
Abstract
The huge progress in whole genome sequencing (genomic revolution) methods including next generation sequencing (NGS) techniques allows one to obtain data on genome sequences of all organisms, ranging from bacteria to plants to mammals, within hours to days (era of whole genome/exome sequencing) (Goodwin et al. in Nat Rev Genet 17:333–351, 2016; Levy and Myers in Annu Rev Genomics Hum Genet 17:95–115, 2016; Giani et al. in Comput Struct Biotechnol J 18:9–19, 2020). Today, within the era of functional genomics the highest goal is to transfer this huge amount of sequencing data into information of functional and clinical relevance (genome annotation project). The World Health Organization (WHO) estimates that more than 10,000 diseases in humans are monogenic, i.e., that these diseases are caused by mutations within single genes (Jackson et al. in Essays Biochem 62:643–723, 2018). NGS technologies are continuously improving while our knowledge on genetic mutations driving the development of diseases is also still emerging (Giani et al. in Comput Struct Biotechnol J 18:9–19, 2020). It would be desirable to have tools that allow one to correct these genetic mutations, so-called genome editing tools. Apart from applications in biotechnology, medicine, and agriculture, it is still not concisely understood in basic science how genotype influences phenotype. Firstly, the Cre/loxP system and RNA-based technologies for gene knockout or knockdown are explained. Secondly, zinc-finger (ZnF) nucleases and transcription activator-like effector nucleases (TALENs) are discussed as targeted genome editing systems. Thirdly, CRISPR/Cas is presented including outline of the discovery and mechanisms of this adaptive immune system in bacteria and archaea, structure and function of CRISPR/Cas9 and its application as a tool for genomic editing. Current developments and applications of CRISPR/Cas9 are discussed. Moreover, limitations and drawbacks of the CRISPR/Cas system are presented and questions on ethical concerns connected to application of genome editing tools are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Development of genome editing tools for targeted genome editing: from basic science to application
Powerful genome editing that allows a specific alteration of DNA sequences even on a single nucleotide scale, without creating unwanted off-target effects, is the basis to transform basic science into personalized medicine [1,2,3,4,5]. The following sections explain the currently used tools for genomic editing and describe how these can be used for applications in medicine, agriculture, and biotechnology. Genome editing includes diverse manipulations of the genome: (a) deletions, (b) insertions, (c) knockouts, (d) activation/repression of transcription, (e) protein targeting in cells, and (f) alteration of the epigenetic state in cells.
The Cre/loxP system
In the past genome engineering was mostly performed using technologies based on homologous recombination. This implies that homologous DNA must be present. Using these methods, it is possible to study how genotype affects phenotype. To achieve gene deletions in living mice or in cells of tissues or organs derived from mice, the Cre/loxP system was most often applied (Fig. 1) [6]. The system, which is based on from the Escherichia coli bacteriophage P1, is a tyrosine recombinase (Cre—causes recombination or cyclization recombination) that binds with high affinity to the loxP sites (loxP—locus of crossing over of bacteriophage P1), a specific DNA sequence. The loxP sites encompass 34 base pairs (bp), in which 13 bp at the termini form inverted palindromic repeats flanking an asymmetric central 8-bp core sequence (5′-ATAACTTCGTATA-NNNTANNN-TATACGAAGTTAT-3′; N any nucleotide) (Fig. 1b). The inverted repeats are the specific recognition sites for Cre and the core sequence is the site in which recombination takes place. One Cre monomer binds to one 13-bp inverted repeat sequence, i.e., a Cre dimer binds to a single DNA double strand and causes bending of that part of the DNA (Fig. 1c). Within its 343 residues (molecular weight of 38 kDa), the Cre recombinase monomer encompasses two domains, an N-terminal domain composed of five α-helices making contacts to the major groove of the DNA and a C-terminal domain composed of nine α-helices as well as three β-strands important for DNA and inter-subunit interactions [7,8,9,10]. The active site is located in the C-terminal domain. For Cre-mediated recombination, two DNA double strands are involved each containing loxP sites (Fig. 1a, c). This results in a Cre tetramer including four active sites binding to the two DNA double strands (Fig. 1c). Depending on the orientation and localization of the loxP sites relative to each other, several types of recombination can be observed: translocation (loxP sites on different DNA double strands), inversion (loxP sites oriented in opposite directions on the same DNA double strand), or deletion (loxP sites oriented in same direction on the same DNA double strand) (Fig. 1). As a site-specific tyrosine recombinase, Cre catalyzes a type I topoisomerase-like reaction (Fig. 1d). In contrast to other recombinases such as Flp recombinase, the active site is not shared between different subunits (in trans) but is entirely present in one subunit (in cis). The family of tyrosine recombinases use a highly conserved active site composed of an arginine-histidine-arginine (Arg-His-Arg; one-letter code R-H-R) catalytic triad, a tyrosine (one-letter code Y) as well as a tryptophan (one-letter code W) residue as nucleophiles. The catalytic triad and the tryptophan are important to coordinate the steps for DNA cleavage. Within the Cre tetramer, one monomer within each dimer becomes active and each tyrosine residue as a nucleophile attacks the phosphodiester bond of a single DNA strand of the bound double strand DNA (in cis) (Fig. 1a, step 1). Thereby, two covalent phosphotyrosine bonds in two single strands of two DNA double strands are formed with the 3′-phosphates of the scissile bonds (Fig. 1, step 2). Subsequently, the liberated 5′-hydroxyl groups attack the phosphotyrosines of the opposed other double strand (in trans) resulting in the formation of a Holliday intermediate (Fig. 1, step 3). The mechanism repeats with the other two Cre molecules of the dimer becoming active and the tyrosines attacking the phosphates in the DNA strands (in cis) and the free 3′-hydroxyl groups attacking the phosphotyrosines. Finally, the recombinant DNA products are formed (Fig. 1, step 4) [9, 10]. Compared to other recombination systems, the Cre recombination system has several advantages. Firstly, it requires only Cre recombinase and two loxP sites. No cofactors such as ATP are needed, no accessory proteins are needed, and no other sequence elements except from loxP sites are needed. Secondly, Cre/loxP can be applied in almost every cell type, i.e., in mammals, yeast, bacteria. Thirdly, it can be used in vitro and in vivo. Cre can be used in cells but also in tissues or organs and even in living organisms. By using specific promotors that drive expression in different cell types or tissues, it is possible to create tissue/organ-specific recombination [6, 11]. Thereby, it is possible to specifically delete genes only in specific tissues/organs (conditional gene knockout). Owing to the high affinity of the Cre recombinase for loxP sites, Cre is very effective and precise. However, generating gene knockouts using the Cre/loxP system is very laborious and time consuming as it involves extensive selection and screening. To this end, it is not applicable for high-throughput screening. Moreover, Cre/loxP carries the risk of creating unwanted side effects and mutagenic effects [12]. It was shown that expression of Cre recombinase occurs sometimes in cells not targeted for genome editing. Although this seems to be at a very low expression level, those Cre protein levels are still sufficient to create unwanted recombination events at loxP sites in the genome [12]. Besides, it was shown that Cre recombinase can also have activity on off-target sites in target genomes, i.e., sites that resemble loxP sites. Approaches to create Cre recombinase variants by protein engineering strategies allow one to increase the specificity of Cre recombinase [13].

Cre/loxP system from Escherichia coli can be used as a tool for genome editing. a Mechanism of homologous recombination used by Cre recombinase. Cre recombinase is a site-specific recombinase using a catalytic tyrosine residue for catalysis. Two Cre molecules bind to loxP sites at one dsDNA molecule that associate to form the recombination synapse (tetrameric Cre complex bound to two loxP sites in the target DNA) (step 1). An active site tyrosine residue (Tyr324) of Cre recombinase attacks a phosphodiester bond within the same DNA strand (in cis) (step 1, cleavage) resulting in formation of a 3′-phosphotyrosine intermediate (step 2). The released 5′-hydroxyl groups attack the phosphotyrosines in trans in a strand exchange/transfer reaction resulting in formation of the Holliday junction intermediate (step 3). Cleavage and strand exchange reactions repeat involving the other DNA strands. As a result, homologous DNA fragments were recombined and recombination products are formed (step 4). (modified figure from [7]). b Sequence of loxP sites. The inverted, palindromic repeats are underlined. The nucleotides highlighted in red show the region of strand exchange or crossover. The arrows show the phosphodiester bonds cleaved during Cre–loxP recombination. c Structure of tetrameric recombination synapse complex of the E. coli bacteriophage P1 Cre recombinase in complex with loxP target DNA. The structure shows the reaction intermediate from step 2 in b, in which two active Cre monomers (orange) are in the 3′-phosphotyrosine covalent intermediate state. The other Cre molecules (green) are in the inactive state. Cre recombinase and loxP are shown in a cartoon-type representation. The figure was prepared using the structure with PDB code 1Q3V. d Close-up of the 3′-phosphotyrosine DNA–protein covalent intermediate. Cleavage occurs at the 3′-phosphate of the sequence 5′-ApT-3′ in the loxP sequence
RNA interference
Another method to achieve targeted gene knockdown is based on RNA interference (RNAi). RNA interference (antisense RNA) for targeted gene knockdown was first applied in 1998 by Andrew Z. Fire and Craig C. Mello using the nematode Caenorhabditis elegans as a model system [14]. Fire and Craig were awarded the Nobel Prize in Physiology and Medicine in 2006 “for their discovery of RNA interference–gene silencing by double-stranded RNA”. Today it is known that besides its important role in applying targeted gene knockdown, RNA interference constitutes an important physiological process in inhibiting gene expression in all eukaryotes: fungi, worms, flies, plants, and mammals [15, 16]. These findings suggest that RNAi is an evolutionary highly conserved mechanism for the regulation of gene expression and gene silencing on the post-transcriptional level.
