
Highlights
-
Basic principles of photo- and electro-catalytic hydrogen peroxide generation.
-
Recent progresses on the design, performance and mechanisms of photo- and electro-catalysts for hydrogen peroxide generation.
-
Scientific challenges and prospects of engineering photo- and electro-catalysts for hydrogen peroxide production.
Abstract
Hydrogen peroxide (H2O2) is a high-demand organic chemical reagent and has been widely used in various modern industrial applications. Currently, the prominent method for the preparation of H2O2 is the anthraquinone oxidation. Unfortunately, it is not conducive to economic and sustainable development since it is a complex process and involves unfriendly environment and potential hazards. In this context, numerous approaches have been developed to synthesize H2O2. Among them, photo/electro-catalytic ones are considered as two of the most promising manners for on-site synthesis of H2O2. These alternatives are sustainable in that only water or O2 is required. Namely, water oxidation (WOR) or oxygen reduction (ORR) reactions can be further coupled with clean and sustainable energy. For photo/electro-catalytic reactions for H2O2 generation, the design of the catalysts is extremely important and has been extensively conducted with an aim to obtain ultimate catalytic performance. This article overviews the basic principles of WOR and ORR, followed by the summary of recent progresses and achievements on the design and performance of various photo/electro-catalysts for H2O2 generation. The related mechanisms for these approaches are highlighted from theoretical and experimental aspects. Scientific challenges and opportunities of engineering photo/electro-catalysts for H2O2 generation are also outlined and discussed.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Hydrogen peroxide (H2O2) is a strong oxidant reagent compared with Cl-containing oxidants such as HOCl. This efficient and environmental-friendly chemical has been selected as one of the 100 most important chemical substances in the world [1, 2]. Owing to its strong oxidation capability, H2O2 has been widely used in a wide range of industrial and household applications, including wastewater treatment, chemical synthesis, industrial bleaching, energy storage, medical disinfection and a promising energy carrier in fuel cells [3,4,5,6,7]. It has been pointed out that H2O2 has advantages of the highest concentration of active oxygen (47.1 wt%) and the cleanest by-product (H2O) without carbon emission. These features make H2O2 a promising fuel alternative to H2 and O2 for one-chamber fuel cells without a membrane [8,9,10]. Although H2O2 fuel cells produce a bit lower theoretical output voltage (1.09 V) than H2/O2 fuel cells (1.23 V), the energy density of H2O2 (2.1 MJ kg−1 for 60% aqueous H2O2) is comparable to that of compressed hydrogen (3.5 MJ kg−1) [11,12,13,14,15]. In addition, H2O2 is a liquid fuel that can be fully soluble in water. It is thus more secure and facilitated to be transported and stored than the conventional H2. Accordingly, H2O2 has been predicted to be produced on a scale of approximately 5.7 million tons by 2027 in the global market, emphasizing its growing influence in the fields of sustainable energy [16].
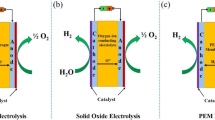
The widely used method to synthesize H2O2 in the industry is anthraquinone (AQ) oxidation, which accounts for more than 95% of the total production of H2O2 [17]. Generally, the AQ oxidation process is a four-step cycle: (i) hydrogenation of AQ using a catalyst, (ii) oxidation of hydrogenated AQ to regenerate AQ and produce H2O2, (iii) extraction of H2O2 and (iv) purification concentration of H2O2. Therefore, this process requires a high energy demandingness and resource consumption and meanwhile produces many harmful by-products in series reactions (Fig. 1a) [18]. In this context, the pursuit of high concentration of H2O2 via AQ oxidation technology brings in more environmental pollutants and explosive substances, being threatened to the safety in fuel storage and transportation [19, 20]. To avoid these shortcomings of the AQ process, the direct preparation of H2O2 through the reaction from H2 and O2 appears more popular. It has been considered as a more energy-efficient and resource-efficient route alternative to the AQ oxidation (Fig. 1b) [21,22,23,24]. This partial hydrogenation reaction can be fully performed at low temperatures, if the highly efficient catalysts are designed and applied. It needs to be pointed out that this process usually selects expensive Pd-based materials as catalysts to limit competing side reactions [25, 26]. More seriously, it requires precise control of the ratio of H2 and O2 to leave for the explosive range, which unavoidably leads to low practice yields of the direct synthesis [27,28,29]. Another potential route to synthesize H2O2 is through a range of enzymatic catalytic processes in nature. It has been reported that some oxidase enzymes (e.g., glucose oxidase, D-amino acid oxidase and cholesterol oxidase) can selectively catalyze the corresponding native substrates to generate H2O2 [30,31,32,33]. Unfortunately, the inferior activity with a low value of turnover frequency occurs in these enzymatic catalytic reactions, which has impeded the rapid development of such biosynthesis of H2O2 [34, 35]. Consequently, environmental-friendly, efficient, safe and convenient alternative routes still need to be explored for H2O2 production.

Schematic illustration of a the anthraquinone (AQ) oxidation process and b the direct synthesis from H2 and O2
In recent years, photo/electro-catalytic synthesis of H2O2 has attracted extensive attention. Comparison with the aforementioned three routes, these new approaches are more economical and environmental-friendly [36,37,38,39]. Belonging to direct synthesis methods, they can be realized using the abundant H2O and O2 on the earth as reactants, but are easier to be handled with lower operating risks [40, 41]. In addition, they can take full advantages of clean and renewable energy sources (e.g., solar energy, wind energy) in a sustainable fashion. To obtain efficient photo/electro-catalytic conversion efficiency, high-performance catalyst platforms need to be established. In other words, the design of advanced photo/electro-catalysts plays a vital role in such reaction conversion [42, 43]. From both fundamental and practical viewpoints, the rational design and controllable synthesis of various photo/electro-catalysts are thus of great importance for H2O2 synthesis.
In this regard, many efforts have been made in the design and synthesis of effective photo/electro-catalysts for active and selective generation of H2O2. It is therefore extremely significant to summarize the progress and state-of-art about design, characterization and application of developed photo/electro-catalysts for active and selective production of H2O2. This review article provides a comprehensive account of the development of photo/electro-catalysts toward H2O2 generation, covering related synthesis mechanisms, state-of-the-art catalysts/materials together with their performance evaluation criterion and typical engineering strategies for H2O2 formation. The future perspectives of photo/electro-catalyst design for H2O2 production are discussed and outlined.
2 Mechanisms of Catalytic H2O2 Synthesis
2.1 Photocatalytic Mechanisms of H2O2 Synthesis
The best advantage of photocatalytic synthesis is the direct utilization of renewable and sustainable solar energy to synthesize various products or degrade series of pollutes [44,45,46]. Most of recent photocatalytic synthesis studies focus on photocatalytic reactions on the semiconductors. During a typical photocatalytic process of H2O2 synthesis, three consecutive fundamental steps undergo on the photocatalysts [47]. In the first step, a semiconductor photocatalyst absorbs the excitation light, of which energy is greater than the band gap of the used photocatalyst, to create the negatively charged electron (e−) on its conduction band (CB), accompanying positively charged hole (h+) on its valence band (VB). In the second step, these photoinduced charge carriers (namely both e− and h+) in the interior are separated and diffuse into the surface of the photocatalyst. Finally, they react with H2O and O2 to generate H2O2 via different redox pathways. Some recombine with each other and do not participate in any chemical reactions.
In the second step of a photocatalytic process of H2O2 synthesis, either oxygen reduction reaction (ORR) or water oxidation reaction (WOR) is involved [15]. As for the process of ORR (Fig. 2a), there are two potential mechanisms: indirect two-step single-electron (O2 → ·O2− → H2O2) and direct one-step two-electron (O2 → H2O2) pathways. For example, the photons are absorbed by the semiconductor photocatalyst to excite the electrons in its VB to its CB, while holes are remained in its VB at the first step. Subsequently, the holes (h+) oxidize H2O to produce O2 (Eq. 1). In the indirect two-step single-electron pathway, one formed O2 molecule reacts with one electron (e−) to form O2·− in the CB of the photocatalyst (Eq. 2a), which spontaneously combines H+ to generate a HO2·− intermediate (Eq. 2b). This radical can also get one electron to produce HO2− intermediate via another one-electron reduction reaction pathway (Eq. 2c). Finally, the synthesized HO2− species reacts with H+, leading to H2O2 evolution in the form of Eq. 2d. In the direct one-step two-electron pathway, one formed O2 molecule directly reacts with two electrons (e−), resulting in the formation of H2O2, as demonstrated in Eq. 3.

