from agglomeration behavior of the current collector material, which may require attention in the recycling process design.
The particle size class < 125 µm was also analyzed by XRD which, due to fluorescent radiation from the Mn present in the sample caused by the Kα energy of Co anode radiation, has a high Mn background in relation to the other peaks. The diffractogram obtained is similar to that previously measured by Rodrigues and Mansur in their characterization work and there is a strong correlation of peak shapes and positions, although not of relative peak strengths [33]. Additionally, unlike Rodrigues and Mansur, the present work was not able to confirm the presence of REE oxides or hydroxides. However, analysis of the x-ray diffractogram did confirm the presence of both Ni hydroxide and metallic mischmetal alloy (Fig. 6). Unless the drying pre-treatment of the battery materials at T = 60 °C utilized by Rodrigues et al. resulted in the oxidation of mischmetal alloys, it is unlikely that La2O3 or La(OH)3 would be the major REE phases in NiMH battery waste. In the current work, the crushing process ensures that complete oxidation of the battery waste does not occur, a claim which is supported by a previous study that detected gas evolution during the sulfuric acid leaching of NiMH waste [19] as a result of H+ ion reduction during the oxidation of the metal alloys. Taken together, these observations suggest that La2O3 and La(OH)3 are most likely present only in small quantities as thin passivation layers. This is not surprising as the metallic alloys are extremely reactive and have been shown to react with moist air according to reaction (6) [34]:

Powder XRD of NiMH battery waste fraction of − 125 µm, showing the presence of two major phases: an amorphous nickel hydroxide and very crystalline mischmetal alloy. (Color figure online)
Water Leaching
The chemical analyses of the water washing are presented in Table 3. The difference between the dissolved metal concentrations is caused by a difference in the amounts of washing water used and the pH of the wash waters was observed to be high, pH > 10. Filtration of the raw material after water washing was shown to be difficult due to the presence of very fine particles in the raw material, which easily prevents passage of the solution through the filtration medium. Additionally, for the test 3W (Table 1), the water washing was performed for 1 h under mixing, after which the solution was let to settle for 24 h. It was found that colloidal particles appeared to be present as some of the particles did not settle even after 24 h of settling. However, unless in-situ precipitation of REEs is desired during leaching, the washing treatment must be performed in order to avoid loss of REEs as shown previously [19] or alternatively, a process for additional leach residue treatment must be designed. Table 1 shows that the predominant dissolved and analyzed impurities were K and Na, whereas Ni was also present in minor amounts.
In each of the wash waters, the color of the water was yellow, indicating that compounds, other than K and Na, were also dissolved. Curiously, when neutralization of the wash water with sulfuric acid was attempted, a reaction was observed that initially led to the formation of a water immiscible phase, after which a yellowish phase precipitated from the solution. The gelatinous form and apparent density seemed to indicate organic origin. Analysis of the precipitate indicated the presence of carbon (51–55 wt%), hydrogen (4.31–4.77 wt%), and nitrogen (0.42–0.45 wt%) in the material. It is possible that a similar reaction will and has occurred in prior studies during the sulfuric acid leaching of the raw material, causing some of the water-washable organic materials to remain in the leach residue instead of the pregnant leach solution. This precipitate represents a new solid waste product from NiMH battery recycling process that consequently requires a more detailed analysis of composition to ensure safety. After washing, the raw material was filtered and dried. The wash water was measured to be basic (pH ca. 10) before the acidification. The findings detailed here show that if the washing of the NiMH waste raw material is undertaken prior to direct NiMH battery waste leaching, several aspects need to be taken into account: (i) the washing step results in a basic washing water rich in Na (500–1000 mg/L) and K (1500–4000 mg/L) and a variety of other elements, which would need a separate unit process for water purification to be considered. Nevertheless, were it not for the unknown exact composition of the precipitating mass, the wash water itself could potentially be used as a precipitating agent in REE double sulfate precipitation.
Extraction of Metals
Leaching experiments 1E and 2E were performed as defined in Sect. 4.3. Firstly, the underflow of the 1000 µm sieve was divided and analyzed by total dissolution and ICP-OES. The chemical analysis was performed by total dissolution and ICP-OES for the sieved raw material (< 1000 µm) that was used in the leaching experiments, Table 4.