Micro-RNAs (miRNAs)
A current field of research in RNA interference was initiated by the discovery of micro-RNAs (miRNAs) in 1993. Micro-RNAs are non-coding RNA molecules with a length of approximately 20–35 nucleotides that are present in all domains of life (eukaryotes, bacteria, and archaea) to silence gene expression [17, 18]. It is known today that about 20,000–25,000 protein-encoding genes are present in humans [4, 19], which makes up only a very small fraction (about 1.5%) of the genome considering the total genome size of approximately three billion bp in the haploid state. The rest of the genome encodes either non-coding RNAs, regulatory DNA sequences, or sequences derived from mobile genetic elements such as LINEs (long interspersed non-coding elements) or SINEs (short interspersed non-coding elements) [19,20,21]. Additionally, for other parts of the genome no real function could be determined. However, it is known today that the human genome encodes more than 2300 miRNAs [22]. This number is continuously increasing and it is very likely that more micro-RNAs will be discovered in the future. It is assumed that 20–30% of all human genes are regulated by miRNAs and that these molecules control fundamental cellular processes: cell division, cell differentiation, cell proliferation, and renewal of stem cells [18, 23]. Dysfunction of miRNAs was correlated with the development of severe diseases such as diverse tumor types. MiRNAs are genomically encoded and are transcribed by DNA-dependent RNA polymerases II or III (RNA polymerase II or III for short) expressed as 500–3000-nucleotide-long primary transcript miRNAs (pri-miRNAs) (Fig. 2) [18, 24]. As messenger RNAs (mRNAs), the pri-miRNAs carry a poly(adenosine monophosphate) tail (poly-A-tail) at the 3′-end and a 7-methylguanosine cap at the 5′-end. The pri-miRNAs form a stem-loop (hairpin) structure, composed of double-stranded RNA (dsRNA) sections connected via single-stranded RNA (ssRNA) loop regions. Within the nucleus, the pri-miRNA is processed to an approximately 70–80-nucleotide-long pre-miRNA by the microprocessor complex consisting of two components: an enzyme with RNase III activity called Drosha and a dsRNA-binding protein DGCR8 [18]. The pre-miRNA is bound in the nucleus by an export receptor called exportin-5 (Fig. 2). Together with the small GTP-binding protein Ran (Ras in the nucleus), pre-miRNA, exportin-5, and Ran·GTP form a ternary complex that transports the pre-miRNA from the nucleus through the nuclear pore complex (NPC) into the cytosol. In the cytosol, the pre-miRNA is further processed by another RNase III called Dicer [24]. Dicer binds and cleaves the pre-miRNAs resulting in approximately 17–24-nucleotide-long ds-miRNA molecules. Dicer forms a complex with the dsRNA-binding protein TRBP. Binding of the ds-miRNA by TRBP results in unwinding and formation of the single-stranded mature miRNA (Fig. 2). Subsequently, the miRNA is bound by a ribonucleoprotein complex called RISC (RNA-induced silencing complex). RISC contains proteins of the Argonaut family, some of which have RNA-binding capacity and others are additionally capable of using the bound miRNA as guide RNA to scan for complementary mRNA sequences [25]. The subsequent steps depend on the similarity of the miRNA sequence and the mRNA sequence. If the sequences show a high degree of sequence complementarity, the target mRNA is degraded by endonucleolytic cleavage catalyzed by the Argonaut protein Ago2. However, if the sequences of miRNA and mRNA show a lower degree of complementarity gene silencing occurs since the binding of initiation factors for the translation is blocked. Both scenarios result in a knockdown of gene expression (Fig. 2). In mammals, the binding of miRNAs occurs in the 3′-untranslated region (UTR) of the target mRNA [18, 24].

Using RNA interference (RNAi) for targeted gene knockdown. RNA interference encompasses gene knockdown mediated by different types of RNA molecules: miRNA, shRNA, and siRNAs as described in the text. The RISC contains the Argonaut protein Ago2, which is composed of an N-terminal domain (N), a PAZ domain, a MID domain, and a C-terminal PIWI domain. The PAZ domain binds to the 3′-end of the miRNA/siRNA and mediates protein–protein interactions. The C-terminal PIWI domain exerts endonuclease activity for cleavage of the target mRNA. The figure is modified from [110]
Small-interfering RNA (siRNA)
Another RNAi category is small-interfering RNA (siRNA). This RNA interference was discovered as endogenous gene silencing mechanism in plants applied as an antiviral defense mechanism [26]. It is also in other eukaryotic organisms like fungi, worms, mice, and human, pointing to an evolutionary ancient defense mechanism [27]. The siRNA molecules are composed of approximately 21–25 nucleotides and have overhangs of two nucleotides at their 3′-ends. They are processed from up to several thousand-nucleotide-long viral dsRNA molecules and cut (“diced”) as part of the cell’s immune system [28, 29]. In contrast to miRNAs, which are encoded by the cellular DNA and for which processing occurs within the nucleus, siRNA is located in the cytosol (Fig. 2). This means that processing in the nucleus and nuclear-cytosolic transport are not needed for siRNA. Besides, the following steps for maturation and processing are highly similar between siRNA and miRNA and involve endonucleolytic processing by the RNase III Dicer, and binding of the single-stranded siRNA by the RISC [30]. This scans the mRNA resulting in decrease of translational efficiency (in the case in which sequence similarity is moderate to low) or degradation of the mRNA (in the case in which sequence similarity is high) by the Argonaut proteins (Fig. 2).
Targeted gene knockdown applying RNA interference
After showing that miRNAs and siRNAs are endogenous, physiologically important regulatory systems to modulate gene expression, these endogenous cellular pathways were used to artificially silence gene expression by exogenous supply of RNA molecules activating the endogenous si/miRNA machinery. Several strategies were developed for targeted gene knockdown via RNAi. One strategy involves the exogenous application of siRNA to cells. This could include the transfection (cellular uptake of nucleic acids using lipid vesicles) or microinjection (injection of nucleic acids into cells using a fine needle) of dsRNA molecules to suppress expression of the gene of interest. Alternatively, the dsRNA can be expressed from the genome to activate the endogenous miRNA maturation and processing pathway [30]. To this end, cells are infected by lentiviral particles, which carry the RNA of interest and a reverse transcriptase, which converts the RNA to DNA, enabling integration into the host cell’s genome. The integrated RNA genes are under the control of endogenous promotors and terminators for transcription (Fig. 2). Their expression results in formation of a short-hairpin RNA (shRNA) that carries a stem-loop (hairpin structure) that is recognized by Drosha in the nucleus [30, 31]. This shRNA is subsequently maturated and processed using the miRNA pathway, including RNase III Drosha in the nucleus and Dicer in the cytosol. These steps result in the formation of a siRNA, a double-stranded RNA molecule that is recognized by RISC and processed by Argonaut proteins (Fig. 2).
The development of RNA-based strategies for gene knockout is a powerful technique to allow high-throughput screening of gene functions. However, there are several drawbacks and limitations of this technology. Firstly, the knockdown of the expression of the desired gene is often not quantitative. There will often be some reminiscent gene product produced. If the amount of this reminiscent product is sufficient for its physiological function, the real phenotype might not be approachable by siRNA/shRNA [31]. Secondly, often considerable variations between experiments and the efficiency of gene knockdown are obtained. Thirdly, as this technology is based on hybridization of complementary RNA sequences a considerable degree of off-target effects must be considered. Fourthly, RNA-mediated gene knockout is not stable. In turn, the stability of the RNA determines the efficiency and duration of gene knockdown. All these drawbacks exacerbate the precise determination of the concise genotype–phenotype relationships.
Zinc-finger nucleases and TALENs for specific genome editing
Afterwards, novel strategies to directly correlate genotype to phenotype took all of the points mentioned above into consideration. These technologies allow one to manipulate every gene in diverse cell types and organisms with a high degree of selectivity, potency, and specificity. As these approaches allow one to edit the genome, they are called technologies for genome editing. These technologies include ZnF-Ns (zinc-finger nucleases) and TALENs (transcription activator-like effector protein nucleases) (Fig. 3) [32,33,34,35,36]. Both technologies are based on a specific DNA-binding module and an engineered restriction endonuclease (nuclease for short) that is able to cleave phosphodiester bonds within DNA. In so-called chimeras, a DNA-binding module was fused to the nuclease module, with which it is possible to efficiently introduce DNA double strand breaks (DSBs) that stimulate mainly two different cellular DNA repair programs in mammals: (a) error-prone, imprecise non-homologous end joining (NHEJ), which operates predominately in the cell cycle phase G1 or (b) non-error-prone, precise homology directed repair (HDR) that is restricted to the late S to G2 phases [32, 36,37,38].