Simplified schematic of photocatalytic H2O2 generation from a ORR, b WOR and c dual-channel pathway. Simplified schematic of electrocatalytic H2O2 production from d ORR, e WOR and f dual-channel pathway
The mechanism of H2O2 synthesis via the WOR (Fig. 2b) can be divided two pathways: direct one-step (H2O → H2O2) and indirect two-step (H2O → ·OH → H2O2) approaches. In the case of direct one-step process, one H2O molecule is directly oxidized by two holes to generate one H2O2 through the one-step reaction (Eq. 4). Regarding indirect two-step WOR, H2O is firstly be oxidized and the ·OH intermediate is formed (Eq. 5a). These intermediates react with each other to produce H2O2 (Eq. 5b).
It is acknowledged that the process of photocatalytic H2O2 synthesis might simultaneously occur through both the ORR and WOR mechanisms (Fig. 2c). However, these strategies have the unavoidable drawbacks [48, 49]. Firstly, the solubility of O2 in liquid phase is low, limiting related reaction rates. Secondly, the recombination of electrons and holes is fast, reducing dramatically the efficiency of electron utilization. Thirdly, thermodynamic feasibility of the 4e− pathway is greater than of the 2e− pathway, leading to low selectivity of such processes. Therefore, it is necessary to design advanced photocatalysts to overcome these shortcomings.
The apparent quantum yield (AQY), defined as the ratio of the electron number in generating H2O2 molecule and the incident photons at a given wavelength, is an important indicator to evaluate the photocatalytic efficiency of various photocatalysts. It is frequently calculated using Eq. 6 [20].
where n is the electron amount of produced H2O2, Naph is the number of incident photons shined on the photocatalysts.
In the section of discussion of photocatalysts (Chapter 3), we summarize recent progress and achievements on representative photocatalysts for H2O2 synthesis, in terms of their morphology and electronic band structures as well as their corresponding visible light-driven performance (e.g., activity, selectivity and stability) for H2O2 production.
2.2 Electrocatalytic Mechanisms of H2O2 Synthesis
Electrocatalytic synthesis of H2O2 via the oxygen electrochemistry has been widely considered as an attractive route for on-site production of H2O2. Such an approach has effectively addressed the disadvantages of the AQ process and the possibility to be coupled with sustainable energy sources [50, 51]. Note that there are two direct pathways to produce H2O2, including two-electron oxygen reduction reaction (2e− ORR, Fig. 2d) and two-electron water oxidation reaction (2e− WOR, Fig. 2e) [52,53,54]. As for the 2e− ORR process, the reduction of O2 to H2O2 proceeds by a two-step consecutive reaction road. This process can be described as an overall electrochemical reaction (Eq. 7), where \(E^{\theta }\) represents the standard thermodynamic equilibrium potential, which is calculated to 0.70 V versus reversible hydrogen electrode (RHE). In more detail, one O2 molecule diffuses to the active surface of an electrocatalyst and gets adsorbed. If it gains one electron from the electrode and further reacts with one H+ ion, the OOH* intermediate is generated (Eq. 7a, noted that * represents an unoccupied active site). Since the formed OOH* possesses high chemical reaction activity, it can combine with another H+ ion and one electron to produce H2O2, accompanying with regeneration of the active site (Eq. 7b). Unfortunately, the OOH* intermediate can undergo a four-electron pathway to decompose and form *O and *OH intermediates, which forms H2O instead of H2O2 [55]. The overall electrochemical reaction for the 4e− ORR process is depicted as Eq. 8, where \(E^{\theta }\) is 1.23 VRHE. In the reaction mechanism, the as-formed OOH* intermediate in the first step (Eq. 8a) reacts with one H+ ion and electron to form O* (Eq. 8b), which is further combined with another H+ ion and electron. The formation of OH* intermediate (Eq. 8c) in this step leads to the undesirable 4e− ORR, which forms H2O, an alternative to H2O2 (Eq. 8d). Therefore, the OOH* intermediate plays a vital role in determining the 2e− or 4e− ORR pathways. For example, retaining the O–O bond in OOH*, meaning weaker oxygen binding energy, is favorable to improve the selectivity of H2O2 production [54, 55]. On the contrary, the stronger oxygen binding energy facilitates the 4e− ORR pathway, resulting in the electrocatalytic synthesis of H2O. The 2e− ORR:
The 4e− ORR:
Similar as photocatalytic process of H2O2 synthesis, both ORR and WOR might simultaneously occur in electrocatalytic process (Fig. 2f).
To evaluate the catalytic selectivity in the electrocatalytic H2O2 synthesis via the ORR approach, the number of electrons transferred (n) needs to be calculated by use of the rotating ring-disk electrode (RRDE) or rotating disk electrode (RDE) technique. For the RRDE measurements, linear sweep voltammetry (LSV) is generally performed on the disk electrode, while a constant potential (e.g., 1.2 VRHE) is applied on the ring electrode. When H2O2 is produced on the disk, it can diffuse to the ring then be detected. Equation 9 has been often employed to calculate the n value, where ID is the current on the disk electrode, IR is the current at the ring electrode, and N is the efficiency of H2O2 collection. The N value can be obtained using Eq. 10. For the RDE measurements, the Koutecky–Levich (K–L) equation is applied to calculate n (Eq. 11), where I is the measured steady-state current derived from the ORR (mA cm−2), IK is the kinetic current of the reaction with active substances on the electrode surface, F is the Faraday constant (96,500 C mol−1), A is the geometric area of the work electrode, D0 is the diffusion coefficient of O2 in the electrolyte (cm2 s−1), ω is the angular rotation speed (ω = 2πN, N is the linear rotation speed), ν is the kinematic viscosity of the electrolyte (cm2 s−1), \({C}_{{O}_{2}}\) is the saturated concentration of O2 in the solution (mol cm−3).
Using as-obtained n value, the selectivity of H2O2 production can be calculated according to Eq. 12.
Another promising route for electrocatalytic synthesis of H2O2 is the 2e− WOR pathway, which can be depicted in an overall electrochemical reaction (Eq. 13) with \(E^{\theta }\) of 1.76 VRHE. In this process, one H2O molecule is firstly electrooxidized by active sites on the electrode to form OH*, providing one electron and H+ ion (Eq. 13a). Another H2O molecule goes through the same reaction (Eq. 13b). Two OH* then combine with each other, leading to the production of H2O2 (Eq. 13c). Similar with the case in ORR, the 2e− WOR pathway faces the influence from the 4e− WOR pathway (Eq. 14), where the thermodynamically equilibrium potential (1.2 VRHE) is lower than that in the 2e− WOR pathway [41, 56]. In more detail, the OH* radicals generated in the first step (Eq. 14a) are further electrooxidized, resulting in the formation of O* intermediate (Eq. 14b). These O* intermediates then lose one electron to form OOH* intermediate (Eq. 14c). Finally, the OOH* intermediate converts into O2 molecular to complete this 4e− WOR process (Eq. 14d).
The 2e− WOR:
The 4e− WOR:
The key factor to dominate the H2O2 synthesis via the 2e− WOR pathway is thus the appropriate interaction between the catalytic surface and O intermediates (e.g., *O, *OH and *OOH) [57]. Specifically, too strong OH binding promotively oxidizes *OH to *O, further to *OOH, thereby prompting the undesirable 4e− pathway to generate O2. On the other hand, too weak binding improves the selectivity of 2e− WOR, but inevitably causes high kinetic barriers to hydrolysis and significantly slows down the reaction rate. It is disappointed that H2O2 produced in the 2e− WOR process is not chemically stable. The undesirable disproportionation (2 H2O2 → 2 H2O + O2) or homolysis (H2O2 → 2 ·OH) spontaneously arises, decreasing the selectivity of H2O2 production. Therefore, the electrocatalysts with proper binding energies are highly pursued to boost 2e− pathway toward H2O2 production.
In general, the selectivity of H2O2 generation via H2O electrooxidation is defined using the term of Faradaic Efficiency (%FE) (Eq. 15).
where \({Q}_{{H}_{2}{O}_{2}}\) is the charge that is used to produce H2O2, and \({Q}_{total}\) is the total charge passed during the whole process for water electrooxidation. In the section of the discussion of electrocatalysts (Chapter 4), we will summarize and evaluate research progress on representative electrocatalysts for synthesizing H2O2, in terms of the morphology, structure, composition as well as the corresponding activity, selectivity and stability.
It should be pointed out that the detection of the H2O2 concentration in the electrolyte is very important for the practical applications. Such a concentration is generally determined by the following methods, including [58]: (1) titration using potassium permanganate (KMnO4) or cerium sulfate [Ce(SO4)2], (2) colorimetric change from Fe2+ to Fe3+, (3) colorimetric determination using titanium (IV) sulfate and (4) high-performance liquid chromatography (HPLC). To separate the generated H2O2 from the electrolyte, the oil–water two-phase systems or distillation can be applied, although few reports have focused on such an issue.
3 Photocatalytic H2O2 Synthesis
3.1 Graphitic Carbon Nitride
Graphitic carbon nitride (g-C3N4) possesses a graphitic stacking of structure aromatic molecules with an alternating arrangement of earth-abundant carbon and nitrogen elements. Since the pioneering work of g-C3N4 for the promotion of photocatalytic hydrogen evolution under visible light irradiation, g-C3N4 has received strongly attention [59]. Such a situation is derived from this metal-free polymer photocatalyst features intrinsic advantages, such as its suitable band gap (~ 2.7 eV) for visible light response, facile preparation, relatively higher conductivity, excellent chemical stability in strong acidic and alkali media and special electronic structure [60,61,62,63,64]. In this context, it has been frequently employed for various photocatalytic applications, such as CO2 reduction, H2 production, and especially for H2O2 production [65,66,67,68,69,70]. Especially, since the CB potential (−1.3 V) of g-C3N4 is more negative than the reduction potential of O2/H2O2 (0.695 V), g-C3N4 is thermodynamically promising to reduce O2 for the H2O2 production using visible light [71]. In addition, the VB potential of g-C3N4 (1.4 V) is lower enough to effectively prevent the oxidative decomposition of H2O2 [64]. For example, Shiraishi’s group employed g-C3N4 for photocatalytic H2O2 production with the selectivity higher than 90%. The generated intermediate generated on the g-C3N4 surface 1,4-endoperoxide promotes the two-electron ORR pathway to produce H2O2 [68]. However, it must stress that the pristine g-C3N4 has many drawbacks that limit its photocatalytic performance toward the H2O2 production, covering its small specific surface area, weak ability to capture visible light and low chemical adsorption capacity of O2 on its surface [72, 73]. To overcome these shortcomings, ultra-thin g-C3N4 nanoplates and hexagonal rosettes of g-C3N4 have been prepared, which showed remarkable photocatalytic activity in the H2O2 production, stemming from their high surface area, rich active sites and strong light-harvesting capability [74, 75]. Unfortunately, the overall photo-conversion efficiencies of these g-C3N4 nano-photocatalysts are still unsatisfactory, due to rapid recombination of photogenerated carriers. Therefore, it is necessary to introduce organic scavengers into these nano-photocatalysts (e.g., ultra-thin g-C3N4 nanoplates and hexagonal rosettes of g-C3N4) to consume holes in the process of H2O2 production [70, 76]. In short, the efficiency for visible-light excitation, the separation of photogenerated carriers and photo-reaction kinetics of g-C3N4 photocatalysts must be further improved. In this regard, surface engineer of g-C3N4 photocatalysts has been proposed and developed to facilitate its performance with the enhanced the photocatalytic production efficiency of H2O2 on g-C3N4 photocatalysts. To optimize the photocatalytic H2O2 synthesis, the reported strategies cover manufacturing of surface defects, loading precious metal nanoparticles, constructing heterojunction composites, polyoxometalate hybridization and metal/non-metal element doping are performed (Table 1). In the following sessions, the details of these strategies are explained.
3.1.1 Manufacturing Surface Defects
Vacancy defects are known to have the ability to capture photogenerated electrons/holes, thereby effective inhibiting the recombination of photogenerated electrons and holes [77,78,79,80]. Meanwhile, they can enhance the adsorption and activation of gas molecules (e.g., O2), due to their abundant local electrons for the adjustment of the electronic structure, thus facilitating a photocatalytic process [81, 82]. In general, two main types of defects appear in (or on) the g-C3N4 photocatalysts: carbon and nitrogen vacancies.
Carbon vacancies The defects derived from carbon element in/on g-C3N4 photocatalysts bring in carbon vacancies. They stand at mid-gap states between VB and CB and thus can capture more visible light [46]. Namely, the optical and electronic structures of g-C3N4 photocatalysts can be optimized. For instance, the introduction of carbon vacancies to the surface of g-C3N4 accompanied the appearance of amino group by simple calcination under argon atmosphere (Fig. 3a). In this case, the generated carbon vacancies (Cv-g-C3N4) reduce the band gap of g-C3N4, thereby expanding the absorption of visible light range and increasing excitable electron longevity [71]. Moreover, carbon vacancies offer more sites to adsorb molecular oxygen, beneficial for the transfer of electrons from g-C3N4 to the surface adsorbed O2. Furthermore, this Cv-g-C3N4 photocatalyst alters the H2O2 generation pathway: from a two-step single-electron indirect reduction pathway to a one-step two-electron direct one. This is attributed to generated amino groups around carbon vacancies (Fig. 3b). The activity of Cv-g-C3N4 for the reduction of O2 to H2O2 is 14-fold higher than that of a pure g-C3N4 photocatalyst under irradiation with visible light (Fig. 3c). Carbon vacancies (Cvs) are then believed to produce more trapping sites, which retards the recombination of photogenerated electrons and holes, eventually resulting in an increased efficiency of the H2O2 generation [83]. Another representative case to strengthen the photocatalytic H2O2 synthesis was achieved via the creation of more vacancies, where an inverse opal (IO) structure was constructed on g-C3N4 accompanying with increased carbon vacancies to improve the separation ability of carriers. This IO g-C3N4 photocatalyst showed an outstanding H2O2 generation yield. For example, 325.74 μM was realized only after 2 h of visible light irradiation. The pathway of the H2O2 production was proposed as following: the electrons on the CB of this photocatalyst are feasible to reduce O2 to form H2O2 because its CB is more negative than that of O2/·O2−.

a Schematic illustration of g-C3N4 with carbon vacancy. b Mechanism diagram of H2O2 production in Cv-g-C3N4. c Concentration of generated H2O2 in g-C3N4 and Cv-g-C3N4 systems. Reproduced with permission from Ref. [71]. Copyright 2016 Elsevier. d Schematic illustration of g-C3N4 with nitrogen vacancies (CN-ND). e Proposed mechanism for H2O2 generation of CN-ND. f H2O2 production of different catalysts. Reproduced with permission from Ref. [86]. Copyright 2021 American Chemical Society
Nitrogen vacancies The defects from nitrogen element in/on g-C3N4 photocatalysts are called nitrogen vacancies. Similar to carbon vacancies, they can also facilitate the photocatalytic H2O2 synthesis. For example, nitrogen vacancies in g-C3N4 have been created via the formation of C≡N functional groups by means of thermal reduction treatment [70]. The as-formed nitrogen vacancies narrow down the band gap and make the band edge positively shift, thus enhancing the visible-light absorption and facilitating photocatalytic H2O2 synthesis [84]. Nitrogen vacancies in carbon nitride also effectively trap photoelectrons, boosting the reduction of N2 to ammonium ions [82]. For the photocatalytic H2O2 synthesis, nitrogen vacancies have been designed and embedded in the g-C3N4 catalyst (PH-CN) using a dielectric barrier discharge (DBD) plasma in H2 atmosphere. Since this photocatalyst improves the separation efficiency of the photogenerated carriers, it displays a H2O2 concentration of 5.2 mmol L−1, 13 times higher than that obtained on a pristine g-C3N4 photocatalyst under the identified conditions [85]. In this case, nitrogen vacancies are assumed not only to serve as active centers for the adsorption of reactive oxygen molecules, but also to alter the electronic band structures of g-C3N4 to harness more visible light in the photocatalytic reaction process. One recently reported the nanostructured g-C3N4 with nitrogen defects was adjusted by cyanuric acid-melamine supramolecular adducts (CN-ND) in the optimum range (Fig. 3d) [86]. Due to the narrowed band gap and the newly formed midgap states, the photocatalytic H2O2 production on CN-ND reached 200 μM under 1 h visible light irradiation, much higher than that obtained bulk g-C3N4 photocatalyst (35 μM) and the pristine nanostructured g-C3N4 photocatalyst (85 μM) (Fig. 3e, f). Again, such improved performance of photocatalytic H2O2 generation was attributed to these nitrogen vacancies since they promote the visible-light-harvesting capability and retard the recombination rate of photogenerated carriers. Meanwhile, the reaction mechanism of photocatalytic H2O2 production on nitrogen deficiency in carbon nitride has been investigated through coupling experiments with the aid of time-dependent density functional theory (TDDFT) and density functional theory (DFT) calculations [87]. It is disclosed that the introduced bicoordinated nitrogen vacancies play an important role in oxidation, reduction and charge recombination, which are beneficial to the generation of h+, ·O2− and 1O2, respectively. Therefore, nitrogen vacancies in carbon nitride can modulate photocatalytic H2O2 generation.
3.1.2 Loading Precious Metal Nanoparticles
The loading of precious metal nanoparticles (NPs) on g-C3N4 forms strong interaction between metallic NPs and g-C3N4, as derived from the delocalization of long electrons in the g-C3N4 matrix. This is one of the most effective approaches to strengthen the photocatalytic activity and selectivity of g-C3N4 since the photogenerated charge separation in such photocatalysts are accelerated, leading to boost photocatalytic reaction in the special pathways [88]. In addition, these photocatalysts suppress the H2O2 decomposition to obtain high yields during photocatalytic H2O2 synthesis. For example, Au NPs deposited on the surface of g-C3N4 by a carbon-layer-stabilized method exhibited a higher photocatalytic yield of H2O2 than the pristine g-C3N4 due to the increased removal of the generated H2O2 from the active reactive sites [89]. In another case, Au NPs were uniformly dispersed on g-C3N4 support (Au/CN) and further utilized to produce H2O2 under visible light irradiation [90]. Its outstanding photocatalytic yield of H2O2 was believed to result from its inertness to catalyze the H2O2 decomposition reaction. The use of 0.01% Au led to the maximum H2O2 production activity (2027 μM) with light irradiation for 30 h, superior to that using other NPs (e.g., Au, Ag, Pd and Pt) on g-C3N4. This high activity of 0.01% Au/CN was attributed to the rapidly reduced recombination of charge carriers. It was derived from the strong interaction between Au and g-C3N4, as testified by obviously quenched PL signal. Photocatalytic H2O2 synthesis for this Au/CN photocatalyst has been further revealed using in-situ electron spin resonance (ESR) technique, indicating so-called direct 2e− reduction mechanism. With similar aim to extend light absorption region and effectively suppress the recombination of electron–hole pairs for photocatalytic H2O2 synthesis, Ag NPs have also been combined with g-C3N4 [91]. For example, Ag NPs have been loaded on ultrathin g-C3N4 nanosheets (U-g-C3N4-NS) by use of a post gas etching (PGE) technology. These Ag NPs that are uniformly deposited on the surface of single U-g-C3N4-NS layers (Ag@U-g-C3N4-NS) possess significantly stronger adsorption intensity in the 200–2000 nm range than the counterparts in their UV–vis diffuse reflectance spectrum (UV–DRS, Fig. 4a). It was suggested that such enhanced adsorption of Ag@U-g-C3N4-NS was attributed to the special structure of the U-g-C3N4-NS and the localized surface plasmon resonance (LSPR) effect from the Ag NPs. Effective separation of photogenerated carriers helped to adsorb light more efficiently, leading to their enhanced photocatalytic H2O2 activity. An optimal structure of the Ag@U-g-C3N4-NS-1.0 nanocomposite showed excellent photocatalytic activity of H2O2 production with a yield of 1.975 × 10–6 M min−1 under visible-light irradiation, even without any sacrificial carbon-containing organic electron donor (Fig. 4b).