Acid consumption in leaching was defined by stepwise addition of H2SO4, and the effect of added acid on pH is shown in Fig. 7. Initially, water containing the raw material was measured to be basic (pH > 10), indicating that alkali hydroxide electrolyte residues existed in the battery material, even after water washing. During mixing of washed battery waste with the acid, vigorous gas evolution was observed and this gas is most likely hydrogen, as indicated in Eq. (2). This phenomenon can cause issues in realizing an industrial leaching operation for NiMH batteries, if raw material is leached by a similar methodology to the one used in this study, although this problem can be overcome by, e.g., roasting [35]. The results (Fig. 7) show that ca. 14 mmol H2SO4 per gram of sample is required to achieve pH < 1. The acid was added stepwise and redox potential was measured along with pH from the reactor. Up to 6.2 mmol of acid per gram of sample, the pH remained around 4 indicating that a notable amount of acid-consuming reactions were still occurring. At higher acid addition, a discernable decrease in pH is evident, suggesting either (i) that most of the leaching reactions were complete or (ii) that reactions other than acid-consuming reactions, such as in Eq. (3), had started to prevail. Furthermore, when compared to previous research on leaching [27], the problem of a viscous surface phase being present in the reactor was not readily apparent in the present study. This may be an indication that water washing had additional benefits like the simultaneous removal of water-soluble organic matter along with the alkali metals, K+ and Na+. In addition, as the anode active material has a very low standard reduction potential—relevant half-reaction has standard reduction potential (vs. SHE) of − 0.704 V (reaction 7) as calculated with HSC Chemistry [26]—it will therefore act as a reductant.

Acid consumption in leaching (1E) of NiMH battery waste (fraction—1 mm). pH and redox as a function of time after sulfuric acid additions. (Color figure online)
Consequently, the total cell reaction would be
After leaching (1E), the solution was filtered and the leach residue weighed. The final wet residue mass was 15.3 g, and its moisture content was measured to be 5.6 g showing that 9.7 g (10.2 wt%) of the original sample remained after leaching. The leach residue was dissolved, before being analyzed by ICP-OES and it was shown to still contain ~ 46 wt% Ni, (see Table 5), which corresponds to > 90% Ni extraction when calculated from the raw materials analysis (Table 4). It is interesting to note that it was not possible to obtain good extraction of Ni, whereas most of the REEs and Co—which are part of the anode active materials—was dissolved. It has been shown in previous research that Ni is present as different phases whose dissolution increases in the presence of oxidative materials [36]. Since both REEs (ca. 0.6 wt%) and alkali metals (ca. 2.2 wt%) were found in the residue, it is possible that some double sulfate precipitation occurred during the leaching as REE double sulfates, e.g., LaNa(SO4)2·H2O has a solubility of 2.34 g/L in H2O in terms of La2O3 [37], however, it is clear that most of the REEs had dissolved as is evident when the leach residue analysis is compared to raw material analysis (Table 5 vs. Table 4). Nevertheless, ensuring a good and thorough washing is mandatory in order to avoid loss of rare earths into the leach residue, otherwise the leach residue must be separately treated for REE extraction.
As is evident from experiment 1E, with the addition of acid, the solution pH is buffered to 3–4 when acid concentration is kept low. Consequently, this pH window was investigated in more detail in order to find out if low acidity leaching could result in both selective and high extractions from the investigated battery waste. The plateau of stable pH visible in Fig. 7 indicates that there is no driving force for reactions remaining—see Fig. 8 and Table 6—and that the buffering could be explained by the onset of, for example, Al and REEs metal hydrolysis. Due to the low redox potential, Fe most likely remains soluble in the form of ferrous ions, as that ferric ions are susceptible to hydrolysis only around a pH of ca. 2—3. This presents a challenge for the subsequent solution treatments as selective iron removal in the ferric form is dependent on being able to oxidize the ferrous ions present to ferric ions. The chemical content of the leach residue (25 g) was also analyzed as shown in Table 7. Large fraction of the leach residue was Ni (60.8%), however, the total mass reduction during leaching was already significant: 70.6 wt%. Based on the results, selective leaching of Ni vs. Fe is possible, at low final Ni yield.

pH and redox potential as a function of time and added acid during NiMH battery waste leaching (2E, T = 50 °C, final S/L = 1/10). (Color figure online)
A comparison between leaching experiments 1E and 2E shows that there is a disparity between REE contents for both residue and leachate. Although the samples used in this research were carefully homogenized and then divided using a rotating sample divider, the small initial sample size of ca. 100 g used is unlikely to be totally homogeneous. This further highlights the importance of developing robust methods of characterization of waste raw materials. It is also recommended to carefully control the acid concentration as well as the solid contents, both battery material feed rate and acid concentration due to the exothermic nature of the reaction and gas evolution. By maintaining the process close to a pH range of 3–5, the intensity of the reactions was observed to be reduced and could be more easily controlled.