Structure of TALENs and ZnF-Ns. a Structure of the transcription activator-like effector (TALE) from Xanthomonas oryzae bound to target DNA (PDB code 3UGM). DNA binding is mediated by tandem repeats of 33–35 amino acids in length. The TALE repeats bind to the major groove of the DNA. b Three repeats of the X. oryzae TALE repeats bound to target DNA. Each TALE repeat forms a left-handed, two-helix bundle presenting RVD (repeat variable diresidues) loop for DNA binding. DNA sequence specificity is created by the second RVD residue contacting a single DNA base (here TR1, Ile; TR2/3, Asp) in the major groove of DNA, while the first (here TR1, Asn; TR2/3, His) is important to stabilize the TALE repeat, as described in the text (PDB code 3UGM). c Schematic representation of a TALE nuclease (TALEN) dimer bound to target DNA. The two TALE DNA-binding sites are separated by a spacer sequence of 12–20 bp. Upon binding of each TALEN monomer to the target DNA site 5′ upstream or 3′ downstream to the spacer sequence facilitates FokI dimerization and thereby FokI activation and cleavage of the DNA within the spacer sequence. d Structure of a zinc-finger protein (ZnF) consisting of four zinc-finger repeats in complex with target DNA (PDB code 2I13). The ZnF binds to the major groove of the DNA. Close-up: each zinc-finger has a typical ββα structure and binds a zinc ion with two Cys and two His residues (Cys2His2). Each zinc-finger binds to 3–4 bp in the major groove of the DNA. Sequence specificity is created by residues 1, 2, 3 and 6 contacting the bases of the DNA. The structure is shown in cartoon representation. The zinc ions are shown in space filling representation with a van der Waals radius of 139 pm. e Domain organization of a zinc-finger nuclease (ZnF-N) dimer bound to target DNA. The DNA target site consists of two ZnF tandem binding sites separated by a 6-bp spacer sequence. This spacer sequence contains the FokI restriction endonuclease cleavage site. FokI is only active upon dimerization. The figure of the structure was created using structure with PDB code 2I13

CRISPR/Cas9 constitutes an adaptive immune system in bacteria and can be used as an efficient targeted genome editing tool. a CRISPR/Cas9 constitutes an adaptive immune system in bacteria. Shown is how bacterial cells develop immunity and how a memory of past infections is created. Thereby, the cells are protected against an infection with the same phage or mobile genetic element. Immunity is created in three phases: (1) adaptation, (2) crRNA maturation/biogenesis, and (3) interference. b The Cas9·crRNA·tracrRNA surveillance complex. Upon binding to the crRNA and tracrRNA conformational changes within Cas9 bring it into a competent state for target DNA binding. The PAM sequence in the target DNA is bound by the PI domain within Cas9. The target dsDNA is melted. The strand non-complementary to the crRNA is cleaved by RuvC and the complementary strand by HNH. Further nucleases result in the degradation of the target DNA. c Structure of Streptococcus pyogenes Cas9 in complex with a small guide RNA (sgRNA) and target DNA (PDB code 4008). Shown is a crystal structure of Cas9 in cartoon representation. RuvC, BH, Rec1, Rec2, HNH, and PI domains are color coded as presented in the diagram showing the domain organization below the structure. d The Cas9 PAM-interaction (PI) domain mediates binding of the target DNA in a state competent for cleavage by HNH and RuvC endonucleases (PDB code 4UN3). Two positively charged arginine residues in the PI domain mediate interactions with the guanine bases within the 5′-NGG-3′ PAM sequence (N any nucleotide; G guanine base or guanosine-5′-phosphate) in the target DNA. e The DNA cleavage is mediated by RuvC and HNH nuclease domains. RuvC uses a two-metal-ion cleavage mechanism for phosphodiester bond cleavage in the target DNA non-complementary to the crRNA (or sgRNA). Mg2+ ion A (MgA) contacts the active site water molecule and the scissile phosphate, while Mg2+ ion B (MgB) contacts the 3′-hydroxyl leaving group and the scissile phosphate. Recent data suggest that a His (His983) acts as catalytic base activating the nucleophile, i.e., a water molecule in the active site, to attack the scissile phosphate. HNH domain uses a one-metal-ion cleavage mechanism for cleavage of the complementary DNA strand three nucleotides upstream from the PAM sequence. HNH uses a His (His840) as catalytic base to activate a water molecule for nucleophilic attack of the scissile phosphate. The figure is from [79]. f CRISPR/Cas9 is programmable. By design of an sgRNA, Cas9 allows one to target specific DNA sequences for genome editing. The sgRNA must contain an approximately 20-nucleotide-long spacer sequence at the 5′-end for target DNA binding and a section with a three-dimensional secondary structure for Cas9 recognition and binding (PDB code 4008)
ZnF-Ns and TALENs contain DNA-binding modules that are programmable and allow one to recognize almost every DNA sequence with high accuracy [39]. The specificity of the DNA-binding module for the target DNA sequence (selectivity) and the affinity for the target sequence (potency) determine how suited these tools are to modify the genome. Therefore, the engineered ZnF-N and TALEN chimeras are the basis for “site-specific nuclease technologies”. Notably, the DNA-binding modules can be combined with a variety of effector domains: nucleases to introduce DSBs (without DNA repair DSBs might be toxic for the cell), recombinases/transposases for DNA integration/excision/inversion (independent from DNA repair), DNA and histone methyltransferases or acetyltransferases (for epigenetic programming). Today, site-specific nucleases are applied in almost all model organisms (Homo sapiens, Mus musculus, Drosophila melanogaster, Caenorhabditis elegans, Arabidopsis thaliana, etc.). Several methods were developed for delivery of ZnF-Ns or TALENs into cells. DNA- or RNA-based methods, i.e., transfection or microinjection of plasmid DNA, or of viral vectors or of in vitro transcribed mRNA into cells, were used for intracellular delivery [40]. However, those techniques have some limitations. Firstly, these delivery techniques often allow targeting of only certain cell types. Secondly, they can show considerable off-target effects (insertional mutagenesis, toxicity, low efficiency of delivery, etc.) [32]. Other ways include microinjection of the purified ZnF-Ns or TALENs proteins into cells. This was shown to result in considerably less off-target effects compared to gene-based delivery. Moreover, these approaches face less regulatory issues as knockouts can be created in cells without using genetic material. Using these approaches for genome editing allows one to therapeutically tackle a disease on the basis of correcting the cause of the disease, i.e., the mutation of the DNA sequence. In comparison to RNA-based knockdown strategies this allows one to permanently eliminate symptoms of the disease [32, 41].
Zinc-finger nucleases (ZnF-Ns)
During evolution all organisms developed proteins that bind to specific DNA or RNA sequences to allow precisely coordinated and accurate gene expression programs. In eukaryotes, the most often used DNA-binding domain is the zinc-finger domain (ZnF; zinc-finger for short). ZnFs consist of approximately 30 amino acids, which structurally form a ββα-conformation (Fig. 3a) [42]. Going from the N- to the C-terminus a β-strand is following a β-strand, which is completed by a C-terminal α-helix (Fig. 3a). The two β-strands form an antiparallel β-sheet, i.e., both strands are in opposite orientation to each other, connected via a β-turn [32]. The structure is stabilized in a finger-like conformation via coordination of a Zn2+ ion by the imidazole rings of two histidine (three-letter code His; one-letter code H) residues and the sulfhydryl groups (SH-R) of two cysteine (three-letter code Cys; one-letter code C) side chains, forming a His2Cys2-ZnF domain (His2-Cys2-ZnF: classic ZnF domain) (Fig. 3a). There are also examples in which the Zn2+ is coordinated by four cysteine residues (Cys4-ZnF) or in which two Zn2+ ions are coordinated by a total of six cysteine residues (Cys6-ZnF) [42]. DNA-binding is achieved by interactions of 3–4 bp of the major groove of the DNA with side chains of the ZnF’s α-helix (Fig. 3a). These interactions determine the DNA sequence specificity of binding [42]. This means that a single ZnF domain can recognize one DNA triplet. This interaction of amino acids within the ZnF domain with bases of the DNA creates the sequence specificity of binding [42]. For DNA-binding domains/proteins contacting the sugar-phosphate backbone of the DNA a sequence-independent binding is observed (e.g., histones). Arranging more than three ZnF domains in tandem by protein engineering enabled the specific binding of DNA patches with a length of 9–18 bp [32]. Recognition of a 18-bp DNA fragment means that a specificity of 1:68 × 109 bp can be created (18 bp with four different nucleotides, i.e., 418 = 68 × 109 bp). Nowadays, individual ZnF domains are designed that are capable of targeting all 64 nucleotide triplets, enabling a combination of ZnFs that target a specific DNA sequence (modular assembly approach). The creation of a specific ZnF-based DNA-binding module can be achieved by rational design, i.e., combining individual ZnF domains with different DNA sequence specificities [43]. Alternatively, a combinatorial genetic library can be constructed encoding diverse ZnF variants. This library can be selected and screened for a desired specificity, i.e., assessing affinity for specific DNA sequences. Protein engineering enables the design of ZnF-based DNA-binding domains that allow binding of almost all DNA sequences. These specific DNA-binding modules are connected with a restriction endonuclease (restriction enzyme for short) forming a single polypeptide chain. One enzyme that shows a very high performance in this context is the restriction enzyme FokI from Flavobacterium okeanokoites. FokI is a restriction endonuclease of type IIS, i.e., it recognizes DNA on a short recognition sequence (FokI: 5′-GGATG-3′; complementary strand: 3′-CCTAC-5′; reverse-complementary: 5′-CATCC-3′) and sets a DSB outside the recognition sequence (FokI: 5′-GGATG-3′(9/13). This means, it cuts nine nucleotides from 3′-end and 13 nucleotides from 5′-end of the complementary strand, resulting in a sticky end with a 5′-overhang (Fig. 3b) [32]. A well-considered reason to use FokI as nuclease module in ZnF-Ns is that its dimerization is a prerequisite for endonuclease activity [44]. Binding of the ZnF modules to DNA target sequences allows FokI to homodimerize and initiate DNA DSBs. This allows one to specifically introduce a restriction site at any desired position within the target DNA (Fig. 3b). As FokI creates a DSB in the target DNA, it will also activate DNA repair pathways, either error-prone NHEJ or non-error-prone HDR [32, 36]. To reduce the occurrence of error-prone NHEJ, optimized ZnF nickases were developed, which introduce single-strand DNA cleavages (nicks) instead of DSBs that increases the induction of non-error-prone HDR [45, 46].
Transcription activator-like effector proteins (TALEs) coupled to nucleases (TALENs)
Similarly, as described for the ZnF-Ns, TALENs also contain sequence-specific nucleic acid (either DNA or RNA) binding domains connected to an endonuclease on a single polypeptide chain (Fig. 3a). TALEs are proteins that were discovered in the plant pathogenic Gram-negative bacteria of the species Xanthomonas. The TALEs are secreted by Xanthomonas into their plant host cells using the type III secretion system to support an efficient infection process [32]. Later, they were also found in the plant pathogenic Gram-negative bacterial species Ralstonia solanacearum and Burkholderia rhizoxhinica [47]. They are structurally not related to ZnF domains [48]. However, functionally TALEs show some similarities to ZnF domains with respect to their capability to bind in a sequence-specific manner to DNA/RNA. TALEs are composed of a central region containing tandem repeats of usually 34 amino acids (only the C-terminal repeat is truncated), which is flanked by an N-terminal sequence encompassing a type III secretion signal and a C-terminal sequence containing a nuclear localization signal (NLS). The Xanthomonas TALE contains 23 full TALE repeats and additionally two degenerated repeats at the N-terminus that also contact the DNA, and a truncated half-repeat at the C-terminus [47, 49, 50]. Each TALE repeat recognizes one single bp in double-stranded DNA. The DNA sequence specificity of TALEs is determined by two hypervariable residues (repeat-variable diresidues, RVDs) (Fig. 3). The RVD is located at position 12 and 13 in the individual 34 amino acids encompassing the TALE repeat (Xanthomonas RVD specificity: His/Asp targets cytosine; Asn/Gly targets thymine; Asn/Ile targets adenine; Asn/Asn targets guanine/adenine; Asn/Ser targets adenine/guanine/cytosine/thymine, Asn* (33-amino acid repeat with only one RVD residue) targets thymine) (Fig. 3b). The first residue of the RVD forms a stabilizing interaction with the backbone residues of the TALE repeat while the second residue creates the DNA sequence specificity by interacting with nucleotide bases within the major groove of the DNA (Fig. 3b) [50]. Ralstonia and Burkholderia TALEs have a similar RVD-mediated DNA-binding mode but they show different nucleotide preferences compared to the Xanthomonas TALE [32, 48]. In analogy to ZnF domains, consecutive TALE repeats can be connected on a single polypeptide chain to create a high DNA sequence specificity for any DNA sequence of choice. In contrast to ZnF-Ns, in the designing of TALENs no linker sequences need to be considered since under physiological conditions they already function as repetitive sequences of connected TALE repeats. Structurally, each TALE repeat forms a left-handed, two-helix bundle that presents a loop containing the RVDs to the DNA (Fig. 3a). Overall, all TALE repeats form a right-handed superhelix that contacts the DNA’s major groove (Fig. 3a) [49, 50]. On the basis of the work on ZnF-Ns, TALEs were fused to a variety of different effector domains with enzymatic activities: (a) endonucleases (i.e., FokI), (b) transcriptional activators, (c) recombinases/integrases or epigenetic modifiers (acetyltransferases, methyltransferases, etc.) [32, 51].