a UV-DRS absorption spectra of different samples. b H2O2 concentration of different catalysts. Reproduced with permission from Ref. [91]. Copyright 2019 Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. c Proposed mechanism of different sites on all-solid-state Z-scheme heterojunctions for photocatalytic production of H2O2. d Concentration of generated H2O2 in NCN and PIx-NCN systems. Reproduced with permission from Ref. [97]. Copyright 2017 Elsevier
3.1.3 Constructing Heterojunction Composites
Using g-C3N4 photocatalysts that contain N and/or C vacancies to prepare H2O2, the organic reagents (such as methanol and ethanol) are frequently added. They usually act as hole scavengers to improve the yield, but contaminate the product of H2O2, and further increase the cost. Note that the potential of VB of g-C3N4 (+1.57 V) is negative than that of ·OH/OH− (+1.99 V) [92,93,94], the H2O2 synthesis is thus hard to be realized through photocatalytic H2O oxidation mechanism. Instead, a two-channel H2O2 generation approach (namely O2 reduction and hole oxidation OH−) on g-C3N4 is highly desirable. To overcome these issues, heterogeneous composites of g-C3N4 with other components were constructed. The separation of photogenerated carriers of these composites were enhanced by introduced electron traps, increased potential of VB and created additional active sites. On these composites, the H2O2 generation was achieved via the H2O oxidation pathway. More importantly, this strategy spatially isolates the oxidation and reduction reaction sites, bringing in minimized deactivation of catalytic ability of these photocatalysts. It has been reported that Z-scheme heterostructures with a wide range of light-trapping properties promoted the separation of photogenerated carriers but without the introduction of hole scavengers and thus rapidly improved the catalytic H2O2 production [95, 96]. For example, the combination g-C3N4 nanosheets (NCN) with perylene imides (PI) has been reported to form an all-solid-state Z-scheme heterojunction: a g-C3N4-based photocatalyst (PIx-NCN), where the excited electrons on the CB of PI are transferred to the VB of NCN, leading to retard recombination of photogenerated carriers (Fig. 4c) [97]. In this case, this PIx-NCN photocatalyst obviously enhanced the charge separation, resulting in an enhanced H2O2 yield where more electrons were found to take participate in producing H2O2 from O2 reduction. In addition, since the holes in the VB of PI moiety exhibited more positive potential (2.08 V) than that of NCN (1.63 V), the H2O2 generation was assumed to be realized through the direct 2e− WOR approach. In other words, the application of scavengers was avoided here. The optimal PI5.0-NCN catalyst showed high activity for the H2O2 production: the generation of about 120 μmol H2O2 in 120 min under visible-light irradiation (Fig. 4d). Meanwhile, other Z-scheme heterojunction catalysts (e.g., Cu2(OH)2CO3/g-C3N4 [98] and Bi4O5Br2/g-C3N4 [99]) have been employed for the two-channel H2O2 production under visible-light conditions, thanks to their suitable CB and VB band structures for realizing completed ORR and WOR pathways.
3.1.4 Polyoxometalate Hybridization
Polyoxometalates (POMs) are commonly constructed using a basic unit of oxo-metal polyhedra of MOx, in which a hole center (O−) and a trapped electron center (Mn+) (n = 5, 6) act as the electron acceptor and donor, respectively [100]. Once POMs are excited by visible light irradiation, they exhibit promising photocatalytic activities and stability. The reason behind is that the reduction of photogenerated-charge recombination is derived from the well-defined HOMO–LUMO band gaps. In this context, POMs have been widely applied in the field of photocatalysis, such as for water oxidation, alcohols oxidation, CO2 reduction and hydrogen evolution [101,102,103,104]. More interestingly, polyoxometalate hybridization strategy has been recently proposed, namely the formation of POM chemical bonds with g-C3N4, which was further utilized to improve the photocatalytic H2O2 synthesis. For instance, the POM cluster of [PW11O39]7− (PW11) has been covalently combined with three dimensionally ordered macroporous graphitic carbon nitride (3DOM g-C3N4), applying for the photocatalytic H2O2 synthesis (Fig. 5a) [105]. The hybrid catalyst of 3DOM g-C3N4-PW11 produced 3.5 μmol H2O2 in 60 min irradiated by λ ≥ 320 nm light, where no organic electron donor was needed (Fig. 5b). Such a H2O2 yield was superior to that of the pure 3DOM g-C3N4 catalyst (1.3 μmol), the K-PW11 catalyst (< 0.1 μmol) and the 3DOM g-C3N4-PW11-IMP catalyst (1.5 μmol). It was explained that the covalent bonding of PW11 clusters with 3DOM g-C3N4 optimizes the VB and the CB of 3DOM g-C3N4-PW11 when compared with 3DOM g-C3N4. Such optimal band structures facilitate both the 2e− WOR pathway and the 2e− ORR pathway for the H2O2 production, accompanying with the decreased separation of photogenerated holes and electrons.

a, d Schematic illustration of the synthetic route to 3DOM g-C3N4-PW11 and g-C3N4-PWO. b, e H2O2 production over different catalysts in 60 min. c, f Scheme of energy levels and charge transfer pathways of 3DOM g-C3N4 and 3DOM g-C3N4-PW11, g-C3N4 and g-C3N4-PWO. Reproduced with permission from Refs. [105, 107]. Copyright 2017 Elsevier and 2018 Elsevier
On the other hand, 3DOM g-C3N4-PW11 hybrid catalyst still has some defects. For instance, the number and reactivity of -NH2 groups of g-C3N4 need to be enhanced to further increase the covalent interaction between g-C3N4 and POMs. Moreover, the photoreduction ability of POMs needs to be improved to further enhance the selectivity of the 2e− ORR pathway. In this context, the surface of g-C3N4 has been modified by its covalent binding with another POM cluster, [SiW11O39]8− (SiW11) via thermal treatment of g-C3N4 mixed with urea [106]. The as-prepared g-C3N4 catalyst has a larger amount of more highly active-NH2 groups than 3DOM g-C3N4. Via an organic linker bride, the POM SiW11 cluster with more negative CB potential than PW11 was effectively covalently combined with g-C3N4. This g-C3N4-SiW11 greatly enhanced the adsorption and activation of O2, thereby promoting the two-electron reduction of O2 for the H2O2 production. It produced 17.8 μmol H2O2 in 60 min under simulated sunlight (AM 1.5) irradiation together with the selectivity as high as 80.1% in the presence of methanol. The high selectivity of O2 reduction to H2O2 was attributed to a positive shift of the CB level in g-C3N4-SiW11.
The hybrid catalysts of g-C3N4 with POM-derived metal oxides were prepared by the thermal decomposition of the mixture of g-C3N4 and POM precursors. These POM-derived metal oxides increase the H2O2 production by enhancing photoinduced generation of electrons. Using such a calcination method, the g-C3N4-PWO [107] and g-C3N4-CoWO [108] have been prepared. Take g-C3N4-PWO as an example (Fig. 5d), it showed a H2O2 yield of 6.3 μmol in 60 min under visible light but in the absence of organic electron donor, outperforming that of the counterparts (Fig. 5e). This is because the incorporation of PWO into the g-C3N4 framework results in the negative shift of the CB edge of g-C3N4. The H2O2 production was achieved formed a two-step single-electron O2 reduction reaction routine, namely from 1e− reduction pathway (from O2 to ·O2−), and sequentially followed by another 1e− reduction pathway (from ·O2− to H2O2) (Fig. 5f).
3.1.5 Metal/Non-metal Element Doping
Doping of metal/non-metal element into g-C3N4 modulates the bandgap of g-C3N4 and further alters the transfer directions of charge carriers, thereby adjusting electronic, optical and other physical properties of g-C3N4. Moreover, the heteroelements on g-C3N4 change the reaction pathways of the H2O2 generation. Therefore, doping of metal/non-metal element into g-C3N4 has been proposed as a potential strategy to improve the performance of photocatalytic H2O2 generation. In a typical case, Cu-doped g-C3N4 microspheres, denoted as Cu-SCN, have been synthesized using a modified template method (Fig. 6a) [109]. The Cu species were inserted into the interstitial position of SCN with a special mesoporous structure, resulting in the formation of the coordinated Cu–N bonds. The Cu-doping not only decreased the bandgap of g-C3N4 to enhance its capture of visible light, but also retarded the recombination of photogenerated carriers, as testified from lower PL intensity of Cu(2)-SCN than that of GCN. In addition, Cu(2)-SCN showed a weaker PL intensity in O2 atmosphere when compared with that in Ar (Fig. 6b). The performance of photocatalytic H2O2 production using the Cu-doped g-C3N4 photocatalyst was thus superior to that of the g-C3N4 photocatalyst. From the X-ray photoelectron spectroscopy (XPS) of Cu-doped g-C3N4 photocatalyst, Cu+ was observed in its XPS spectrum of Cu 2p. Such Cu(I)-N site thus served as the adsorption center for molecular O2 to increase the O2 adsorption. Moreover, it changes the transfer pathways of photoelectrons from the catalyst to the adsorbed O2, altering in a two-step single-electron reaction pathway to a one-step two-electron reduction process. In the other study, K+ and Na+ ions have been co-doped into g-C3N4 through a molten salt method [110]. The incorporation of alkali metals in g-C3N4 tuned the band gap of g-C3N4, increased its specific surface area and reduced the size of g-C3N4 layered structure. Therefore, this alkali metals co-doped g-C3N4 photocatalyst can enhance the visible-light adsorption and promote the separation of photoelectron-hole pairs to boost photocatalytic performance toward H2O2 production. In more detail, the VB potential of GCN(20) is +2.05 V, more positive than that (+1.99 V) of ·OH/OH−. This was derived from alkali metal incorporation. In this context, the reaction process of photocatalytic H2O2 generation was changed from a single pathway (O2 → H2O2) to a dual channel pathway (O2 → H2O2 and OH− → ·OH → H2O2), a very attractive method for practical solar-to-chemical applications (Fig. 6c). The MCN(20) produced the H2O2 concentration of 4.6 mmol L−1, more than 9 times higher than that produced by GCN (Fig. 6d). Furthermore, a crystalline g-C3N4 modified with K+, Ni and N-doped carbon (KNiCN) was fabricated to overcome the inherent structural defects and low carrier separation efficiency [111]. The synergistic effects between K+, Ni species and N-doped carbon increased the light adsorption, improved charge separation and boosted O2 adsorption and selectivity for the H2O2 generation. A two-electron reduction pathway was suggested (Fig. 6e). This photocatalyst displayed a photocatalytic H2O2 yield of as high as 79.6 μM in O2-saturated pure water (Fig. 6f).