Bulk Recovery of Rare Earths
After leaching experiment 1E, the resulting PLS was collected by vacuum filtration and precipitation experiments were performed. The PLS was analyzed for metals content and the results are presented in Table 8. Some soluble Na+ and K+ were found to remain even after water washing and consequently may have caused precipitation during leaching. Nonetheless, the solution concentration of La far exceeds that which was achieved in our previous research, as a much greater quantity of REEs was dissolved into the solution [19].
In this study, conditions similar to those used during leaching—pH of the initial PLS was ≪ 1, T = 50 °C—with less concentrated Na2SO4 additions were utilized. The rare earth and alkali metal concentrations determined after the precipitation experiments are presented in Table 9. It can be seen that amount of Y decreases only at the highest Na2SO4 concentration utilized. It can also be observed that the potassium content decreased with increasing sodium content, indicating that potassium co-precipitation was assisted by the sodium addition. The pH was measured to be 1.08 after precipitation experiments. These results could potentially be improved by adopting a higher temperature [38] and by using longer reaction time as the process operates close to the limits of supersaturation, and 1 h most likely is not enough to achieve equilibrium in precipitation–dissolution reactions [39].
Finally, the Na + K to REE ratios were calculated and are shown in Table 10. The results show that a higher than 1.82 Na/(La + Ce + Pr + Y + Sm) ratio should be used in order to achieve a good extraction of La + Ce + Pr + Sm. Interestingly, it would appear that the Y double salt has a higher solubility in this mixed solution when compared to the other REEs. This finding is similar to the results obtained by Kul et al. [40], which also indicated that Y double salt has a higher solubility than other REEs and the recent thermodynamic investigation by Das et al. [38] on the solubility of REE double sulfates. Consequently, these results indicate that it is not viable to recover the small quantity of Y present in solution by the double salt method.
Losses during sieving amount to 7% of La, and therefore REE, being lost due to size separation (93% recovery). In terms of REE extraction during leaching, > 99% of La, Ce, and Pr were leached as well as > 95% of Sm (Table 5). It is most probable that a majority of the REEs dissolved during the stepwise addition of acid, which suggests that REEs are mostly in a form that are easily dissolvable, i.e., as metallic active materials. In contrast, Ni can be present as several different compounds, some of which are not easily dissolvable under reducing conditions, therefore limiting its dissolution. Finally, after all the losses during sieving and leaching, precipitation of REEs may incur a loss of 1–10% of REEs, depending on the time given for the precipitation process to proceed towards equilibrium, and also on the amount of Na2SO4 added. Excessive addition of Na2SO4 can result in sodium balance issues. The PLS and REE double sulfates produced here can be further treated such that it can be accommodated into current state-of-the-art industrial processes [30, 31].
Conclusions
NiMH batteries were obtained from an industrial operator in a crushed state. The obtained battery waste was characterized by sieving and pycnometry and the fractions obtained were chemically analyzed. The separation of valuable fine powders was achieved with sieving, such that a majority of the steel pieces and plastics remained in the overflow (> 1 mm). During sieving, it was seen that the fibrillar material present from the polymeric separators could pass even through the smaller sieve sizes, although with prolonged sieving, agglomeration of these fibers was also observed. The < 1 mm fraction was used in the hydrometallurgical experiments and this raw material was washed. This wash water was analyzed, found to be highly alkaline, and contains Na (500–1000 mg/L) and K (1500–4000 mg/L) originating from the alkaline electrolyte present in these batteries. Additionally, the presence of water immiscible organic compounds in the wash water was observed for the first time. When neutralization of the wash water was undertaken, these organic compounds precipitated to yield a clear solution containing Na and K.
In leaching experiments, the consumption of acid was investigated along with the redox behavior of the solution. It was determined that the particular raw material stream used in this study required ca. 14 mmol of H2SO4 per gram of sample in order to complete the leaching reactions and decrease the solution a pH 1. The redox conditions generated by the raw material were very reducing; potentials within range of − 400 to 0 mV (vs. Ag/AgCl) were recorded during leaching. These results suggest that the waste has highly reductive properties due to gas evolution, further supporting the suggested leaching mechanism. Behavior of the raw material in low acid concentrations was investigated in order to more fully understand the behavior under such conditions. Slightly selective dissolution was found when the acidity available was limited and the dissolution conducted at pH 3–4. Fe and Al were observed to begin to dissolve only after significant dissolution of REEs, Ni, and Co. REEs were precipitated as double salts and these results correlate with the findings of other researchers, which also show that efficient recovery Y is not possible without a high excess level of Na (Na:REE = 9).