Genome editing using CRISPR/Cas9
Next to site-specific nucleases such as ZnF-Ns and TALENs, the CRISPR/Cas9 system was more recently described as a novel efficient tool for genome editing. CRISPR (clustered regularly interspaced short palindromic repeats) arrays were identified in 1987 in the genome of the Gram-negative bacterium E. coli [32, 52, 53]. In 1993 CRISPR arrays were also identified in the archaeon Haloferax mediterranei [54] and later CRISPR arrays were found in up to 50% of all bacteria and in 90% of all archaea [54]. CRISPR arrays contain short palindromic and identical sequences, which are interspaced by unique spacer sequences, resulting in a “repeat–spacer–repeat” arrangement. The identification of CRISPR loci in so many bacterial species and in archaea opened the question of their physiological relevance (Fig. 4a). In 2005 this miracle was solved owing to the progress in DNA sequencing technologies [55]. It was discovered that the spacer sequences within the CRISPR arrays show homologies to sequences from mobile genetic elements such as bacteriophages (phages for short) (Fig. 4a). It turned out that bacteria and archaea can take up foreign DNA and insert it into the CRISPR arrays as part of a defense mechanism against mobile genetic elements including phages [55]. Interestingly, the bacterial strains were not infected by phages if they contained fragments of the phage DNA in the host cell’s CRISPR loci. This finding suggested that CRISPR arrays mediate a defense mechanism against phage infection, i.e., they constitute an adaptive immune system in prokaryotes [56, 57]. The CRISPR array is an archive and a memory of past infections, protecting and “immunizing” the bacteria against an infection with the same phages. This was experimentally shown by bacteriophage infection of a Streptococcus thermophilus bacterial culture resulting in expansion of the CRISPR array [58]. This shows that the CRISPR system constitutes a bacterial acquired/adaptive immune system as it is expandable to new infections. Moreover, it also constitutes a bacterial innate immune system as it is inheritable and bacterial cells are immunized and protected against another infection with the same phages. According to the red queen hypothesis from Leigh van Valen (1973), based on the novel Through the Looking-Glass and What Alice Found There (1871, by Lewis Carroll), this shows that the adaptation of host and pathogen occurs in direct interplay with each other. The aim of this process is not to drive adaptation to perfection but only to maintain adaptation to be able to exist in the environment. This is in analogy to Alice who had to run very fast together with the red queen in order to stay at the same place, i.e., without reaching a destination. This shows that coevolution for more than one billion years resulted in the development of complex and versatile defense and evasion mechanisms in prokaryotes to fight viral invaders. Today, it is known that CRISPR systems are extremely versatile but it is still not understood in all detail how the different systems work on the molecular and mechanistic level. It was found that additional genes, the cas (CRISPR-associated) genes are located in direct vicinity upstream of the CRISPR array (Fig. 4a) [56, 59,60,61]. These genes encode enzymes and proteins that are important for mediating the defense mechanism and for establishment of immunity in the three steps: (1) adaptation, (2) CRISPR-RNA (crRNA) biogenesis, and (3) interference [61]. The systems are categorized into two classes, class 1 and class 2, which are subdivided into a total of six types (class 1: types I, III, IV; class 2: types II, V, VI). The classification was done on the basis of the encoded cas genes and the nature of the interference complex used for viral defense [61]. The following section will focus on CRISPR/Cas9, belonging to class 2, type II, as this system is the best studied and most often applied CRISPR/Cas type for genome editing approaches [62].
Adaptation (spacer acquisition)
The spacer acquisition, i.e., integration of foreign DNA into the CRISPR array, occurs in several steps (Fig. 4a):
-
1.
The phage DNA/mobile genetic element is detected
-
2.
The protospacer, i.e., the non-processed spacer DNA, is selected
-
3.
The protospacer is processed to form the mature spacer DNA
-
4.
The spacer is integrated into the CRISPR array
Each of the steps involves several proteins and/or RNA. The spacer acquisition begins with the detection of the foreign DNA. This is processed and integrated into the CRISPR array [59, 61]. To ensure that the CRISPR/Cas machinery is not directed against cell endogenous DNA in the sense of an autoimmune reaction, the system has to distinguish between foreign and self. Studies in E. coli showed that a main source of protospacers are DNA fragments that are generated during repair of DNA DSBs. The RecBCD complex (Rec—recombination) is recruited to the DSBs (in Gram-positive bacteria: AddAB) and unwinds the DNA using its helicase activities and subsequently degrades it until a chi sequence (chi—crossover hotspot investigator) is reached. It was found that sequences that are close to chi sequences, which are sequences that result in stalling of the replication fork during recombination, and sequences that are located at exposed DNA ends are major sources of protospacer sequences (spacer sampling). Foreign DNA carry fewer chi sequences compared to chromosomal E. coli DNA. This ensures RecBCD-mediated degradation of major sections of foreign DNA resulting in the integration of predominant foreign DNA into the CRISPR arrays. The type I and type II CRISPR/Cas systems select protospacers on the basis of the presence of a PAM (protospacer adjacent motif) sequence within the viral/mobile genetic element DNA: the selected protospacer sequences are always in direct vicinity of DNA sequences composed of 3–6 bp with the sequence 5′-NGG-3′ (N any nucleotide) [61, 63]. Notably, the PAM sequence is not present in the CRISPR array [62, 63]. Thereby, the cell can discriminate between self and foreign DNA. The proteins Cas1 and Cas2, as genes encoded upstream of the CRISPR array in class 2, type II CRISPR/Cas, are sufficient for PAM recognition in foreign DNA (Fig. 4a). Structurally, two Cas1 dimers are bridged by one Cas2 dimer [61, 62]. The Cas1–Cas2 complex forms an integrase. Cas1 binds to the PAM complementary sequence and has catalytic activity, while Cas2 is of structural importance (Fig. 4a) [61]. Additionally, Cas9 and a tracrRNA, both encoded upstream of the cas1/2 genes, as well as Csn1, encoded downstream of cas1/2 genes, are necessary for spacer acquisition (Fig. 4a). Cas9 is important for selection of protospacers that carry a PAM sequence [61]. The integration of the spacer DNA occurs mainly at the 5′-end of the CRISPR array, which is ensured by a AT-rich leader sequence preceding the CRISPR array. In this way a memory of the chronology of infection is created. During replication of the bacterial DNA, the DNA breaks, resulting as well in the activation of the RecBCD complex. RecBCD repairs the DNA break generating a 3′-OH overhang that can be used for insertion of a new repeat–spacer unit. Subsequently, RecA mediates homologous recombination repair of the DNA lesion.
CRISPR-RNA (crRNA) biogenesis
The memory of past infections is built into the CRISPR array and transcribed to generate a long precursor CRISPR-RNA (pre-crRNA) (Fig. 4a). The transcription initiation occurs within the leader sequence preceding the CRISPR array. This pre-crRNA is processed within the repeat sequences to build the mature crRNAs. The mature crRNAs contain a repeat segment (5′-GUUUUAGAGCU(A/G)UG (C/U)UGUUUUG-3′), which is recognized by the Cas9 protein in a mechanism dependent on structure and sequence [61, 64]. Moreover, it contains a spacer sequence, which is important for binding to the target DNA. Type II systems need a tracrRNA (trans-activating cr RNA) for CRISPR-mediated immunity. The tracrRNA is encoded at the 5′-end of the type II CRISPR/Cas9 locus. The tracrRNA forms duplexes with the pre-crRNA [65]. The duplexes are bound by the effector nucleases, i.e., Cas9 for the type II system. In this complex, the crRNA:tracrRNA is processed by RNase III, which is recruited to the complex [61, 66]. A second cleavage is done by a so far unknown RNase, which removes the tag derived from the 5′ repeat sequence. Afterwards, the mature effector complex is formed containing Cas9, the mature crRNA and a trans-activating crRNA (crRNA:tracrRNA) [61]. This ternary complex is capable of mediating the interference (Fig. 4a, b).
Interference
The interference machinery is guided by the crRNA containing information of the invading virus or mobile genetic element (Fig. 4a, b). The crRNA guides the machinery to cleave complementary sequences, the so-called protospacers, located within the foreign DNA of invading genetic material. This ultimately protects prokaryotes against a viral infection [61]. Cas9 is an RNA-guided DNA endonuclease that has been structurally and functionally characterized in great detail up to atomic resolution and subtypes of Cas9 are known, i.e., II-A, II-B, II-C (Fig. 4c). Next to the crRNA, Cas9 binds to the tracrRNA, which shows sequence complementarity towards the repeat region of the crRNA [61, 65, 67]. Cas9 identifies the target DNA by recognition of the PAM sequence within the foreign DNA and the base pairing of the approximately 20-bp spacer region within crRNA (guide RNA) with the viral target DNA (Fig. 4b). Binding of the RNA to Cas9 results in substantial conformational changes within the protein [68,69,70] that orders the PAM interaction site as well as the RNA interaction site in Cas9 (Fig. 4d) [70, 71]. In this state, Cas9 is competent for binding of the target DNA and for recognition of the PAM sequence within the complementary strand of target DNA, which is not bound by crRNA (Fig. 4b). This Cas9·crRNA·tracrRNA ternary complex forms the surveillance complex that scans the DNA (Fig. 4b). If a PAM sequence in the target DNA strand binds to the PAM interaction domain within Cas9, the Cas9·crRNA·tracrRNA complex starts to melt the base pairs immediately upstream of the PAM sequence in the target DNA (Fig. 4d) [72]. The complementary region within the target DNA base pairs with the spacer region of the crRNA. This activates Cas9 endonuclease activity which creates a blunt-end DSB in the target DNA three base pairs upstream, i.e., 5′, to the PAM sequence using its HNH and RuvC nuclease domains (Fig. 4e). This finally results in degradation of the phage/mobile genetic element DNA (Fig. 4b) [68, 70, 73].