a Elemental mapping images of Cu(2)-SCN. b PL intensities of GCN and Cu(2)-SCN. Reproduced with permission from Ref. [109]. Copyright 2017 Elsevier. c Band gap structures of various catalysts. d H2O2 concentration over catalysts under visible light. Reproduced with permission from Ref. [110]. Copyright 2018 Elsevier. e Proposed mechanism of H2O2 production over the KNiCN catalyst. f Photocatalytic H2O2 production on the catalysts in pure water. Reproduced with permission from Ref. [111]. Copyright 2021 Royal Society of Chemistry
In addition to metal doping of g-C3N4, halogen doping, another classic non-metal doping has been used to synthesize photocatalysts for photocatalytic H2O2 production. In these cases, the incorporation of halogen into g-C3N4 not only reduces the bandgap (to enhance the visible-light absorption and improve the electrical conductivity), but also boost charge carrier transmission rates (without damaging of main structures of the g-C3N4 polymer). These features benefit much photocatalytic H2O2 synthesis [112, 113]. For example, halogen doped g-C3N4, prepared by means of a hydrothermal method in saturated NH4X (X = Cl, Br) solution followed with a post-treatment, has more negative CB potential than g-C3N4 [114]. This catalyst is thus beneficial for the O2 reduction into H2O2. For example, a Br-doped g-C3N4 photocatalyst showed higher photocatalytic H2O2 production capacity (1.99 mmol L−1) in comparison with the counterparts. The incorporation of multiple heteroelements into the g-C3N4 framework with various configurations was also demonstrated to tune the bandgap of g-C3N4 [115]. One co-doped g-C3N4 photocatalyst exhibited an outstanding photocatalytic activity for the H2O2 generation, which is 17–25 times higher than that of bare g-C3N4.
3.2 TiO2 Photocatalysts
TiO2 is the most famous photocatalyst since the discovery of its photocatalytic activity by Fujishima and Honda for the first time in 1972 [128]. It possesses many advantages for photocatalytic reactions, such as appropriate site for the CB, good physical and electrical properties, stable chemical structures and outstanding biocompatibility [129]. The potential of the CB of TiO2 (\(E^{\theta }\)) is − 0.5 VNHE, which is negative enough to drive a 2e− ORR for the H2O2 production [16]. The generated photoelectrons in their CBs reduce O2 to form H2O2, following the reaction of O2 + 2 H+ + 2 e− → H2O2. While these simultaneously generated holes in their VBs oxidize water to generate O2 and H+, following the process of H2O + 2 h+ → 1/2 O2 + 2 H+. The free Gibbs Energy (ΔG) of such a photocatalytic process is 117 kJ mol−1. An uphill reaction indicates thermodynamical unfavorablity [13, 120]. TiO2 photocatalysts also have been confirmed to feature low cost, low toxicity, high chemical- and photo-stability during the H2O2 production [130,131,132].
However, the concentration of H2O2 produced by pure TiO2 photocatalyst has been only confined to the micromolar level (< 0.2 mM) [133]. This is because TiO2 photocatalyst can be excited only by UV light irradiation to generate photocarriers, due to the large band gap of TiO2 photocatalyst [47]. Besides, the formed H2O2 on the TiO2 surface is not stable enough and is easy to be converted to Ti-OOH complexes when it is in touch with the Ti–OH groups on the TiO2 surface. Subsequently, Ti-OOH is decomposed, following the reduction reaction of Ti-OOH + H+ + e− → Ti–OH + OH− [134]. In order to address the drawbacks of these TiO2 photocatalysts, surface modification strategies on TiO2 photocatalysts have been validated and applied to facilitate the photocatalytic H2O2 production, covering loading precious metal nanoparticles, modifying graphene quantum dots and complexing with cation and anion surfaces (Table 2).
3.2.1 Loading Precious Metal Nanoparticles
The NPs from individual precious metals have been loaded on the TiO2 surface to enhance the photocatalytic activity of TiO2 photocatalysts. In 2010, the synthesis of the Au/TiO2 photocatalyst has been reported using a deposition–precipitation (DP) method [133]. The as-obtained Au/TiO2 photocatalyst exhibited a high yield in the photocatalytic H2O2 production. The reported H2O2 concentration reached 10 mM under UV-irradiation. The mechanism of the H2O2 formation on this Au/TiO2 photocatalyst was explained as following (Fig. 7a). The oxidation and reduction sites on the Au/TiO2 photocatalyst are separated to TiO2 and Au NPs, respectively. The activity for photocatalytic H2O2 formation can be adjusted by kf (the formation rate constant) and kd (the decomposition rate constant). In another work, Au NPs with a bimodal size distribution were loaded on rutile TiO2 (BM-Au/TiO2), which was further modified with carbonates to obtain BM-Au/TiO2-CO32− (Fig. 7b) [135]. The UV/Vis absorption spectra of BM-Au/TiO2-CO32− indicated that the loading of Au NPs results in strong and extensive absorption in visible light region because of the LSPR of Au NPs. This BM-Au/TiO2 photocatalyst yielded 640 ± 60 μM H2O2 under visible-light irradiation for 1 h when 4% HCOOH was presented as the sacrificial agent. Such a yield was much higher than that of single small Au/TiO2 photocatalyst (50 μM) and single large Au/TiO2 photocatalyst (75 μM) (Fig. 7c). The improvement on the H2O2 yield on the BM-Au/TiO2-CO32− was believed to be mainly originated from the occurred long-range charge separation by the visible-light-induced, vectorial interfacial electron transfer in the way of S–Au → CB-(TiO2) → L-Au. It is known that the generated H2O2 on a pure TiO2 photocatalyst is easily degraded since it can be reduced by the peroxide species (Tis-OOH) on the TiO2 surface. However, the modification of CO32− onto the TiO2 surface efficiently inhibits such reductive degradation of H2O2. The concentration of generated H2O2 on this BM-Au/TiO2-CO32− thus reached about 1 mM. The mechanism of the H2O2 generation on the BM-Au/TiO2-CO32− photocatalyst includes plasmonic effect and photocatalytic O2 reduction (Fig. 7d). It has been further reported that the photocatalytic kinetics for the H2O2 formation is dependent on temperature and pH value for the deposition of Au NPs on anatase TiO2 when a heating temperature-varied deposition–precipitation technique was applied [136]. The amount of generated H2O2 under the irradiation was increased with a decrease of the temperature and pH in the reaction system. The highest H2O2 concentration produced on the Au/anatase TiO2 photocatalyst was about 17 mM at the temperature of 5 °C in the solution with a pH value of 2. In conclusion, these introduced metal Au NPs on the TiO2 surface prohibit the H2O2 decomposition that is induced by the Ti–OH species. Meanwhile, they capture photogenerated electrons in the CB of TiO2, leading to retarded recombination of photogenerated carriers. The output of the H2O2 production on these photocatalysts was obviously increased. The H2O2 concentration even reached the millimolar level. Nevertheless, the generated H2O2 molecules are inclined to be adsorbed on Au NPs due to their strong interactions. Subsequently, spontaneous decomposition of H2O2 occurs: H2O2 + e− → ·OH + OH− [137, 138].

a Mechanism of the H2O2 formation over the Au/TiO2 photocatalyst. Reproduced with permission from Ref. [133]. Copyright 2010 American Chemical Society. b TEM image of the BM-Au/TiO2 photocatalyst. c Photocatalytic H2O2 production on various photocatalysts. d Proposed mechanism of the BM-Au/TiO2-CO32− plasmonic catalyst in the photocatalytic H2O2 generation. Reproduced with permission from Ref. [135]. Copyright 2016 Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. e HRTEM image of the Au0.1Ag0.4/TiO2 photocatalyst. f Schematic energy-band and mechanism of the H2O2 production on the AuAg/TiO2 photocatalyst. Reproduced with permission from Ref. [139]. Copyright 2012 American Chemical Society
To promote the H2O2 formation and simultaneously inhibit its decomposition on the TiO2 photocatalysts, the NPs from bimetal alloys have been deposited on the TiO2 surface to enhance the photocatalytic activity. For example, a bimetal alloy catalyst (AuAg/TiO2) has been designed and applied for photocatalytic H2O2 generation (Fig. 7e) [139]. The H2O2 concentration produced by the Au0.1Ag0.4/TiO2 photocatalyst was as high as 3.4 mM, approximately 7 times higher than that produced on a pure TiO2 photocatalyst. This excellent photocatalytic performance has been attributed to the following two aspects (Fig. 7f). Firstly, the work function of the alloy AuAg is located between Au and Ag. The alloy/TiO2 photocatalyst produces a barrier, of which energy is larger than that of the Ag/TiO2 photocatalyst but smaller than that of Au/TiO2 photocatalyst. Effective separation of hole-electron pairs is thus possible. Secondly, electron transfer from Ag to Au takes place, because Au atoms have a higher electronegativity than Ag atoms. In other words, the electron density of Au is increased, suppressing the H2O2 adsorption on Au atoms. Such statements were further proved using the DFT simulations. The affinity between the H2O2 and Au atoms was confirmed to be weakened via the alloy effect. It was taken place that H2O2 is inclined to be adsorbed on Ag atoms, thus inhibiting the H2O2 decomposition. In another case, the midgap state in the Ag2Au2@TiO2 (101) photocatalyst was reveal to own a suitable position for the H2O2 production via a photocatalytic reaction, as demonstrated by the DFT analysis. More importantly, the photoresponse of these bimetal alloy photocatalytic systems is rapidly promoted in visible and infrared light region, improving the H2O2 yield [140].
3.2.2 Modifying Graphene Quantum Dots
Graphene quantum dots (GQDs) feature advantages of high chemical stability, good biocompatibility, large surface area and high extinction efficiencies (that are derived from the atomically thin sp2 carbon structure of graphene). GQDs exhibit unique luminescent characteristic. Therefore, GQDs have attracted extensive attention in various photocatalytic applications. Especially, the sensitization of GQDs with other wide band gap photocatalysts (e.g., TiO2) can extend light-adsorption range from ultraviolet to the visible region [141, 142]. For example, sulfur and nitrogen co-doped graphene quantum dots (SN-GQDs) have been combined with the TiO2 photocatalyst (SN-GQD/TiO2) and further applied for photocatalytic H2O2 production [143]. As confirmed from related UV–vis diffuse reflectance spectra (DRS), SN-GQD/TiO2 exhibited obviously enhanced adsorption in visible light region when compared with the pure TiO2 photocatalyst. On its surface, more than 82.8 μM H2O2 was produced under visible light irradiation only for 90 min. Such a yield was about 5.3 and 3.1 times larger than that generated on the GQD/TiO2 photocatalyst and the N-GQD/TiO2 suspensions under the same conditions, respectively (Fig. 8a). Theoretical calculation and free energy diagram analysis showed that the H2O2 generation on the SN-GQD/TiO2 photocatalyst followed a proton-coupled electron transfer (PCET) mechanism. Namely, high selectivity of the H2O2 generation on the SN-GQD/TiO2 photocatalyst was realized via a 2e− ORR pathway (Fig. 8b, c). A Nafion layer was further introduced into the SN-GQD/TiO2 (Nf-SNG/TiO2) to facilitate the H2O2 generation [144]. The GQDs coupled with a Nafion layer were found not only to improve the visible light adsorption, but also to significantly hinder the H2O2 decomposition (Fig. 8d). The yield of photocatalytic H2O2 production on the Nf-SNG/TiO2 photocatalyst with 3.5% Nafion content was 141 μM after 120 min visible light irradiation than that obtained on the counterparts (Fig. 8e). Related photocatalytic reaction mechanism of the H2O2 formation on Nf-SNG/TiO2 was testified to follow a two-electron-dominated ORR pathway (Fig. 8f). It was impacted by GQDs sensitization, different from photocatalytic H2O2 production on the pure TiO2 photocatalyst.

a Photocatalytic H2O2 generation on the catalysts. b Proposed mechanism of photocatalytic H2O2 formation over the SN-GQD/TiO2 photocatalyst. c Free energy diagrams of reaction pathways. Reproduced with permission from Ref. [143]. Copyright 2018 Elsevier. d Photocatalytic decomposition and e production of H2O2 on different catalysts under visible light. f Photocatalytic reaction mechanism for H2O2 formation on the Nf-SNG/TiO2 photocatalyst. Reproduced with permission from Ref. [144]. Copyright 2019 American Chemical Society
3.2.3 Complexing with Cation and Anion Surfaces
The surface states are important during photocatalytic H2O2 generation, since photocatalytic reactions are surface-controlled processes. Therefore, various surface modification approaches have been developed to modulate photocatalytic activities of TiO2 photocatalysts. Among them, complexing cations or anions onto the TiO2 surface has been wildly accepted as the simplest one to effectively boost the photocatalytic H2O2 production. For example, the inner spherical surface of the metal cations can modulate interfacial electron transfer via suppressing the surface trapping sites for photogenerated carriers during the photocatalytic process. Taking the modification of TiO2 by Cu2+ ion as an example, it has been revealed that complexing with a small amount of Cu2+ onto the TiO2 surface significantly promoted the photocatalytic activity of H2O2 production [145]. A 20-fold increment in the amount of generated H2O2 was achieved under UV irradiation in comparison with that on the TiO2 photocatalyst without Cu2+ modification. When the concentration of Cu2+ was in the range of 30–40 μM, the performance for photocatalytic H2O2 generation remained stable. Such enhanced activity of photocatalytic H2O2 production was proposed to be dependent on the optimization of the TiO2 surface state through Cu2+ modification, eventually promoting the H2O2 formation on the TiO2 surface via a 2e− ORR pathway. In another case, Zn2+ ions were complexed onto the TiO2 photocatalyst [146]. This Zn2+ modified TiO2 photocatalyst exhibited obviously improved performance of photocatalytic H2O2 generation. Since Zn2+ ions block the ≡Ti–OH sites, the complexation of peroxide/superoxide species on the TiO2 surface is thus much limited, resulting in reduced H2O2 decomposition.
In addition to cations, complexing with anions onto the TiO2 photocatalyst has been also applied for the photocatalytic H2O2 production. For instance, the fluorinated TiO2 photocatalyst exhibited a boosted reaction toward photocatalytic H2O2 generation. Stemming from surface state modulation via F− modification, this fluorinated TiO2 photocatalyst suppresses the Ti-OOH formation. It produced H2O2 with a concentration of 1.3 mM under UV light irradiation, one of the best activities among the reported TiO2 photocatalysts for the H2O2 generation [147].
3.3 BiVO4 Photocatalysts
In addition to TiO2 photocatalysts, some more complex inorganic oxides have also been employed for photocatalytic H2O2 generation. Among them, bismuth vanadate (BiVO4) is one classic representative of bimetallic oxide photocatalyst [156,157,158,159]. Again, it is well known a pure TiO2 photocatalyst has a wide bandgap and thus can be excited only under UV light irradiation, resulting in insufficient activity for photocatalytic H2O2 generation in sunlight [160, 161]. Distinguished from a TiO2 photocatalyst, BiVO4 has its appropriate band structure and is thus active in the visible light region. In other words, it can promote the H2O2 production via a 2e− ORR pathway under the visible-light irradiation [162]. Unfortunately, pure BiVO4 is not conducive to the efficient photocatalytic H2O2 production, due to its lack of active sites for the 2e− ORR. In this context, nano-Au cocatalyst was introduced onto the BiVO4 surface. Stemming from the strong interaction between Au and BiVO4, the activation of the special d-band electrons promoted the selectivity of the 2e− ORR pathway to efficiently generate H2O2 under visible light irradiation (Fig. 9a) [163]. This is because of the edge in the CB of BiVO4 was disclosed to be 0.02 V, more positive than that (−0.13 V) of one-electron reduction of O2 and more negative than that (0.68 V) of two-electron reduction of O2 (Fig. 9b). The inhibition of single electron reduction over the Au/BiVO4 surface was further identified by electron spin resonance (ESR) spectra, where Au0.2/BiVO4 exhibits almost no signal, while Au0.2/TiO2 displays distinctive signals (Fig. 9c). The latter was ascribed to the DMPO − ·OOH spin adduct formation.