References
Liu H, Zhang L, Sun A (2011) Electrochemical technologies for energy storage and conversion. Wiley, Weinheim
Dehghani-Sanij AR, Tharumalingam E, Dusseault MB, Fraser R (2019) Study of energy storage systems and environmental challenges of batteries. Renew Stustain Energy Rev 104:192–208
Reuter MA, Hudson C, Van Schaik A, Heiskanen K, Meskers C, Hagelüken C (2013) Metal recycling: opportunities, limits, infrastructure, a report of the working group on the global metal flows to the international resource panel. UNEP.
European Comission: Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions on the 2017 list of Critical Raw Materials for the EU. (2017). https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=COM:2017:0490:FIN. Accessed 1 Aug 2019
Stahl H, Baron, Y, Hay D, Hermann A, Mehlhart G, Baroni L, Rademaekers K, Williams R, Pahal S (2018) Evaluation of the Directive 2006/66/EC on batteries and accumulators and waste batteries and accumulators. Trinomics. https://ec.europa.eu/environment/waste/pdf/Published%20Supporting%20Study%20Evaluation.pdf. Accessed 13 Nov 2019
Rotter VS, Maehlitz P, Korf N, Chancerel P, Huisman J, Habib H, Herreras L, Söderman, ML, Hallberg A. Waste flow studies. https://www.prosumproject.eu/sites/default/files/ProSUM%20D4.1%20Waste%20Flow%20Studies_0_0.pdf. Accessed 13 Nov 2019
Tunsu C, Petranikova M, Gergorić M, Ekberg C, Retegan T (2015) Reclaiming rare earth elements from end-of-life products: a review of the perspectives for urban mining using hydrometallurgical unit operations. Hydrometallurgy 156:239–258
Larsson K, Ekberg C, Ødegaard-Jensen A (2013) Dissolution and characterization of HEV NiMH batteries. Waste Manage 33:689–698
Innocenzi V, Ippolito NM, De Michelis I, Prisciandaro M, Medici F, Vegliò FV (2017) A review of the processes and lab-scale techniques for the treatment of spent rechargeable NiMH batteries. J Power Sources 362:202–218
Chagnes A, Swiatowska J (2015) Lithium process chemistry: resources, extraction, batteries, and recycling. Elsevier, Amsterdam
Pietrelli L, Bellomo B, Fontana D, Montereali M (2005) Characterization and leaching of NiCd and NiMH spent batteries for the recovery of metals. Waste Manage 25:221–226
Polyakov EG, Sibilev AS (2015) Recycling rare-earth-metal wastes by pyrometallurgical methods. Metallurgist 59:368–373
Tirronen T, Sukhomlinov D, O'Brien H, Taskinen P, Lundström M (2017) Distributions of lithium-ion and nickel-metal hydride battery elements in copper converting. J Clean Prod 168:399–409
Nickelhütte. https://www.nickelhutte.com. Accessed 15 Oct 2019
Ojanen S, Lundström M, Santasalo-Aarnio A, Serna-Guerrero R (2018) Challenging the concept of electrochemical discharge using salt solutions for lithium-ion batteries recycling. Waste Manage 76:242–249
Salgado AL, Veloso AMO, Pereira DD, Gontijo GS, Salum A, Mansur MB (2003) Recovery of zinc and manganese from spent alkaline batteries by liquid–liquid extraction with Cyanex 272. J Power Sources 115:367–373
Ebin B, Petranikova M, Ekberg C (2018) Physical separation, mechanical enrichment and recycling-oriented characterization of spent NiMH batteries. J Mater Cycles Waste Manage 20:2018–2027
Bertuol DA, Bernardes AM, Tenório JAS (2006) Spent NiMH batteries: characterization and metal recovery through mechanical processing. J Power Sources 160:1465–1470
Porvali A, Wilson BP, Lundström M (2018) Lanthanide-alkali double sulfate precipitation from strong sulfuric acid NiMH battery waste leachate. Waste Manage 71:381–389
Innocenzi V, Vegliò F (2012) Recovery of rare earths and base metals from spent nickel-metal hydride batteries by sequential sulphuric acid leaching and selective precipitations. J Power Sources 211:184–191
Li L, Xu S, Ju Z, Wu F (2009) Recovery of Ni, Co and rare earths from spent Ni–metal hydride batteries and preparation of spherical Ni (OH)2. Hydrometallurgy 100:41–46
Nan J, Han D, Yang M, Cui M (2006) Dismantling, recovery, and reuse of spent nickel–metal hydride batteries. J Electrochem Soc 153:A101–A105
Petranikova M, Herdzik-Koniecko I, Steenari B, Ekberg C (2017) Hydrometallurgical processes for recovery of valuable and critical metals from spent car NiMH batteries optimized in a pilot plant scale. Hydrometallurgy 171:128–141
Wang R, Yan J, Zhou Z, Gao X, Song D, Zhou Z (2002) Regeneration of hydrogen storage alloy in spent nickel–metal hydride batteries. J Alloys Compd 336:237–241
Meshram P, Pandey BD, Mankhand TR (2016) Process optimization and kinetics for leaching of rare earth metals from the spent Ni–metal hydride batteries. Waste Manage 51:196–203
Outotec, HSC Chemistry, https://www.outotec.com/products/digital-solutions/hsc-chemistry/ 9.4.1 (2018)
Porvali A, Lundström M (2017) Leaching of crushed NiMH batteries. Eur Metall Conf 1:139–148
Bertuol DA, Bernardes AM, Tenório JAS (2009) Spent NiMH batteries—the role of selective precipitation in the recovery of valuable metals. J Power Sources 193:914–923
Davis JR (1998) Metals handbook, desk edition (2nd edition).