RNA-guided endonuclease Cas9
Cas9 (CRISPR-associated sequence 9) is an RNA-guided endonuclease. This means that Cas9 is a ribonucleoprotein of which several can be found in eukaryotic and prokaryotic cells, i.e., ribosomes, RNase P, telomerase, factors of the splicing machinery, translation initiation factors, and the RISC. These ribonucleoproteins perform important tasks that are fulfilled by RNA molecules in order to maintain cellular function. As such, these important functions support the hypothesis that RNA was important for exerting catalytic function during evolution in the early RNA world. Similarly, defense mechanisms against phages or other mobile genetic elements evolved very early, several billion years ago.
Cas9 from S. pyognenes consists of 1368 amino acids and has a molecular weight of 158 kDa. Cas9 has a bilobed structure, i.e., it consists of two lobes, the recognition (REC) lobe and the nuclease (NUC) lobe (Fig. 4c) [71, 72]. The REC lobe can be subdivided into a long α-helix (bridge helix), the REC1 domain, and the REC2 domain. The NUC lobe is formed by the HNH (name based on characteristic His-Asn-His residues) domain, the RuvC domain, and the PALM interacting (PI) domain that is located at the C-terminus of Cas9 (Fig. 4c) [68, 71, 72]. The REC lobe and the NUC lobe are connected via an unfolded linker and by the highly conserved Arg-rich bridge helix. This bridge helix forms several contacts to the bound RNA. The REC lobe is primarily involved in binding to the crRNA:tracrRNA (guide RNA) and to the target DNA. However, as mutational approaches demonstrate, it also has an effect on the Cas9 endonuclease activity and maybe plays a structural role. The NUC lobe’s HNH domain has endonuclease activity cleaving the DNA complementary to the crRNA, i.e., the target DNA strand that is hybridized to the crRNA [68, 74, 75]. The RuvC domain cleaves the non-complementary strand in the target DNA [68]. The name RuvC is derived from the E. coli protein, which plays a role in repair of DNA damage induced by UV radiation as a nuclease/resolvase resolving Holliday junctions during homologous recombination [76]. For cleavage of target DNA, a PAM sequence within the target DNA is essential. For correct positioning of the DNA cleavage site, Cas9 has a PI domain at its C-terminus [71]. In contrast to restriction endonucleases, which cleave phosphodiester bonds in DNA within specific recognition sequences or outside of these sequences, Cas9 is a guided nuclease, i.e., Cas9 recognizes its specific DNA cleavage sites by the bound crRNA:tracrRNA duplex. Binding of the crRNA:tracrRNA to Cas9 results in substantial conformational changes in the protein resulting in ordering of residues in the PI domain and residues in the crRNA binding region [70, 77, 78]. Thereby, Cas9 is in a competent state for target DNA binding and PAM recognition [71]. If a target DNA binds to the crRNA spacer region, the HNH domain (complementary strand to crRNA) and RuvC (non-complementary strand to crRNA) each cleave the phosphodiester bond of one single strand of the target DNA after the third nucleotide upstream of the PAM sequence within the target DNA [73, 74, 79]. RuvC and HNH are both nucleases that perform cleavage of a DNA single strand in a Mg2+-dependent manner.
RuvC in Cas9 cleaves the single-stranded target DNA using a two-metal-ion catalytic mechanism to cleave the non-complementary strand
RuvC shows structural homologies to ribonuclease H (RNase H) and exerts a two-metal-ion mechanism of phosphodiester bond cleavage [76]. It consists of a six-stranded mixed β-sheet (β1, β2, β5, β11, β14, β17) surrounded by α-helices (α33, α34, α39, α45) and two additional two-stranded antiparallel β-sheets (β3/β4, β15/β16). The RuvC domain in Cas9 shows similarity to RuvC nucleases involved in resolving Holliday junctions during DNA repair and homologous recombination. However, while RuvC resolvases act as dimers, the RuvC domain in Cas9 works as monomer and has further structural elements which mediate interactions with the RNA duplex. The two catalytic metal ions are coordinated by three carboxylates, i.e., Asp10 (D10), Asp986 (D986), and Glu762 (E762) within the active site (numbering is based on S. pyogenes Cas9) (Fig. 4e). This DDE (or DEDD) motif is a highly conserved motif in two-metal-dependent nucleases [79,80,81]. One Mg2+, Mg2+ ion A (MgA), binds to the scissile phosphate on the nucleophile side (nucleophile is a water molecule), while Mg2+ ion B (MgB) also binds to the scissile phosphate on the leaving group side (3′-hydroxyl end of cleaved DNA). The bound Mg2+ ions are important for binding and orientation of the reactants, i.e., the negatively charged DNA sugar-phosphate backbone and the active site residues at the cleavage site. Two-metal-ion catalysis is also found in polymerases and other nucleases to enhance substrate recognition and catalytic specificity [80]. Moreover, the Mg2+ ions are essential to neutralize the highly negatively charged pentacovalent phosphate intermediate emerging during catalysis. MgA is coordinated by a catalytic water molecule. To this end, it is directly involved in formation of the nucleophile. The phosphodiester bond is kinetically very stable, i.e., spontaneous hydrolysis occurs very slowly. However, hydrolysis is thermodynamically favorable, making the hydrolysis reaction an energetically favorable process [82,83,84]. The Mg2+ ions in the RuvC lower the activation energy (Gibbs energy of activation) to reach the transition state of catalysis, thereby affecting the kinetics for the reaction. Furthers studies by molecular dynamics simulations in addition to experimental results suggest that the RuvC-catalyzed cleavage reaction occurs in an associative mechanism, via a nucleophilic substitution of type SN2 [85]. For many enzymes using a two-metal-ion catalytic mechanism the Mg2+ ions are acting as a general base activating the nucleophilicity of the attacking water molecule. Two-metal-ion catalytic enzymes show a stringent requirement for two Mg2+ ions for their activity, i.e., these cannot be replaced by other ions such as Ni2+, Zn2+, Ca2+, Cu2+, Mn2+, or Cd2+ [80]. The steric environment forms the basis for catalytic specificity of two-meal-ion enzymes. Structural and mutational studies on the RuvC domain of Cas9 suggests that a His [S. pyogenes: His983 (H983)] acts as a general base abstracting a proton from a catalytic water molecule, increasing its nucleophilicity (Fig. 4e). Mutation of His983 to Ala converts Cas9 into a nickase, i.e., it only performs cleavage of the complementary single strand of DNA by the HNH nuclease domain. This water molecule attacks the phosphodiester bond three nucleotides upstream from the PAM sequence.
HNH nuclease in Cas9 uses a one-metal-ion catalytic mechanism to cleave the complementary strand
Several endonucleases use a one-metal-ion catalytic mechanism. The HNH (His-Asn-His) domain of Cas9 also uses an one-metal-ion catalytic mechanism (Fig. 4e) [80, 81, 85]. Interestingly, recent data suggests that the catalytic triad is not composed of an His-Asn-His catalytic triad. Instead structural and functional studies show that the catalytic triad is formed by the residues D839-H840-N863 (numbering for S. pyogenes Cas9), rather than previously suggested D839-H840-D861, D837-D839-H840, or D839-H840-D861-N863 [75]. Structurally, it consists of a two-stranded antiparallel β-sheet (β12, β13) flanked by four α-helices (α35–α38). The single metal ion, a Mg2+ in the HNH domain of Cas9, is bound by a ββα-metal fold (ββα-Me) [74, 75]. This fold forms a V-shaped Mg2+-binding site with the antiparallel β-sheet forming one arm of a V and the following α-helix the other arm. The Mg2+ ion is bound at the opening of the V and it is coordinated by active-site residues, the scissile phosphate, and the 3′-OH leaving group [68]. The single Mg2+ ion in the HNH nuclease of Cas9 is spatially and structurally equivalent to MgB in the RuvC domain. It is important for binding to the single-stranded DNA substrate; it neutralizes the negative charges emanating in the pentacovalent phosphate intermediate during catalysis, thereby enabling nucleophilic attack of an activated water molecule. The Mg2+ ion is coordinated by three protein ligands and two oxygen atoms of the scissile phosphate. This coordination by two oxygen atoms of the scissile phosphate results in an unfavorable coordination angle (O–Me–O), smaller than 90°, resulting in destabilization of the scissile bond and facilitating the nucleophilic attack of a water molecule [80]. Moreover, the Mg2+ ion accelerates product release and the turnover rate by rebinding of water ligands. The nucleophile in the HNH nuclease is activated by a His residue (His840) acting as a strong catalytic base (Fig. 4e). This histidine is only marginally involved in Mg2+ binding but it is in hydrogen bonding distance of the catalytic water molecule to polarize this water molecule, i.e., to deprotonate the water and form an hydroxyl ion, resulting in an increase in its nucleophilicity. Moreover, the His orients the catalytic water molecule for an in-line attack of the scissile phosphate bond. This finally results in cleavage of the scissile phosphate bond. One-metal-ion catalysis is less stringent on the type of metal ion bound. These enzymes often can tolerate other divalent cations than Mg2+, such as Ni2+, Zn2+, Ca2+, Cu2+, Mn2+, or Cd2+ [80]. The HNH nuclease in Cas9 is highly sequence specific. The specificity is created by substrate binding due to the ββα-Mg2+ motif and other protein domains [68, 75].