a TEM image of an Au0.2/BiVO4 photocatalyst. b Energy diagram of the Au/BiVO4 photocatalyst and reduction potential of O2. c ESR spectra of an Au0.2/BiVO4 photocatalyst and an Au0.2/TiO2 photocatalyst in an EtOH/water/O2 system with DMPO. Reproduced with permission from Ref. [163]. Copyright 2016 American Chemical Society. d Energy diagram of a Cu@Au/BiVO4 photocatalyst. e Mechanism of H2O2 generation on a Cu@Au/BiVO4 photocatalyst. Reproduced with permission from Ref. [166]. Copyright 2021 American Chemical Society. f Energy diagram and the reaction mechanism of a Cu(hfacac)2/ms-BiVO4 photocatalyst. Reproduced with permission from Ref. [167]. Copyright 2020 American Chemical Society
Nevertheless, the activity of the BiVO4 photocatalyst toward photocatalytic H2O2 production still remains unsatisfactory. This is mainly attributed to the formed built-in field between Au and BiVO4, which inhibits the transfer of photogenerated electrons as well as accumulates negative charges of Au to deteriorate the 2e− ORR pathway [164, 165]. In order to overcome this impediment, one Cu@Au/BiVO4 photocatalyst was designed and obtained using combined photodeposition and galvanic displacement methods [166]. The Cu species was found to facilitate the transfer of photogenerated electrons from BiVO4 to Au. The accumulation of negative charges on Au was then reduced, resulting in the enhanced activity for the photocatalytic H2O2 production (Fig. 9d, e). Specifically, the ohmic contact was expected to be produced between Cu and BiVO4 since their work functions (Φ) are different: \({\Phi }_{Cu}\)= 4.7 eV and \({\Phi }_{{BiVO}_{4}}\)= 4.9 eV. Transferring of the photogenerated electrons is thus beneficial. The photogenerated electrons that are transferred from Cu to Au eventually boost the two-electron O2 reduction pathway, namely the H2O2 formation. On the other hand, photogenerated charges have been accumulated on the nano-Au cocatalyst in the Cu@Au/BiVO4 photocatalyst, leading to the generation of stronger adsorption of O2 and HOO* on the Au surface of Cu@Au/BiVO4. Consequently, O2 reduction to form H2O2 is accelerated. For a bis(hexafluoroacetylacetonato) Cu(II) adsorbed monoclinic scheelite (ms)-BiVO4 photocatalyst (Cu-(hfacac)2/ms-BiVO4), it exhibited an outstanding activity of photocatalytic H2O2 generation [167]. An external quantum yield of 0.47% was reported under visible light irradiation (λex = 470 nm). Such performance was attributed to the enhanced charge separation by the interfacial electron transfer from ms-BiVO4 to the surface complex and the O2-enriching effect near the surface of ms-BiVO4, as well as outstanding electrocatalysis for a 2e− ORR pathway (Fig. 9f).
3.4 CdS Photocatalysts
CdS has exhibited promising photocatalytic activities in various reactions under visible light irradiation, since the potential of its VB is positive enough to drive water oxidation and meanwhile the potential of its CB is more negative to promote O2 reduction. CdS thus has great potential to be applied as a catalyst for photocatalytic H2O2 production [168,169,170]. Unfortunately, CdS exhibits relatively low photocatalytic capability toward H2O2 generation, originating from its weak adsorption capacity for reactants and its poor photostability. More seriously, its easy aggregation can cause the severe recombination of photogenerated carriers [171].
To improve the performance of CdS for photocatalytic H2O2 production, many efforts have been made and several strategies have been proposed. One strategy is to complex organic polymers onto CdS. For example, the hybrids of CdS and reduced graphene oxide (CdS-RGO) prepared through a hydrothermal process significantly boosted the kinetics of photocatalytic H2O2 generation when compared with pure CdS photocatalyst (Fig. 10a) [172]. The CdS-RGO photocatalyst with a 20 wt% RGO content (CdS-G2) produced the H2O2 concentration of as high as 128 μM under sunlight irradiation for 12 h. Such enhancement was mainly attributed to accelerated separation of photogenerated carriers due to the enhanced electron transfer from the photoexcited CdS to RGO, the increment of visible light absorption and more active reaction sites (as demonstrated by their PL spectra (Fig. 10b)). The mechanism of photocatalytic H2O2 production on the CdS-RGO photocatalyst was also discussed (Fig. 10c). In the first step, electron–hole pairs are generated on the CdS-RGO photocatalyst upon its excitation by light irradiation. Water molecules are then oxidized by photogenerated holes, leading to the production of H2O2 and protons. Simultaneously, O2 is reduced to form H2O2, following a 2e− ORR pathway (step 1–4). These photogenerated electrons have been demonstrated to play a vital role in the process of photocatalytic H2O2 generation. The H2O2 yield from a 2e− ORR pathway is the decisive factor when compared with that from the 2e− WOR pathway in the whole photocatalytic process.

a H2O2 concentration produced on different catalysts. b PL spectra of the CdS and CdS-G2 photocatalysts. c Mechanism for H2O2 production on the CdS-G hybrid photocatalyst under sunlight illumination. Reproduced with permission from Ref. [172]. Copyright 2016 Elsevier. Schematic illustration of H2O2 formation over a CdS photocatalyst and a CdS@S-doped carbon photocatalyst. e H2O2 concentration for 24 h with 20% of 2-propanol. Reproduced with permission from Ref. [174]. Copyright 2020 Elsevier
Noble metals have also been complexed onto CdS to improve the photocatalytic performance of CdS catalysts. For example, the CdS-Pt and CdS-Au nanorods exhibited enhanced photocatalytic activity to produce H2O2 when compared with a pure CdS photocatalyst [173]. The Au tip with a smaller size was found to be more efficient as reactive site to form H2O2 than a Pt tip. This was attributed to different surface reactivity and selectivity related to the metal tip composition. On these CdS-metal photocatalysts, the H2O2 production through ORR pathway has been confirmed, instead of the WOR pathway. A CdS/sulfur-doped carbon nanocomposite, synthesized by thermal annealing of Cd(EDDA) MOF, exhibited outstanding activity toward photocatalytic H2O2 production [174]. This is due to the presence of sulfur-doped carbon, which can effectively hinder the H2O2 decomposition, as experimentally and theoretically testified (Fig. 10d). The H2O2 concentration generated on a nanocomposite based photocatalyst (M-CdS-10 h) reached 17.1 mM under visible light irradiation when 2-propanol was used as a sacrificial agent (Fig. 10e).
3.5 Metal–organic Frameworks Photocatalysts
Metal–organic frameworks (MOFs) own unique porous structures and strong metal–ligand interactions [175]. Similar to semiconductors, MOFs can produce electron–hole pairs upon light irradiation. They thus exhibit great potential in photocatalysis [176,177,178,179]. The Ni/MIL-125-NH2, obtained via depositing NiO on the MIL-125-NH2 was the first MOF photocatalyst used in the photocatalytic H2O2 synthesis [180]. The mechanism of the H2O2 production on the Ni/MIL-125-NH2 (Fig. 11a) was described as following. Nano NiO effectively inhibits the H2O2 decomposition through a 2e− ORR pathway under visible light irradiation when benzylalcohol is accompanied as an electron donor. Specifically, MIL-125-NH2 produces Ti8O8(OH)4·− and a hole upon its excitation by light irradiation. Once the hole is trapped by triethanolamine (TEOA), O2 is reduced to O2·− by Ti8O8(OH)4·−, followed by rapid H2O2 formation via the O2·− disproportionation reaction in the presence of NiO. Unfortunately, in this case the product was a mixture of H2O2 and benzaldehyde dissolved in acetonitrile. In other words, further energy-consumption for product separation and purification is required. Later, a two-phase system containing benzylalcohol/water (BA/water), was employed to separate these products [181]. The hydrophilic MIL-125-NH2 was conversed to hydrophobic MIL-125-Rn (n = 1, 4 and 7) through the growth of the alkyl chains on the catalyst surface, as clarified using water adsorption isotherms and water contact angles measurements (Fig. 11b). A pristine MIL-125-NH2 was proved to exist in the water phase during the photocatalytic H2O2 process, while alkylated MIL-125-Rn is located in the BA phase (Fig. 11c). During the photocatalytic H2O2 process (Fig. 11d), O2 is firstly reduced to O2·− on the hydrophobic MOFs in the BA phase. Subsequently, O2·− is transferred to the water phase where H2O2 is rapidly produced through a disproportionation reaction in the presence of H+ or Na+. Although this two-phase system inhibited the further reaction of MOFs with H2O2, its photocatalytic activity of the two-phase system was not satisfying. This is because the grafting of the alkyl chains blocks the pores of the MIL-125-R7, thus greatly reducing its photocatalytic activity. In this regard, a hydrophobic MOF, namely OPA/MIL-125-NH2, was developed. In this case, MIL-125-NH2 retained most of its pores using octadecylphosphonic acid (OPA) treatment [182]. Its surface area was 1242 m2 g−1, comparable to that (1500 m2 g−1) of MIL-125-NH2. Under visible light irradiation for 3 h, the concentration of generated H2O2 on this OPA/MIL-125-NH2 photocatalyst was approximately 3 times larger than that of MIL-125-R7. The enhanced activity was attributed to the rapid diffusion of O2·− through the unblocked pores of the OPA/MIL-125-NH2 photocatalyst, which prevents the H2O2 decomposition (Fig. 11e). A hydrophobic titanium doped zirconium-based MOF (OPA/Zr100-xTix-MOF) also exhibited a high rate of H2O2 production (9.7 mmol L−1 h−1), where the Ti species played a role in effectively promoting electron transfer from photoexcited linkers of the MOF to Ti, inhibiting the recombination of photogenerated electron–hole pairs in the hydrophobic MOF matrix (Fig. 11f) [183].