Porvali A, Agarwal V, Lundström M (2019) Circulation of sodium sulfate solution produced during NiMH battery waste processing. Min Metall Explor 36:979–991
Agarwal V, Khalid MK, Porvali A, Wilson BP, Lundström M (2019) Recycling of spent NiMH batteries: integration of battery leach solution into primary Ni production using solvent extraction. Sustain Mater Technol 22:e00121
Tsunekawa M, Ito M, Furuya H, Hiroyoshi N (2007) The recovery of electrode compounds from waste nickel metal hydride batteries by physical separation techniques. Mater Trans 48:1089–1094
Rodrigues LEOC, Mansur MB (2010) Hydrometallurgical separation of rare earth elements, cobalt and nickel from spent nickel–metal–hydride batteries. J Power Sources 195:3735–3741
Srivastava S, Srivastava ON (1999) Synthesis, characterization and hydrogenation behaviour of composite hydrogen storage alloys, LaNi5/La2Ni7, LaNi3. J Alloys Compd 282:197–205
Korkmaz K, Alemrajabi M, Rasmuson Å, Forsberg K (2018) Recoveries of valuable metals from spent nickel metal hydride vehicle batteries via sulfation, selective roasting, and water leaching. J Sustain Metall 4:313–325
Liu F, Peng C, Porvali A, Wang Z, Wilson BP, Lundström M (2019) Synergistic recovery of valuable metals from spent nickel-metal hydride batteries and lithium-ion batteries. ACS Sustain Chem Eng 7:16103–16111
Lokshin EP, Tareeva OA, Ivlev KG, Kashulina TG (2005) Solubility of double alkali metal (Na, K) rare-earth (La, Ce) sulfates in sulfuric-phosphoric acid solutions at 20 C. Russ J Appl Chem 78:1058–1063
Das G, Lencka MM, Eslamimanesh A, Wang P, Anderko A, Riman RE, Navrotsky A (2019) Rare earth sulfates in aqueous systems: Thermodynamic modeling of binary and multicomponent systems over wide concentration and temperature ranges. J Chem Thermodyn 131:49–79
Myerson AS (2002) Handbook of industrial crystallization. Elsevier, Oxford
Kul M, Topkaya Y, Karakaya İ (2008) Rare earth double sulfates from pre-concentrated bastnasite. Hydrometallurgy 93:129–135
Acknowledgements
Open access funding provided by Aalto University. The authors acknowledge METYK project for providing the context where the work was performed. METYK project was financially supported by TEKES, EU, Vipuvoimaa EU:lta 2014-2020, Norilsk Nickel, CrisolteQ, Akkuser, Kuusakoski, Jyväskylän Energia, Luvata, Boliden Harjavalta and municipalities of Pori and Harjavalta. This work has also been supported by the MineWEEE Project (Grant Number 7100/31/2016), BATCircle (Grant Number 4853/31/2018), both by TEKES, and by the Strategic Research Council at the Academy of Finland, project CloseLoop (Grant Number 303454). Miamari Aaltonen is acknowledged for undertaking the leaching sample division. Hannu Revitzer is acknowledged for the total dissolution and chemical analysis of the samples with ICP-OES and AAS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there are no conflict of interest.
Additional information
The contributing editor for this article was T. Hirato.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Porvali, A., Ojanen, S., Wilson, B.P. et al. Nickel Metal Hydride Battery Waste: Mechano-hydrometallurgical Experimental Study on Recycling Aspects. J. Sustain. Metall. 6, 78–90 (2020). https://doi.org/10.1007/s40831-019-00258-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40831-019-00258-2