Using CRISPR/Cas9 as an efficient tool for genome editing
Functional and structural studies on CRISPR/Cas9 revealed that it constitutes an RNA-guided antiviral immune system. The fact that its nuclease activity is RNA-guided opened the possibility to use it as a sequence-specific tool for targeted genome editing [66]. Studies by Emmanuelle Charpentier and Jennifer A. Doudna revealed that CRISPR/Cas is indeed programmable [66] and both scientists advanced the understanding of the CRISPR/Cas system in such detail that it can now be used as a tool for genome editing. For this remarkable work Emmanuelle Charpentier and Jennifer A. Doudna were jointly awarded the Nobel Prize in Chemistry 2020 for “the development of a method for genome editing”. It was shown that the Cas9 target specificity was created by an RNA–DNA Watson–Crick base pairing and by recognition of the PAM sequence (5′-NGG-3′) within the target DNA. Artificial sgRNAs were designed, which fulfill the requirements of showing complementarity to a target DNA sequence and carrying a PAM sequence adjacent to it (Fig. 4d, f) [63, 68, 70]. These artificial sgRNAs were indeed bound by Cas9 and functionally replaced the physiological crRNA:tracrRNA duplex (Fig. 4e). In 2012 it was confirmed that CRISPR/Cas9 is programmable [66]. The artificial sgRNAs are, in contrast to the crRNA:tracrRNA, single-stranded RNA molecules that need only two inherent properties: (1) an approximately 20-nucleotide-long spacer-sequence at the 5′-end, which recognizes the target DNA by Watson–Crick base pairing and (2) a characteristic three-dimensional secondary structure that is recognized and bound by Cas9 (Fig. 4f). This interaction then results in the formation of a simple two-component system (sgRNA and Cas9) that allows the introduction of specific DSBs in any target DNA sequence as long as it is located next to a PAM sequence [86, 87]. With this artificially designed sgRNA, Cas9 becomes a programmable nuclease, with great potential for applications in biomedicine, pharmaceuticals, biotechnology, and agriculture. Active research in ongoing into the safe, efficient, and targetable in vivo delivery of the engineered Cas9 into cells or tissues (Fig. 5) [88,89,90]. This includes infection with engineered viral particles (based on adenovirus, lentivirus, retrovirus) via electroporation/microinjection/lipid particles of Cas9·mRNA and the sgRNA, of linear DNA/plasmid DNA encoding Cas9·sgRNA, and of the purified Cas9·sgRNA complexes [89]. As the target DNA is located in the nucleus in eukaryotic cells Cas9 is genetically modified so that it carries an NLS [86]. This ensures that Cas9 is targeted from the cytosol, where translation occurs, into the nucleus, where the target DNA is located in eukaryotic cells. This transport occurs in a Ran GTP-binding protein-dependent process. Engineering of the Cas9 creates diverse systems used for various applications in genome editing including nucleotide deletions (including gene knockouts), insertions, and inversions. For genome editing using Cas9, after introducing DNA DSBs, it is desirable to activate the precise HDR pathway instead of the error-prone NHEJ pathway. HDR depends on the presence of a homologous DNA sequence. For genome editing this homologous DNA sequence can be supplied as a plasmid donor DNA. This can include a DNA sequence of interest that is flanked by homology arms [61, 91] (Fig. 5). Apart from applications in genome editing various other applications use the possibility to target Cas9 to specific DNA sequences. In these applications beyond genome editing, often catalytically inactive Cas9 variants (i.e., D10A in RuvC, H840A in HNH) that are still capable of sgRNA-guided DNA targeting are used. These Cas9 variants are then coupled to diverse protein-targeting domains or with various enzymatic activities: activation/repression of transcription, epigenome editing (acetylation, methylation of histones/DNA methylation), cellular/chromatin imaging, RNA targeting, subcellular targeting of fusion proteins, and high-throughput screenings to conduct genotype–phenotype correlations [60, 61].

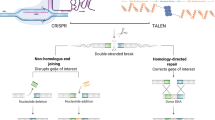
Using Cas9 sgRNA for targeted genome editing. Cas9, small guide RNA, and a DNA fragment with homology arms complementary to the target DNA are delivered into the cell. Diverse delivery methods are under investigation as described in the text. Cas9 forms a complex with the sgRNA, which is homologous to the target DNA in its spacer sequence. Cas9 creates a double strand break (DSB) in the target DNA. This DSB evokes DNA repair programs, either precise homology directed repair (HDR) or imprecise/error-prone non-homologous end joining. HDR enables one to correct mutations in the target DNA by using the DNA construct with homology arms for homologous recombination. The figure was created with BioRender.com
Consequences of inducing CRISPR/Cas9-mediated DSBs in target DNA
CRISPR/Cas9 induces DNA DSBs in the target DNA. In eukaryotic cells this activates the cellular DNA repair programs NHEJ or HDR (Fig. 5). NHEJ is not dependent on the presence of a homologous DNA sequence that can be used as a template for repair. It is error-prone and often results in imprecise repair resulting in insertions and deletions (indels for short) and often generating frameshifts that (results in the production of non-sense proteins and) switches off protein function [92]. In contrast, HDR is not error-prone, i.e., it is precise, and it depends on the presence of a homologous DNA template used for DNA repair. To this end, mostly HDR needs to be activated for efficient and precise genome editing. It is not completely understood which program is elicited by a DNA DSB, opening the possibility to generate substantial off-target effects using Cas9. Moreover, as Cas9 targeting depends on the sequence of the spacer sequence in the sgRNA, binding of Cas9 to similar but not identical DNA sequences can result in DSBs at unwanted positions within the genome. Recent data suggest that introducing a single-strand break in the target DNA, instead of a DSB, predominantly activates HDR [93]. With this in mind, new Cas9 variants were designed that have nickase (single-strand nuclease) activity, i.e., either the RuvC or the HNH nuclease activity is switched off by introducing inactivating mutations (i.e., D10A in RuvC, H840A in HNH) [94, 95].
Conclusions and perspectives
CRISPR/Cas has enormous potential as a genome editing tool with various applications in biomedicine and biotechnology. Medical applications include the treatment of genetic diseases caused by single-nucleotide mutations such as sickle cell anemia, or by gene insertion as found in the ocular disease retinitis pigmentosa and diseases caused by frameshift or point mutations as found in beta-thalassemia by correcting the disease-causing mutations [60]. Even complex genetic diseases are targeted by development of CRISPR/Cas-based editing strategies. Preclinical models of these diseases show promising results. However, it might take more basic research to be able to apply CRISPR/Cas for therapeutic applications in humans and for biotechnology. As stated above, CRISPR/Cas can result in severe off-target effects by targeting similar or identical sequences within the genome and it can result in activation of the error-prone non-homologous end joining DNA repair pathway also resulting in severe mutations [96, 97]. Efforts were made to develop more specific Cas9·sgRNA systems and systems that activate homology-directed repair instead of non-homologous end joining [93, 98,99,100]. Recent approaches reduce off-target effects by co-delivery of short sgRNAs directed against off-target loci [101]. Other strategies involve using Cas9 orthologues from other organisms, using directed evolution and protein engineering to develop improved Cas9 variants or applying different CRISPR/Cas types [60, 102,103,104,105]. In a therapeutic application to treat Duchenne muscular dystrophy (DMD), which is caused by deletions in the gene resulting in frameshift mutations, the correct reading frame of the DMD gene was achieved by specifically activating the NHEJ DNA repair pathway [60]. All of these preclinical applications need to be transferred into the clinics. One of the biggest difficulties in this process is the in vivo delivery of the Cas system [89]. Several approaches are under investigation: gold/polymer nanoparticles, lipid nanoparticles/viral particles, viral-based delivery methods (adeno-associated virus/lentivirus/retrovirus) [89]. The most promising strategy involves ex vivo manipulation of cells of interest and back-administration of the cells into the donor [90]. This has been successfully applied in adoptive T cells immunotherapy [106]. Besides these promising advances in the field of genome editing, CRISPR/Cas has several limitations apart from the off-target effects stated above. It was found that many human individuals show immunogenicity to Cas proteins originating from S. pyogenes or Staphylococcus aureus as these bacteria are highly prevalent in the human environment [60, 107]. Future work needs to focus on development of CRISR/Cas systems of less prevalent bacterial species to reduce the potential to induce an immune response upon Cas-mediated therapies. Additional data suggest that CRISPR/Cas9 is more efficient in cells that have a loss-of-function of the tumor suppressor protein p53 [108, 109]. In turn, those cells that are edited by CRISPR/Cas9 could be prone to the development of tumors. These results show that the usage of CRISPR/Cas9 needs to be critically discussed in terms of the values and the risks connected with it. Of course, applications of genome editing tools have not only a technical dimension but there are also ethical questions that need to be addressed within society, particularly before applications on the human germ line or even in embryos.
Data availability
The authors declare that all structural data used in this article is available in the Protein Data Bank (www.rcsb.org).
Code availability
The figures were prepared with Adobe Illustrator version 24.2.1 and PyMol version 2.3.4.