a Photocatalytic mechanism of H2O2 formation on the MIL-125-NH2 photocatalyst. Reproduced with permission from Ref. [180]. Copyright 2018 ROYAL SOCIETY OF CHEMISTRY. b Water adsorption isotherms for various catalysts. c Digital photographs utilizing the two-phase system. d Mechanism illustration of H2O2 generation by the two-phase system. Reproduced with permission from Ref. [181]. Copyright 2019 Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. e Photocatalytic processes of MIL-125-R7 and OPA/MIL-125-NH2 photocatalysts. Reproduced with permission from Ref. [182]. Copyright 2019 ROYAL SOCIETY OF CHEMISTRY. f Mechanism of photocatalytic H2O2 production using the Zr100-MOF and OPA/Zr92.5Ti7.5-MOF photocatalyst. Reproduced with permission from Ref. [183]. Copyright 2020 ROYAL SOCIETY OF CHEMISTRY. g Photocatalytic O2 reduction to H2O2 over a NH2-MIL-125@ZnS heterojunction. Reproduced with permission from Ref. [187]. Copyright 2021 Wiley–VCH GmbH
The construction of the heterojunction by combining MOFs with other semiconductors also effectively strengthens H2O2 production since the separation of photogenerated electron–hole pairs can be promoted [184,185,186]. For example, on the MOF@MS heterostructure (NH2-MIL-125@ZnS) the photocatalytic synthesis of H2O2 was reported via a 2e− ORR pathway (Fig. 11g) [187]. This heterostructure presents a spatially separated architecture, where ZnS nanocages are selectively adhered on the four corners of a cake-like NH2-MIL-125 body. The coverage of heterojunction was controlled by altering the diameter of ZIF-8/ZnS. In this way, the regulation of the absorption of light, the generation of electron–hole pairs, the charge separation and accessibility were doable, finally leading to adjusted performance of photocatalytic H2O2 production. During the photocatalytic process, photogenerated electrons transfer from ZnS to NH2-MIL-125, while holes migrate from NH2-MIL-125 to ZnS, resulting in excellent performance. When the heterojunction coverage is ≈45.1%, the NH2-MIL-125@ZnS photocatalyst displays a H2O2 yield of as high as 120 mM g−1 h−1.
4 Electrocatalytic H2O2 Synthesis
4.1 Electrocatalytic H2O2 Synthesis through a 2e− ORR Pathway
2e− ORR has been considered as one of the most vital routes in electrocatalytic H2O2 synthesis [188, 189]. However, its selectivity is limited by the competition from the 4e− reduction pathway, namely the reduction of O2 to generate H2O. The fact behind is that O2 has a more negative standard potential to form H2O than that for the H2O2 generation [52, 190, 191]. Further electro-reduction of H2O2 to H2O is thus thermodynamically favorable, which severely hinders the yield of H2O2 from a 2e− ORR pathway. In addition, the spontaneous H2O2 disproportionation to produce H2O and O2 prohibits the 2e− ORR pathway, lowering the selectivity of H2O2 production. Therefore, it is prerequisite to design electrocatalysts to promote the preferential H2O2 formation as well as to facilitate rapid diffusion of H2O2 away from the reactive interface. The reported electrocatalysts using the 2e− ORR pathway for the H2O2 production can be divided into two categories: noble-metal-based and carbon-based electrocatalysts. In the following session, these reported electrocatalysts are systematically classified and their performance toward the H2O2 is detailed (Table 3).
4.1.1 Noble Metal-based Electrocatalysts
According to Sabatier’s principle, the binding energy between an OOH* radical and an electrocatalyst should be neither too strong nor too weak for efficient H2O2 production, since an OOH* radical is the only intermediate in the process of a 2e− ORR pathway [41]. Meanwhile, the integrity of an O–O bond must be well maintained during the conversion process since the splitting of O–O bond is conductive to form water. It has to highlight here that the manners of oxygen adsorption on a catalyst mainly follow three modes (Fig. 12a), namely Pauling model (“end-on”), Griffiths model and Bridge model (“side-on”) [55, 192]. Only Pauling model is beneficial for the H2O2 generation via two-electron reduction of O2. This is due to its low influence on the O–O bond splitting. Differently, other two models induce the O–O bond splitting and facilitate the 4e− ORR pathway. Note that it is more thermodynamically inclined to the pathway of the 4e− ORR to form H2O in comparison with H2O2 generation via the 2e− ORR pathway. To address this issue, the key is to design and synthesize advanced catalysts with high activity for O2 reduction, while well remaining O–O bond during the 2e− ORR process. In the next section, the electrocatalysts on the selective reduction of O2 to H2O2 are reviewed in combination with some typical examples.

a Three typical modes for O2 adsorption. Reproduced with permission from Ref. [192]. Copyright 2021 Elsevier. b Sabatier volcano plots for electrochemical ORR for pure metal slabs and M–N/C. Reproduced with permission from Ref. [193]. Copyright 2018 American Chemical Society. c Proposed ORR pathways on the pristine Pt/C and carbon-coated Pt electrocatalysts. Reproduced with permission from Ref. [200]. Copyright 2014 American Chemical Society. d H2O2 selectivity of the Au1‑xPdx electrocatalyst with different palladium contents at potentials of 0, -0.1 and -0.2 V. Reproduced with permission from Ref. [208]. Copyright 2011 American Chemical Society
Single noble metal materials have been applied for electrocatalytic H2O2 synthesis. For example, DFT calculations have been firstly used to reveal the relationship between the ORR activity and various metallic catalysts. The Sabatier volcano plots were then established (Fig. 12b), namely the electrocatalytic ORR activity of different metals and metal-nitrogen/carbon (M–N/C) [193]. Taking the volcano curves of single metals (the left-hand side of the volcano diagram) as the example, the limiting potential of a 4e− ORR process is more positive than that of a 2e− ORR process, thermodynamically indicating that the 4e− reduction reaction is more likely to occur to obtain H2O than the 2e− reduction reaction to generate H2O2. While on the right-hand side of the volcano plots, the limiting potential of 2e− and 4e− ORR processes are located at the same position, which illustrates that they simultaneously take place to produce H2O2 and H2O, respectively. In more detail, as the cases that the strong interaction occurs between metals and O2 molecules, the selectivity of 2e− ORR can be improved once the metal surface is modified, or its structure is regulated. Taking Pt metal as an example, O2 is adsorbed on the Pt surface mainly through a “side-on” mechanism, instead of the “end-on” mechanism. The H2O2 production on Pt catalysts is thus not conducive to be promoted [194]. In literature, the effects of the size and morphology of metal catalysts have been extensively investigated to explore the activity for electrocatalytic H2O2 production, namely via altering the O2 adsorption mode on the Pt electrocatalysts [195,196,197]. Note that the choice of suitable supports also modulates the selectivity of the Pt electrocatalysts toward electrocatalytic H2O2 production [198, 199]. For example, an amorphous carbon layer coated Pt catalyst was prepared by use of a chemical vapor deposition method with acetylene as the precursor [200]. This amorphous carbon layer presumably produces steric hindrance on the Pt surface, which induces O2 to be adsorbed through the end-on configuration, instead of side on configuration over the pristine Pt/C electrocatalyst (Fig. 12c). The 2e− ORR pathway happened dominantly on the C(Pt)/C-3 h and C(Pt)/C-4 h electrocatalysts. Their H2O2 selectivity reached 41% at a potential of 0.1 VRHE. Similarly, sulfur-doped carbon using zeolite-templated was used to support catalyst, where this carbon material supported and dispersed 5 wt% Pt atoms [197]. This Pt atomic electrocatalyst followed a 2e− ORR pathway. Its H2O2 selectivity was as high as 96%. Modified Pd species have been also designed and obtained to assess selective ORR [201, 202]. For instance, using a nano-Pd electrocatalyst, obtained through in-situ electrochemical deposition from Pd ions, high selectivity for the 2e− ORR pathway to form H2O2 was achieved. Even when Pd is derived from the lowest precursor concentration, related nano-Pd electrocatalyst exhibited high selectivity, which was surpassed 95% [203].
In contrast, the noble metals (e.g., Au) that possess weak adsorption capability toward O2 molecules have been applied to reduce O2 for efficient, selective and stable H2O2 production [204,205,206]. Unfortunately, the adsorption of O2 molecules on the crystal plane of noble metals is different. Namely, the ORR pathway on different metal facets is different. For example, the too weak bond between an Au (111) surface and O2 molecules makes it difficult to form the OOH* intermediate [207]. The H2O2 production on the Au (111) surface via the 2e− ORR pathway exhibits a low efficiency and high energy consumption is associated. In this regard, the crystal plane of noble metals is applied as the reactive interface, since its binding energy with O2 molecule in a high index crystal plane is stronger than that occurred in an (111) facet. For example, an Au (211) facet effectively promotes the activity of a 2e− ORR pathway for the H2O2 production.
Although the H2O2 selectivity of single metal electrocatalysts can be effectively adjusted via various means as discussed above, simultaneous improvement of the activity and selectivity for the 2e− ORR pathway on single metal electrocatalysts is still challenging. This is mainly originated from the fact those with weaker interactions with O2 often need high overpotential to boost the 2e− ORR, while those owning stronger interactions with O2 are prone to bring the direct the 4e− ORR. Combining such metals (namely those weakly interact with O2 and those strongly interact with O2) to form metal alloys is thus a potential strategy to facilitate both activity and selectivity of metal electrocatalysts toward the H2O2 generation. For example, the Au1-xPdx nanoalloys supported on Vulcan XC-72 exhibited the superior selectivity toward the H2O2 production (nearly 95%) when the Pd content in the alloy arrived at 8%, surpassing that using single Au and Pd metal electrocatalysts (Fig. 12d) [208]. The Pd monomer surrounded by Au atoms on the Au–Pd nanoalloy surface was suggested to promote the H2O2 formation via an “end-on” O2 adsorption model. When the percentage of Pd was increased to 15%, the selectivity of the H2O2 generation was decreased. This is because the presence of continuous Pd atoms (e.g., two adjacent Pd atoms) altered the O2 adsorption model and facilitated the H2O formation. In a theoretical work, the tactics was provided based on DFT calculations to screen for metal alloy electrocatalysts that feature excellent activity toward electrocatalytic H2O2 production [5]. These metal alloys are prepared by surrounding individual elements (e.g., Pt, Ir, Pd, Rh and Ru) that are active for the ORR by inert metallic elements (e.g., Hg and Au). Since the active elements exhibit strong adsorption toward O2 molecule, these metal alloys can effectively reduce O2 to OOH*. On the other side, they do not break the O–O bond, because of their difficulty in dissociating OOH* in combination with the neighboring inert sites. In short, the activity and selectivity of metal alloy catalysts toward the 2e− ORR can be regulated by using the advantages of the isolated active sites. Theoretical models have even predicted that the isolated active site of the Pt-Hg alloy can effectively activate O2 molecules and boost the H2O2 synthesis. Experimentally, several Hg-based bimetallic alloys have shown high activity and selectivity for the H2O2 production [209]. Among them, the Pd-Hg alloy exhibited the highest current for the H2O2 production at the same overpotential, as predicted from the DFT calculations. In more detail, the activities of Pt-Hg, Ag and Ag-Hg alloys are one order of magnitude higher than that of individual Au electrocatalyst, while the activity of a Pd-Hg alloy is two orders of magnitude higher than that of individual Au electrocatalyst. The AuPd alloy nanoparticles have been used to disclose the relationship between the activity of electrocatalytic H2O2 production and molar ratio of Au to Pd. Modulating the composition of alloy catalysts is thus recognized as a new way to promote the 2e− ORR for electrocatalytic H2O2 production [210].
4.1.2 Carbon-based Electrocatalysts
It is clear that the noble metals and their alloys feature high activity and selectivity for electrocatalytic H2O2 synthesis via the 2e− ORR pathway. Unfortunately, they are expensive and scarce, hindering their large-scale applications. Many advanced electrocatalysts with reduced costs have been thus explored for electrocatalytic H2O2 synthesis. Carbon-based electrocatalysts have been considered to be one of the most promising substitutes of noble metal-based catalysts. This is due to their unique surface and structural properties, abundant availability and low cost for their synthesis [211,212,213,214]. Note that the pristine carbon materials only exhibit low catalytic activity for the 2e− ORR, thus the reconstruction of carbon materials or their surface modification (Fig. 13) have been developed to optimize their electronic properties for electrocatalytic H2O2 generation [18]. The widely reported approaches include manufacturing porous structures and defects, heteroatom doping and surface (oxygen) functionalization are provided.

Schematic demonstration of tuning electronic structures of carbon electrocatalysts for electrocatalytic H2O2 production via the 2e− ORR pathway. Reproduced with permission from Ref. [18]. Copyright 2020 American Chemical Society
Among diverse carbon materials, porous and defective carbon materials are highly suitable for the 2e− ORR. This is due to their unique advantages, such as their high surface areas, tunable porosities and high electronic conductivities. It has been widely recognized that carbon electrocatalysts with a porous structure can provide abundant active sites and shorten diffusion distance for the reactive species, producing positive contributions for their electrocatalytic H2O2 generation. For example, various hierarchically porous carbon (HPC), produced by hydrothermal treatment and subsequent carbonization, have been applied for selective H2O2 formation (Fig. 14a) [215]. The HPC-H24 (hydrothermal treatment for 24 h and carbonization under H2) exhibited the most positive peak potential and the highest selectivity for the 2e− ORR, namely the best performance of selective H2O2 formation. The remarkable electrocatalytic performance of HPC-H24 was assumed to be originated from high contents of active sites (e.g., sp3-C bonds, vacancy defects and edge defects), more exposed and accessible active sites and fast mass transport of H2O2 inside a porous structure. On the contrary, it was acknowledged that edge-rich graphene presented the typical 4e− ORR mechanism [216]. Later, two different types of porous carbon, namely microporous carbon (denoted as MicroC) and mesoporous carbon (denoted as MesoC) have been developed toward the H2O2 production in alkaline conditions (Fig. 14b) [217]. From the broad π* resonance of near-edge X-ray absorption fine structure (NEXAFS) spectra, the conclusion that both MicroC and MesoC have a larger number of sp2 carbon defective sites than HOPG has been drawn. Electrochemical experiments suggested that both MicroC and MesoC had superior activity and selectivity for the 2e− ORR, where the onset potential was very close to the thermodynamic equilibrium potential (0.7 VRHE), and the selectivity of the H2O2 production even outperformed 70% in some cases (Fig. 14c). In addition, DFT calculations have illustrated the sp2-type defects are the active sites for O2 reduction, which is beneficial for the H2O2 generation. Similar optimization of defects and pore size in carbon materials have been reported for electrocatalytic H2O2 production. For example, the graphitic carbon with rich edge sites has been applied as an electrocatalyst to promote the H2O2 synthesis. Its activity was about 28 times higher than that of a basal plane-rich carbon nanotube (CNT) [218]. This enhanced activity was explained by the fact that graphitic ordered mesoporous carbon (GOMC) has a larger portion of defective carbon and reaction sites (sp2-C) when compared with CNT. Such a statement was solidly supported from the experimental results by means of Raman spectroscopy (Fig. 14d) and XPS (Fig. 14e) characterization. Compared with CNTs, the GOMC showed the H2O2 selectivity higher than 90% via the 2e− ORR pathway and excellent stability (e.g., 90% of the initial current is retained) (Fig. 14f).