Change history
19 February 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40828-021-00131-4
References
Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17:333–351. https://doi.org/10.1038/nrg.2016.49
Levy SE, Myers RM (2016) Advancements in next-generation sequencing. Annu Rev Genomics Hum Genet 17:95–115. https://doi.org/10.1146/annurev-genom-083115-022413
Giani AM, Gallo GR, Gianfranceschi L, Formenti G (2020) Long walk to genomics: history and current approaches to genome sequencing and assembly. Comput Struct Biotechnol J 18:9–19. https://doi.org/10.1016/j.csbj.2019.11.002
Jackson M, Marks L, May GHW, Wilson JB (2018) The genetic basis of disease. Essays Biochem 62:643–723. https://doi.org/10.1042/EBC20170053
Suwinski P, Ong C, Ling MHT, Poh YM, Khan AM, Ong HS (2019) Advancing personalized medicine through the application of whole exome sequencing and big data analytics. Front Genet. https://doi.org/10.3389/fgene.2019.00049
Kim H, Kim M, Im SK, Fang S (2018) Mouse Cre-loxP system: general principles to determine tissue-specific roles of target genes. Lab Anim Res 34:147–159. https://doi.org/10.5625/lar.2018.34.4.147
Guo F, Gopaul DN, van Duyne GD (1997) Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature 389:40–46. https://doi.org/10.1038/37925
Gopaul DN, Guo F, Van Duyne GD (1998) Structure of the Holliday junction intermediate in Cre-loxP site-specific recombination. EMBO J 17:4175–4187. https://doi.org/10.1093/emboj/17.14.4175
Martin SS, Pulido E, Chu VC, Lechner TS, Baldwin EP (2002) The order of strand exchanges in Cre-loxP recombination and its basis suggested by the crystal structure of a Cre-loxP Holliday junction complex. J Mol Biol 319:107–127. https://doi.org/10.1016/S0022-2836(02)00246-2
Ennifar E, Meyer JE, Buchholz F, Stewart AF, Suck D (2003) Crystal structure of a wild-type Cre recombinase-loxP synapse reveals a novel spacer conformation suggesting an alternative mechanism for DNA cleavage activation. Nucleic Acids Res 31:5449–5460. https://doi.org/10.1093/nar/gkg732
Bouabe H, Okkenhaug K (2013) Gene targeting in mice: a review. Methods Mol Biol 1064:315–336. https://doi.org/10.1007/978-1-62703-601-6_23
Song AJ, Palmiter RD (2018) Detecting and avoiding problems when using the Cre-Iox system. Trends Genet 34:333–340. https://doi.org/10.1016/j.tig.2017.12.008
Eroshenko N, Church GM (2013) Mutants of Cre recombinase with improved accuracy. Nat Commun 4:2509. https://doi.org/10.1038/ncomms3509
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811. https://doi.org/10.1038/35888
Setten RL, Rossi JJ, Han SP (2019) The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov 18:421–446. https://doi.org/10.1038/s41573-019-0017-4
Agrawal N, Dasaradhi PV, Mohmmed A, Malhotra P, Bhatnagar RK, Mukherjee SK (2003) RNA interference: biology, mechanism, and applications. Microbiol Mol Biol Rev 67:657–685. https://doi.org/10.1128/mmbr.67.4.657-685.2003
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854. https://doi.org/10.1016/0092-8674(93)90529-y
Dexheimer PJ, Cochella L (2020) MicroRNAs: from mechanism to organism. Front Cell Dev Biol 8:409. https://doi.org/10.3389/fcell.2020.00409
Frazer KA (2012) Decoding the human genome. Genome Res 22:1599–1601. https://doi.org/10.1101/gr.146175.112
Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, Dunham I, Elnitski LL, Farnham PJ, Feingold EA, Gerstein M, Giddings MC, Gilbert DM, Gingeras TR, Green ED, Guigo R, Hubbard T, Kent J, Lieb JD, Myers RM, Pazin MJ, Ren B, Stamatoyannopoulos JA, Weng Z, White KP, Hardison RC (2014) Defining functional DNA elements in the human genome. Proc Natl Acad Sci USA 111:6131–6138. https://doi.org/10.1073/pnas.1318948111
Stamatoyannopoulos JA (2012) What does our genome encode? Genome Res 22:1602–1611. https://doi.org/10.1101/gr.146506.112
Alles J, Fehlmann T, Fischer U, Backes C, Galata V, Minet M, Hart M, Abu-Halima M, Grasser FA, Lenhof HP, Keller A, Meese E (2019) An estimate of the total number of true human miRNAs. Nucleic Acids Res 47:3353–3364. https://doi.org/10.1093/nar/gkz097
Osada H, Takahashi T (2007) MicroRNAs in biological processes and carcinogenesis. Carcinogenesis 28:2–12. https://doi.org/10.1093/carcin/bgl185
O’Brien J, Hayder H, Zayed Y, Peng C (2018) Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne) 9:402. https://doi.org/10.3389/fendo.2018.00402
Hock J, Meister G (2008) The Argonaute protein family. Genome Biol 9:210. https://doi.org/10.1186/gb-2008-9-2-210
Hamilton AJ, Baulcombe DC (1999) A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 286:950–952. https://doi.org/10.1126/science.286.5441.950
Obbard DJ, Gordon KHJ, Buck AH, Jiggins FM (2009) The evolution of RNAi as a defence against viruses and transposable elements. Philos Trans R Soc B-Biol Sci 364:99–115. https://doi.org/10.1098/rstb.2008.0168
Hu B, Zhong L, Weng Y, Peng L, Huang Y, Zhao Y, Liang XJ (2020) Therapeutic siRNA: state of the art. Signal Transduct Target Ther 5:101. https://doi.org/10.1038/s41392-020-0207-x
Qureshi A, Tantray VG, Kirmani AR, Ahangar AG (2018) A review on current status of antiviral siRNA. Rev Med Virol 28:e1976. https://doi.org/10.1002/rmv.1976
Dana H, Chalbatani GM, Mahmoodzadeh H, Karimloo R, Rezaiean O, Moradzadeh A, Mehmandoost N, Moazzen F, Mazraeh A, Marmari V, Ebrahimi M, Rashno MM, Abadi SJ, Gharagouzlo E (2017) Molecular mechanisms and biological functions of siRNA. Int J Biomed Sci 13:48–57
Moore CB, Guthrie EH, Huang MT, Taxman DJ (2010) Short hairpin RNA (shRNA): design, delivery, and assessment of gene knockdown. Methods Mol Biol 629:141–158. https://doi.org/10.1007/978-1-60761-657-3_10
Gaj T, Gersbach CA, Barbas CF 3rd (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31:397–405. https://doi.org/10.1016/j.tibtech.2013.04.004
Hubbard BP, Badran AH, Zuris JA, Guilinger JP, Davis KM, Chen L, Tsai SQ, Sander JD, Joung JK, Liu DR (2015) Continuous directed evolution of DNA-binding proteins to improve TALEN specificity. Nat Methods 12:939–942. https://doi.org/10.1038/nmeth.3515
Remy S, Tesson L, Menoret S, Usal C, Scharenberg AM, Anegon I (2010) Zinc-finger nucleases: a powerful tool for genetic engineering of animals. Transgenic Res 19:363–371. https://doi.org/10.1007/s11248-009-9323-7
Paschon DE, Lussier S, Wangzor T, Xia DF, Li PW, Hinkley SJ, Scarlott NA, Lam SC, Waite AJ, Truong LN, Gandhi N, Kadam BN, Patil DP, Shivak DA, Lee GK, Holmes MC, Zhang L, Miller JC, Rebar EJ (2019) Diversifying the structure of zinc finger nucleases for high-precision genome editing. Nat Commun 10:1133. https://doi.org/10.1038/s41467-019-08867-x
Lee J, Chung JH, Kim HM, Kim DW, Kim H (2016) Designed nucleases for targeted genome editing. Plant Biotechnol J 14:448–462. https://doi.org/10.1111/pbi.12465
Ma AC, Chen Y, Blackburn PR, Ekker SC (2016) TALEN-mediated mutagenesis and genome editing. Methods Mol Biol 1451:17–30. https://doi.org/10.1007/978-1-4939-3771-4_2
Mao Z, Jiang Y, Liu X, Seluanov A, Gorbunova V (2009) DNA repair by homologous recombination, but not by nonhomologous end joining, is elevated in breast cancer cells. Neoplasia 11:683–691. https://doi.org/10.1593/neo.09312
Chandrasegaran S, Carroll D (2016) Origins of programmable nucleases for genome engineering. J Mol Biol 428:963–989. https://doi.org/10.1016/j.jmb.2015.10.014
Ul Ain Q, Chung JY, Kim YH (2015) Current and future delivery systems for engineered nucleases: ZFN, TALEN and RGEN. J Control Release 205:120–127. https://doi.org/10.1016/j.jconrel.2014.12.036
Zhang HX, Zhang Y, Yin H (2019) Genome editing with mRNA encoding ZFN, TALEN, and Cas9. Mol Ther 27:735–746. https://doi.org/10.1016/j.ymthe.2019.01.014
Cassandri M, Smirnov A, Novelli F, Pitolli C, Agostini M, Malewicz M, Melino G, Raschella G (2017) Zinc-finger proteins in health and disease. Cell Death Discov 3:17071. https://doi.org/10.1038/cddiscovery.2017.71
Mani M, Kandavelou K, Dy FJ, Durai S, Chandrasegaran S (2005) Design, engineering, and characterization of zinc finger nucleases. Biochem Biophys Res Commun 335:447–457. https://doi.org/10.1016/j.bbrc.2005.07.089
Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I (1998) FokI dimerization is required for DNA cleavage. Proc Natl Acad Sci USA 95:10570–10575. https://doi.org/10.1073/pnas.95.18.10570
de Souza N (2012) Improving gene-editing nucleases. Nat Methods 9:536. https://doi.org/10.1038/nmeth.2060
Ramirez CL, Certo MT, Mussolino C, Goodwin MJ, Cradick TJ, McCaffrey AP, Cathomen T, Scharenberg AM, Joung JK (2012) Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res 40:5560–5568. https://doi.org/10.1093/nar/gks179
Moore R, Chandrahas A, Bleris L (2014) Transcription activator-like effectors: a toolkit for synthetic biology. ACS Synth Biol 3:708–716. https://doi.org/10.1021/sb400137b
de Lange O, Wolf C, Thiel P, Kruger J, Kleusch C, Kohlbacher O, Lahaye T (2015) DNA-binding proteins from marine bacteria expand the known sequence diversity of TALE-like repeats. Nucleic Acids Res 43:10065–10080. https://doi.org/10.1093/nar/gkv1053
Gao H, Wu X, Chai J, Han Z (2012) Crystal structure of a TALE protein reveals an extended N-terminal DNA binding region. Cell Res 22:1716–1720. https://doi.org/10.1038/cr.2012.156
Mak AN, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL (2012) The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 335:716–719. https://doi.org/10.1126/science.1216211
Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186:757–761. https://doi.org/10.1534/genetics.110.120717
Nemudryi AA, Valetdinova KR, Medvedev SP, Zakian SM (2014) TALEN and CRISPR/Cas genome editing systems: tools of discovery. Acta Nat 6:19–40
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433. https://doi.org/10.1128/jb.169.12.5429-5433.1987
Mojica FJ, Rodriguez-Valera F (2016) The discovery of CRISPR in archaea and bacteria. FEBS J 283:3162–3169. https://doi.org/10.1111/febs.13766
Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60:174–182. https://doi.org/10.1007/s00239-004-0046-3
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. https://doi.org/10.1126/science.1138140
Pourcel C, Salvignol G, Vergnaud G (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology (Reading) 151:653–663. https://doi.org/10.1099/mic.0.27437-0
Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S (2008) Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190:1390–1400. https://doi.org/10.1128/JB.01412-07
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. https://doi.org/10.1038/nature09886