a Schematic illustration of the synthetic route to HPC. Reproduced with permission from Ref. [215]. Copyright 2015 Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. b NEXAFS spectra of the C-K edge in HOPG, MesoC and MicroC. c H2O2 selectivity of the MesoC and MicroC electrocatalysts. Reproduced with permission from Ref. [217]. Copyright 2018 American Chemical Society. d Raman spectra, e C 1s XPS spectra and f H2O2 selectivity of GOMC and CNT. Reproduced with permission from Ref. [218]. Copyright 2019 Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim
It has been reported that oxygen-containing groups play an important role on the activity and selectivity of carbon catalysts toward the 2e− ORR [219,220,221,222,223,224,225]. For instance, oxygen-doped microporous polypyrene carbons (OMPC) exhibited the enhanced activity and selectivity for electrocatalytic H2O2 generation [220]. In basic electrolyte, the OMPC4 (oxygen content is 6.52 at%) showed obviously stronger ability to H2O2 production from the Iring than MPC, as well as the higher limited oxygen reduction current (Idisk) (Fig. 15a). Meanwhile, the selectivity of the H2O2 production on OMPC-4 was remarkably increased (e.g., from 40 to 87% at 0.42 VRHE) and the calculated electron transfer number (n) was close to 2 through oxygen doping on OMPC (Fig. 15b). In addition, the selectivity of MPC with the different oxygen content is displayed in Fig. 15c, where OMPC-4 exhibited the highest value. In another case, oxygenated carbon (namely activated carbon black, denoted as aCB) electrocatalyst showed higher ORR activity meaning the stronger capacity of H2O2 generation than CB (Fig. 15d) [222]. The selectivity above 94% was obtained on the aCB catalyst in the potential range from 0.4 to 0.7 V as depicted in Fig. 15e. The DFT calculations further disclosed the activity difference of these oxygen-containing groups toward the H2O2 production (Fig. 15f) is originated from the fact that C–O–C and -CHO are the most active sites toward the H2O2 synthesis compared to other functional groups.

a ORR performance, b selectivity and electron transfer number of OMPC4 and MPC in O2-saturated 0.1 M KOH. c Relationship between the selectivity and oxygen content of various OMPC-samples. Reproduced with permission from Ref. [220]. Copyright 2020 Elsevier. d ORR performance and e selectivity of aCB and CB in 0.1 M O2-saturated KOH. f Activity volcano plot of the 2e− ORR. Reproduced with permission from Ref. [222]. Copyright 2018 American Chemical Society
In addition to structured carbon materials, heteroatom (e.g., N, S, B) doped carbon materials have been widely studied for the electrocatalytic H2O2 production via the 2e− ORR pathway [226,227,228,229,230]. Many studies have confirmed that N-doping of a carbon material can effectively promote the catalytic performance for the ORR, because the higher electronegativity of N atoms than carbon atoms can activate π electrons in carbon system. The charge redistribution is then induced or the electronic properties of carbon materials are changed, leading to varied adsorption capacity of carbon catalysts toward O2 and generated intermediates [55, 231, 232]. It must be pointed out that common N-doped carbon catalysts are conducive to the 4e− ORR pathway to produce water [233,234,235]. It is thus extremely important to bridge the relationship of a nano-structured carbon catalyst and the content of doped N inside with the ORR selectivity. For example, a simple hard-templating strategy was adopted to obtain mesoporous nitrogen-doped carbon (meso-BMP-800) catalyst and its nitrogen content was 14.2 wt% (as determined from XPS analysis) or 17.17 wt% (as evaluated from elemental analysis) [236]. On this electrocatalyst, selective H2O2 production was enhanced (Fig. 16a). This 2e− ORR process has been experimentally and theoretically doable. In such situations, the strategy of N-doping of carbon materials helped to achieve high conductivities of carbon materials and their high selectivity for electrocatalytic H2O2 production. It is so-called a pure two-electron mechanism. However, the regulatory mechanism of the structure of a carbon catalyst on its reactivity is still unclear. In this context, the effect of porous structures of carbon catalysts on their catalytic properties toward the H2O2 production has been explored [237]. Mesoporous nitrogen-doped carbon (MNC) exhibited higher H2O2 selectivity (> 90%, Fig. 16b) than activated nitrogen-doped carbon (ANC). Enhanced mass transport into a mesoporous structure of this catalyst was suggested to be responsible for such high H2O2 selectivity of MNC (Fig. 16c). Later, systematic studies have been explored to reveal the correlations between the performance of electrocatalytic H2O2 production on carbon catalysts and their structural characters and surface physicochemical properties [238]. The selectivity of the H2O2 production on N-doped carbon catalysts is more dependent on the nitrogen doping effect rather than zeta potentials and defect sites of used carbon catalysts. In other words, the tuning of N-doping in carbon materials is very important and even more critical than structuring of carbon materials to obtain the high performance for electrocatalytic H2O2 production. In addition to N-doped carbon materials, B/S-doped ones were synthesized for electrocatalytic H2O2 generation. For example, B-doped mesoporous carbon materials have been synthesized via a convenient hydrothermal method, where F127 and boric acid were the template and boron source, respectively [239]. The obtained B-MC-F2 exhibited enhanced performance for electrocatalytic H2O2 formation in alkaline solution. In another case, a hollow, porous and S-doped carbon spheres presented higher activity and selectivity (> 70%) via the 2e− ORR pathway than carbon spheres without S-doping. The introduced S–S bond is believed to feature low overpotential for the 2e− ORR, leading to high activity for electrocatalytic H2O2 production [240]. In addition to the monoatomic-doped carbon materials, multiple heteroatoms co-doped carbon materials have been utilized for electrocatalytic H2O2 generation. For example, B,N co-doped (BN-C) carbon materials prepared through a simple heat-treatment (Fig. 16d) showed higher activity and selectivity for the H2O2 production via the 2e− ORR pathway than the N-doped carbon materials [241]. Once again, there are various facts (e.g., heteroatom doping) affect the 2e− and 4e− pathways on carbon materials (Fig. 16e). However, the H2O2 formation on B,N-doped carbon materials was preferentially driven by 2e− ORR pathway, resulting from unique catalytic behavior at the interface of h-BN domains and the host graphene lattice.

a The number of electrons transferred based on the Koutecky–Levich plot. Reproduced with permission from Ref. [236]. Copyright 2012 American Chemical Society. b RRDE voltammograms toward ORR over the MNCs and ANC. c Schematic illustration of the ORR on different N-doped carbon. Reproduced with permission from Ref. [237]. Copyright 2014 American Chemical Society. d HRTEM and elemental mapping images of BN-C1. e Schematic illustration of the types of doping motifs and ORR mechanisms on these active sites in the B,N co-doped electrocatalysts (C: gray, N: blue, B: red; O: white). Reproduced with permission from Ref. [241]. Copyright 2018 American Chemical Society
As an interesting type of electrocatalysts, N-doped carbon materials with atomically dispersed transition metals (M–N-C) have been paid close attention in recent years for various catalytic applications, especially as unique catalytic systems for the selective H2O2 production via the 2e− ORR pathway [242,243,244,245]. For example, the relation between physicochemical properties of transition metal (e.g., Mn, Fe, Co, Ni and Cu) single-atom anchored in N-doped graphene and their performance of electrocatalytic H2O2 production has been systematically explored by the means of theoretical calculations and experimental tools [246]. The DFT calculations indicated that the intermediates of *OOH, *O and *OH that are adsorbed on the top site of the metal (M) atoms are the most stable configurations. According to related activity-volcano plot (Fig. 17a), Ni and Cu single-atom catalysts (SACs) tend to follow the 2e− ORR pathway during O2 reduction, suggesting their high selectivity toward the H2O2 generation. On the other side, a large barrier exists between the reduction of *OOH and the activation of O2 molecule. A large overpotential for the ORR is thus indispensable, suggesting low activity of these SACs for electrocatalytic H2O2 production. Differently, the 4e− ORR pathway becomes more favorable on the Mn and Fe SACs during the ORR, resulting in poor selectivity for electrocatalytic H2O2 production. Remarkably, only Co SACs exhibit optimal d-band centers near the apex of the volcano plots, indicated the appropriate adsorption for the intermediates to facilitate the 2e− ORR pathway. Its high activity and selectivity for the 2e− ORR were confirmed by electrochemical ORR tests, where this Co-NC catalyst showed high activity and selectivity for the H2O2 production in acidic media (Fig. 17b, c). Similarly, the activity and selectivity of M–N–C (M = Mn, Fe, Co, Ni and Cu) SACs for electrocatalytic H2O2 production have been investigated [247]. Among these catalysts, the Co–N–C catalyst exhibited the highest ORR selectivity (80%) at 0.1 VRHE in 0.5 M H2SO4 than that of Fe–N–C (28%), Cu–N–C (36%), Mn–N–C (43%), N–C (45%) and Ni–N–C (52%) catalysts under the same conditions. Such high performance was assigned to the fact that the binding free energy of Co–N–C is close to that at the top of the H2O2 production in the volcano plot (Fig. 17e). The number of electrons transferred of a Co–N–C catalyst was much closer to 2 (Fig. 17d). This result indicates that the Co–N–C catalyst is more favorable for 2e− ORR pathway to produce H2O2 (Fig. 17f). The H2O2 selectivity of a Co–N–C catalyst is increased with the potential decreased in acidic solutions, while remained well in the whole potential range under neutral and alkaline electrolyte. One optimal Co–N4 moiety incorporated in N-doped graphene exhibited excellent H2O2 productivity and durability [248]. In the volcano plot of ORR activity, none of M-N4/graphene catalysts are located at the peak position (ΔGOOH* = 4.2 eV) for the H2O2 production. As for the Co-N4, its ΔGOOH* can be modulated once functional groups are attached. For example, when one O* is adsorbed near the Co-N4 moiety (denoted as Co–N4(O)), the ΔGOOH* value increases from 3.9 to 4.1 eV, very close to the optimal ΔG value for the H2O2 production. This phenomenon can be clarified according to the difference in the charge states of cobalt atoms. Specifically, the charge states of cobalt atoms in the Co–N4(O) and Co–N4(2O) moieties are 0.05 e− and 0.10 e− more positive than Co–N4. Therefore, a reasonable design strategy of electrocatalytic H2O2 synthesis is to slightly enlarge ΔGOOH* value of the Co–N4/graphene electrocatalyst by constructing electron-rich oxygen species near the Co–N4 moiety. In this way, the charge states of cobalt atoms are thus gently increased. With aid of this strategy, the Co1-NG(O) SAC were prepared. Among them, the Co1-NG(O) catalyst showed excellent activity and high selectivity for electrocatalytic H2O2 generation.

a Activity-volcano plot of ORR through 2e− or 4e− route. b Linear sweep voltammograms (LSVs) of various catalysts in O2-saturated 0.1 M HClO4. c Faradic efficiencies of the H2O2 formation at different potentials. Reproduced with permission from Ref. [246]. Copyright 2019 Elsevier. d H2O2 selectivity and the number of electrons transferred (n) at +0.1 VRHE. e Activity-volcano plot for the ORR. f H2O2 selectivity and the number of electrons transferred (n) at different potentials. Reproduced with permission from Ref. [247]. Copyright 2019 American Chemical Society
4.2 Electrocatalytic H2O2 Synthesis Through a 2e− WOR Pathway
In addition to the 2e− ORR pathway, the 2e− WOR has been considered as another promising approach toward electrocatalytic H2O2 production. It is worth mentioning that in a 2e− WOR process only H2O is involved as the reactant for the electrocatalytic H2O2 production. Such systems are thus more convenient to be operated than those using the 2e− ORR pathway. However, the H2O2 generation via the 2e− WOR pathway usually displays relatively low Faradaic efficiency. This is because the competitive 4e− WOR (namely O2 evolution process) is thermodynamically more favorable than the 2e− WOR (namely the H2O2 formation) accompanying with the autocatalytic decomposition from H2O2 into O2. In this regard, there are limited studies on this infant field.
4.2.1 Metal Oxides
Noble metal oxides (e.g., RuO2 and IrO2) have been frequently applied for various catalytic applications, due to their high catalytic activities and long-term stability at high potentials [257, 258]. However, their prices are high to limit the large-scale applications [259]. Many alternative noble-free metal oxides thus became more popular in the H2O2 production via the 2e− WOR pathway (Table 4). For example, the free energies of OH* and O* were identified as key parameters to confirm selectivity and activity of different electrocatalysts toward the 2e− WOR [260]. The metal oxide catalysts that simultaneously meet two conditions for OH* and O* binding energies (ΔGO ≳ 3.5 eV and ΔGOH ≲ 2.4 eV) are favorable for selective H2O2 production [261]. In the summarized relationship between the selectivity of 2e− WOR and the binding energies of O/OH for a number of different metal oxides (Fig. 18a), metal oxides (e.g., WO3, BiVO4, MnO2 and SnO2) are located in the region for weak O adsorption energies (green region). They thus promote the 2e− WOR to form H2O2. In verse, metal oxides (e.g., IrO2, RhO2 and PtO2) that feature strong O adsorption energies (blue region) are favorable to generate O2 as the major product. This study provides a good theoretical guide to rationally design metal oxide catalysts in the process of 2e− WOR to promote the H2O2 production. Meanwhile, the activity trends of various metal oxides (e.g., WO3, ZnO, CaSnO3, BiVO4, SnO2 and TiO2) for the H2O2 production via the WOR pathway were summarized as the activity volcano plots. They were based on the relationship between limiting potentials and the free energy of OH* (Fig. 18b) [54]. It was proved again that the catalysts possessing suitable ΔGOH* value (e.g., from 1.6 to 2.4 eV) and low overpotential for the 2e− WOR exhibits high selectivity and activity toward the H2O2 production.