Pickar-Oliver A, Gersbach CA (2019) The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol 20:490–507. https://doi.org/10.1038/s41580-019-0131-5
Hille F, Richter H, Wong SP, Bratovic M, Ressel S, Charpentier E (2018) The biology of CRISPR-Cas: backward and forward. Cell 172:1239–1259. https://doi.org/10.1016/j.cell.2017.11.032
Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. https://doi.org/10.1126/science.1258096
Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62–67. https://doi.org/10.1038/nature13011
Chylinski K, Le Rhun A, Charpentier E (2013) The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol 10:726–737. https://doi.org/10.4161/rna.24321
Chylinski K, Makarova KS, Charpentier E, Koonin EV (2014) Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res 42:6091–6105. https://doi.org/10.1093/nar/gku241
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. https://doi.org/10.1126/science.1225829
Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV (2011) Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9:467–477. https://doi.org/10.1038/nrmicro2577
Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156:935–949. https://doi.org/10.1016/j.cell.2014.02.001
Josephs EA, Kocak DD, Fitzgibbon CJ, McMenemy J, Gersbach CA, Marszalek PE (2016) Structure and specificity of the RNA-guided endonuclease Cas9 during DNA interrogation, target binding and cleavage. Nucleic Acids Res 44:2474. https://doi.org/10.1093/nar/gkv1293
Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343:1247997. https://doi.org/10.1126/science.1247997
Anders C, Niewoehner O, Duerst A, Jinek M (2014) Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513:569–573. https://doi.org/10.1038/nature13579
Huai C, Li G, Yao R, Zhang Y, Cao M, Kong L, Jia C, Yuan H, Chen H, Lu D, Huang Q (2017) Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nat Commun 8:1375. https://doi.org/10.1038/s41467-017-01496-2
Josephs EA, Kocak DD, Fitzgibbon CJ, McMenemy J, Gersbach CA, Marszalek PE (2015) Structure and specificity of the RNA-guided endonuclease Cas9 during DNA interrogation, target binding and cleavage. Nucleic Acids Res 43:8924–8941. https://doi.org/10.1093/nar/gkv892
Zuo Z, Liu J (2017) Structure and dynamics of Cas9 HNH domain catalytic state. Sci Rep 7:17271. https://doi.org/10.1038/s41598-017-17578-6
Zuo Z, Zolekar A, Babu K, Lin VJ, Hayatshahi HS, Rajan R, Wang YC, Liu J (2019) Structural and functional insights into the bona fide catalytic state of Streptococcus pyogenes Cas9 HNH nuclease domain. Elife. https://doi.org/10.7554/eLife.46500
Gorecka KM, Komorowska W, Nowotny M (2013) Crystal structure of RuvC resolvase in complex with Holliday junction substrate. Nucleic Acids Res 41:9945–9955. https://doi.org/10.1093/nar/gkt769
Sashital DG, Jinek M, Doudna JA (2011) An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat Struct Mol Biol 18:680–687. https://doi.org/10.1038/nsmb.2043
Palermo G, Miao Y, Walker RC, Jinek M, McCammon JA (2017) CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc Natl Acad Sci USA 114:7260–7265. https://doi.org/10.1073/pnas.1707645114
Palermo G (2019) Structure and dynamics of the CRISPR-Cas9 catalytic complex. J Chem Inf Model 59:2394–2406. https://doi.org/10.1021/acs.jcim.8b00988
Yang W (2008) An equivalent metal ion in one- and two-metal-ion catalysis. Nat Struct Mol Biol 15:1228–1231. https://doi.org/10.1038/nsmb.1502
Dupureur CM (2010) One is enough: insights into the two-metal ion nuclease mechanism from global analysis and computational studies. Metallomics 2:609–620. https://doi.org/10.1039/c0mt00013b
Westheimer FH (1987) Why nature chose phosphates. Science 235:1173–1178. https://doi.org/10.1126/science.2434996
Westheimer FH (1997) Why nature chose phosphates. 2. Abstracts of Papers of the American Chemical Society 214:126-Orgn
Kamerlin SCL, Sharma PK, Prasad RB, Warshel A (2013) Why nature really chose phosphate. Q Rev Biophys 46:1–132. https://doi.org/10.1017/S0033583512000157
Palermo G, Casalino L, Jinek M (2020) Two-metal ion mechanism of DNA cleavage in CRISPR-Cas9. Biophys J 118:64a–64a
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826. https://doi.org/10.1126/science.1232033
Xu J, Lian W, Jia Y, Li L, Huang Z (2017) Optimized guide RNA structure for genome editing via Cas9. Oncotarget 8:94166–94171. https://doi.org/10.18632/oncotarget.21607
Wang HX, Li M, Lee CM, Chakraborty S, Kim HW, Bao G, Leong KW (2017) CRISPR/Cas9-based genome editing for disease modeling and therapy: challenges and opportunities for nonviral delivery. Chem Rev 117:9874–9906. https://doi.org/10.1021/acs.chemrev.6b00799
Tong S, Moyo B, Lee CM, Leong K, Bao G (2019) Engineered materials for in vivo delivery of genome-editing machinery. Nat Rev Mater 4:726–737. https://doi.org/10.1038/s41578-019-0145-9
Wilbie D, Walther J, Mastrobattista E (2019) Delivery aspects of CRISPR/Cas for in vivo genome editing. Acc Chem Res 52:1555–1564. https://doi.org/10.1021/acs.accounts.9b00106
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153:910–918. https://doi.org/10.1016/j.cell.2013.04.025
Bachu R, Bergareche I, Chasin LA (2015) CRISPR-Cas targeted plasmid integration into mammalian cells via non-homologous end joining. Biotechnol Bioeng 112:2154–2162. https://doi.org/10.1002/bit.25629
Sansbury BM, Hewes AM, Kmiec EB (2019) Understanding the diversity of genetic outcomes from CRISPR-Cas generated homology-directed repair. Commun Biol 2:458. https://doi.org/10.1038/s42003-019-0705-y
Satomura A, Nishioka R, Mori H, Sato K, Kuroda K, Ueda M (2017) Precise genome-wide base editing by the CRISPR nickase system in yeast. Sci Rep 7:2095. https://doi.org/10.1038/s41598-017-02013-7
Fu BXH, Smith JD, Fuchs RT, Mabuchi M, Curcuru J, Robb GB, Fire AZ (2019) Target-dependent nickase activities of the CRISPR-Cas nucleases Cpf1 and Cas9. Nat Microbiol 4:888–897. https://doi.org/10.1038/s41564-019-0382-0
Wang Y, Wang M, Zheng T, Hou Y, Zhang P, Tang T, Wei J, Du Q (2020) Specificity profiling of CRISPR system reveals greatly enhanced off-target gene editing. Sci Rep 10:2269. https://doi.org/10.1038/s41598-020-58627-x
Hendel A, Fine EJ, Bao G, Porteus MH (2015) Quantifying on- and off-target genome editing. Trends Biotechnol 33:132–140. https://doi.org/10.1016/j.tibtech.2014.12.001
Devkota S (2018) The road less traveled: strategies to enhance the frequency of homology-directed repair (HDR) for increased efficiency of CRISPR/Cas-mediated transgenesis. BMB Rep 51:437–443
Schiermeyer A, Schneider K, Kirchhoff J, Schmelter T, Koch N, Jiang K, Herwartz D, Blue R, Marri P, Samuel P, Corbin DR, Webb SR, Gonzalez DO, Folkerts O, Fischer R, Schinkel H, Ainley WM, Schillberg S (2019) Targeted insertion of large DNA sequences by homology-directed repair or non-homologous end joining in engineered tobacco BY-2 cells using designed zinc finger nucleases. Plant Direct 3:e00153. https://doi.org/10.1002/pld3.153
Lin S, Staahl BT, Alla RK, Doudna JA (2014) Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3:e04766. https://doi.org/10.7554/eLife.04766
Coelho MA, De Braekeleer E, Firth M, Bista M, Lukasiak S, Cuomo ME, Taylor BJM (2020) CRISPR GUARD protects off-target sites from Cas9 nuclease activity using short guide RNAs. Nat Commun 11:4132. https://doi.org/10.1038/s41467-020-17952-5
Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar VV, Aryee MJ, Joung JK (2017) CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods 14:607–614. https://doi.org/10.1038/nmeth.4278
Lazzarotto CR, Nguyen NT, Tang X, Malagon-Lopez J, Guo JA, Aryee MJ, Joung JK, Tsai SQ (2018) Defining CRISPR-Cas9 genome-wide nuclease activities with CIRCLE-seq. Nat Protoc 13:2615–2642. https://doi.org/10.1038/s41596-018-0055-0
Akcakaya P, Bobbin ML, Guo JA, Malagon-Lopez J, Clement K, Garcia SP, Fellows MD, Porritt MJ, Firth MA, Carreras A, Baccega T, Seeliger F, Bjursell M, Tsai SQ, Nguyen NT, Nitsch R, Mayr LM, Pinello L, Bohlooly YM, Aryee MJ, Maresca M, Joung JK (2018) In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 561:416–419. https://doi.org/10.1038/s41586-018-0500-9
Tang LC, Gu F (2020) Next-generation CRISPR-Cas for genome editing: focusing on the Cas protein and PAM. Yi Chuan 42:236–249. https://doi.org/10.16288/j.yczz.19-297
Bailey SR, Maus MV (2019) Gene editing for immune cell therapies. Nat Biotechnol 37:1425–1434. https://doi.org/10.1038/s41587-019-0137-8
Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK, Vakulskas CA, Collingwood MA, Zhang L, Bode NM, Behlke MA, Dejene B, Cieniewicz B, Romano R, Lesch BJ, Gomez-Ospina N, Mantri S, Pavel-Dinu M, Weinberg KI, Porteus MH (2019) Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med 25:249–254. https://doi.org/10.1038/s41591-018-0326-x
Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, Kommineni S, Chen J, Sondey M, Ye C, Randhawa R, Kulkarni T, Yang Z, McAllister G, Russ C, Reece-Hoyes J, Forrester W, Hoffman GR, Dolmetsch R, Kaykas A (2018) p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med 24:939–946. https://doi.org/10.1038/s41591-018-0050-6
Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J (2018) CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 24:927–930. https://doi.org/10.1038/s41591-018-0049-z
Borel F, Kay MA, Mueller C (2014) Recombinant AAV as a platform for translating the therapeutic potential of RNA interference. Mol Ther 22:692–701. https://doi.org/10.1038/mt.2013.285
Funding
Open Access funding enabled and organized by Projekt DEAL.. Open Access funding enabled and organized by Projekt DEAL.. Open Access funding enabled and organized by Projekt DEAL.. The group is funded by the German Research Foundation Grant numbers LA2984/5-1 and LA2984/6-1.
Author information
Authors and Affiliations
Contributions
SS and ML wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare neither a conflict of interest nor competing interests.
Ethics approval
The authors declare that there are no ethical concerns.
Consent to participate
The authors declare consent to participate.
Consent for publication
The authors declare consent to publish.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schulze, S., Lammers, M. The development of genome editing tools as powerful techniques with versatile applications in biotechnology and medicine: CRISPR/Cas9, ZnF and TALE nucleases, RNA interference, and Cre/loxP. ChemTexts 7, 3 (2021). https://doi.org/10.1007/s40828-020-00126-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40828-020-00126-7