In 2012, a MnOx electrocatalyst was applied for the H2O2 formation via the 2e− WOR pathway [50]. In this system, 77% of the H2O2 production was achieved. Unfortunately, the activity and yield of H2O2 were too low. This low efficiency was attributed to the competing process of the 4e− ORR and/or continuous loss of H2O2 from the disproportionation. Three BiVO4 catalysts with different morphologies containing different surface ratios of (−121) and (040) facets (so-called seed, nanoneedles and truncated BiVO4) have been applied for electrocatalytic H2O2 production [263]. The high-index plane (−121) was disclosed to be favorable for the H2O2 formation in 1 M KHCO3 electrolyte rather than the O2 formation. Unfortunately, BiVO4 has poor stability in H2O2 synthesis [264]. The Bi2WO6 catalysts were then selected as alternatives to BiVO4 for the H2O2 generation via the 2e− WOR pathway [265]. Among a series of Bi-based oxides (Fig. 19a), Bi2O3, BiVO4 and Bi2WO6 displayed preference for electrocatalytic H2O2 production. On the Bi2WO6 electrocatalyst, the best performance for H2O2 generation was achieved. The current density on the Bi2WO6 electrocatalyst was further increased by means of optimizing the morphology and Mo doping of the Bi2WO6 electrocatalyst. The faradaic efficiency of a FTO/Bi2WO6:5%Mo electrocatalyst was 79% at 3.2 VRHE and its maximum yield rate of electrocatalytic H2O2 generation rate was as high as 300 μmol h−1 cm−2 at 3.4 VRHE (Fig. 19b). The durability test of this electrocatalyst is very stable for such electrocatalytic H2O2 production (Fig. 19c).

a Faradaic efficiencies (FEs) and productivity of Bi-based oxides at 2.6 VRHE. b FE and H2O2 generation rate of a FTO/Bi2WO6:5%Mo electrocatalyst at various potentials. c Stability measurement of a FTO/Bi2WO6:5%Mo electrocatalyst under different potentials. Reproduced with permission from Ref. [265]. Copyright 2020 Wiley–VCH GmbH. d FE, H2O2 generation rates and e the densities of different catalysts at different potentials. f Durability test of CaSnO3 catalyst under various potentials. Reproduced with permission from Ref. [267]. Copyright 2019 American Chemical Society. g Free energy diagram of the WOR for the most active (10 \(\overline{1}\) 0) ZnO catalyst. h Volcano plots of 4e− WOR to O2 and 2e− WOR to H2O2 as a function of OH* binding free energies (ΔGOH*). i Overall current density and FE for H2O2. Reproduced with permission from Ref. [268]. Copyright 2019 American Chemical Society
Other metal oxides such as CaSnO3 and ZnO have been also used for the H2O2 production via the 2e− WOR pathway [266,267,268]. For example, CaSnO3 nanoparticles, prepared via the colloidal synthesis method, have been employed to evaluate H2O2 production in 2 M KHCO3 electrolyte [267]. Its Faraday efficiency (FE) was 76% at 3.2 VRHE, higher than that of F:SnO2 and SnO2 (Fig. 19d). The H2O2 generation rates are outstanding at applied potentials (Fig. 19e). Meanwhile, it exhibited excellent stability during this 2e− WOR process at 2.0 VRHE within 12 h. Slight decrease occurred when a potential of at 2.2 or 2.8 VRHE is applied (Fig. 19f). In another case, metal oxide of ZnO was identified as an ideal catalyst for electrocatalytic H2O2 synthesis. The (10 \(\overline{1}\) 0) facet on the ZnO catalyst is more favorable than the (0001) facet, as demonstrated by the DFT calculations (Fig. 19g, h) [268]. Experimental results further indicated that ZnO nanoparticles with a high fraction of (10 \(\overline{1}\) 0) facets own outstanding performance for catalytic H2O2 generation: the overpotential of 40 mV at a current density of 0.1 mA cm−2 and an FE of 81% during this 2e− WOR process (Fig. 19i). Mn-alloyed TiO2 coatings, prepared by atomic layer deposition, also exhibited the high performance of electrocatalytic H2O2 generation via a 2e− WOR pathway: e.g., an FE of higher than > 90% at the overpotential smaller than 150 mV [269]. It has to highlight here that the principles in designing the electrocatalysts with high performance toward the 2e− WOR is still unclear at this stage. More explorations on reaction mechanism for WOR need be provided in future.
4.2.2 Boron-Doped Diamond
As discussed, various noble metal oxides have shown the capability to generate H2O2 through a 2e− WOR pathway. However, these noble metals are expensive and rare. Up to date, the efficiency of the H2O2 production via 2e− WOR is low. On the other side, noble metal-free oxides are not adapted to be operated at the high current densities and high potentials that are frequently required for the large-scale applications. Among the materials that can endure the corrosion in such a harsh conditions for water oxidation, some molecular metal porphyrins (e.g., AlTMPyP, SiTPyP, ZnTMPyP, SnTMPyP) [273,274,275,276] and carbon fibers [57, 277] are promising and thus have been tried for H2O2 production via the 2e− WOR pathway, but their performance was still poor.
Boron-doped diamond (BDD), as a unique kind of carbon material with excellent physical–chemical stability [278], has been employed for the H2O2 generation via the 2e− WOR pathway. Since the H2O2 production through 2e− WOR on the BDD electrode was reported for the first time in 2003 [279, 280], the catalytic performance of electrochemical H2O2 formation using synthetic BDD films that were coated on a titanium substrate has stimulated extensive attention [281]. The BDD exhibited a H2O2 yield of as high as 29.0 mmol dm−3 at 3.47 VRHE. The H2O2 production rate was 19.7 μmol min−1 cm−2. However, the FE was only 28% at 3.17 VRHE and total current densities surpassed 120 mA cm−2, suggesting a high energy consumption process. To promote the electrocatalytic H2O2 generation on BDD via the 2e− WOR pathway as well as to decrease the overpotential at a high applied current density, BBD films were optimized by tailoring their boron content, thickness and crystal size [282]. On the BDD-4 (with the B doping level of 12,600 ppm, film thickness of 2 μm), the biggest FE of 87% was reached and the H2O2 production rate was 76.4 μmol cm−2 min−1. This BDD-4 was also superb durable (e.g., electrocatalytic H2O2 production for 10 h at a constant current density of 200 mA cm−2). An electrogenerated chemiluminescence (ECL) system was developed via in-situ coreactant production at a BDD electrode [283], where peroxydicarbonate (C2O62−) was produced by the oxidation of carbonate (CO32−), subsequently reacted with water in basic media to form H2O2. Based on experimental and no-economic examination, the efficiency of anodic H2O2 production on a BDD electrode was explored in an electrochemical H2/H2O2 process using sodium carbonate as an electrolyte [284]. The calculated FE of producing H2O2 was to up to 31.7% at 2.90 V (vs. SHE) and the corresponding production rate reached 3.93 μmol min−1 cm−2. This study provided guidance to further implement selective oxidation of water to H2O2 on a BDD electrode.
Note that great progress has been made in the H2O2 preparation using photo- and photoelectro-catalytic approaches for wastewater treatment at a pilot scale, even an industrial scale. This is because the efficient H2O2 production is necessary to guarantee Fenton’s reaction [285, 286]. For example, a carbon-PTFE electro-oxidative system promoted the oxidative power of Fenton and Fenton-like processes based on the enhanced H2O2 generation with a higher current efficiency [287]. Solar photoelectro-fenton (SPEF) with a BDD anode and a cathode of the (Co, S, P)-decorated multiwalled carbon nanotubes showed the excellent performance for bronopol removal due to the high efficiency of H2O2 production via the 2e- WOR on a BDD [288].
5 Conclusions and Perspectives
This article provides an overview of current advances on photo/electro-catalytic H2O2 production using different pathways. Different photo/electro-catalysts have been detailed. Compared with traditional preparation methods, such as AQ oxidation, based on fossil resources, the direct preparation of H2O2 through photo/electro-catalytic processes appears to be more promising in that these processes are simple to be operated, green and safe, sustainable and clean, especially when solar or wind energy is integrated. The used catalyst is the prerequisite and technical core to determine the efficiency of such catalytic H2O2 generation among these technologies, especially in the application beyond the laboratory. In more detail, many advanced photo/electro-catalysts must be constructed under theoretic guidance, including the strategies of their synthesis, design of their components/structures. Once their photo/electro-chemical properties are explored and discussed, their catalytic H2O2 production is possible to be boosted. However, the mechanism comprehension of various catalysts for their photo/electro-chemical H2O2 production is still in the preliminary stage of research, and there is still much room for further improvement.
In the field of the photocatalytic synthesis of H2O2 by means of either the 2e− ORR pathway or the 2e− WOR process, it is concluded that an ideal catalyst needs simultaneously satisfy good photoresponse (e.g., an appropriate band gap), superior separation capacity of photogenerated carriers (namely electrons and holes), as well as high catalytic ability for the H2O2 yield. However, the reported photocatalysts up to date still suffer from relatively low activity or selectivity of H2O2, which remains a great challenge for practical applications. Future studies should propose the innovative ideas for applying cooperative modulation of light adsorption, photogenerated charge separation and catalytic reaction actives in photocatalysts to enhance catalytic H2O2 production. In these regards, supramolecular semiconductors, including MOFs, covalent organic frameworks (COFs) and conjugated polymers can be added as the co-catalysts to improve the H2O2 yield. This is because the energy band structures of these supramolecular semiconductors can be optimized. In other words, their light absorption, charge separation and redox capability can be tuned during the photocatalytic H2O2 production. Another promising strategy is to combine inorganic semiconductors with biomolecules to strengthen the response to the solar light and the selectivity of electrocatalytic H2O2 production. It must be pointed out that although standard photocatalysts (e.g., g-C3N4 and TiO2 materials) have achieved selective H2O2 synthesis under visible light irradiation, organic sacrificial agents are usually used to improve the separation of photogenerated electrons and holes in these cases. Such an approach inevitably leads to the contamination of H2O2. Additional cost is then required to separate and obtain pure H2O2. Consequently, the strategies based on electrochemical assistance, oil–water phase solution and all-solid-state Z-scheme photocatalytic systems appear to effectively facilitate the photogenerated charge separation. Note that the output of H2O2 produced by the currently reported catalysts is only at the mM or μM level, too far for large-scale and industrial applications. Porous photocatalysts and/or new photocatalytic reaction systems with enhanced mass transfer rates are needed to boost the formed H2O2 removal from the active sites and to avoid the H2O2 decomposition. A deep understanding of the photocatalytic mechanism will open up new opportunities for the design of high efficiency photocatalysts. Various high-quality and in-situ characterization technologies (e.g., in-situ HRTEM, IR, XANES) are helpful to disclose the distinct dynamic behavior and reaction mechanism of different photocatalytic H2O2 formation systems. In short, exploring effective strategies to promote the ability to harvest wide solar spectrum, improving the lifetime of photogenerated electrons and holes and propelling the 2e− ORR or water oxidation via the design of advanced photocatalysts have been becoming a hot issue for photocatalytic H2O2 production.
Like photocatalytic H2O2 synthesis, electrocatalytic H2O2 synthesis includes two pathways: 2e− ORR and 2e− WOR processes, accompanying with significant achievements acquired in the past few years. Various electrocatalysts with high selectivity (or Faraday efficiencies) and stability have been developed. However, more efforts are still needed to explore and disclose the mechanisms behind. Take the 2e− ORR process as an example, previous reports have revealed that noble metal-based electrocatalysts (e.g., Pt- and Pd-based electrocatalysts) exhibited decent performance for electrocatalytic H2O2 production. Unfortunately, owing to their high costs and scarce reserves, it is unrealistic to use them for industrial H2O2 production. To address this issue, noble-based alloys, noble catalysts with core–shell structures, and porous or single atomic noble metal catalysts are highly desirable for high-performance H2O2 production. Alternatively, carbon-based materials are recognized as potential ones to noble catalysts for the large-scale H2O2 generation. Note that pristine carbon is generally inert for H2O2 generation due to its lack of active sites. Its catalytic performance can be integrated and improved by the modification of surface carbon atoms such as the manufacture of porous structures and defects, the introduction of heteroatom doping and oxygen functionalization. Nevertheless, most carbon electrocatalysts are only applicable for the H2O2 production under alkaline conditions. Their further research is thus needed to improve catalytic performance in acidic media. Moreover, the hurdle of their instability that is caused by the H2O2 decomposition as well as their low selectivity over a wide potential range inhibits the practical applications of carbon electrocatalysts. Some new electrochemical and photochemical electrode systems need to be designed and developed. For example, BDD, a robust sp3 hybridized carbon material, has exhibited extremely high durability for the H2O2 production in harsh conditions. It is thus highly promising for industrial H2O2 production once the manufacturing cost of BDD films is dramatically reduced. Moreover, the structure–activity relationship of these electrode systems, especially when they are at nanoscales, is critical for the H2O2 production. Only on their optimized interfaces, the highly efficient H2O2 synthesis can be realized. To reduce the H2O2 decomposition, it is better to construct porous structures or multi-site active centers inside carbon electrocatalysts, which can prohibit the formed H2O2 from further reaction. One more promising strategy is to couple external means (e.g., ultrasound and microwave) with electrochemical approaches to firm up the H2O2 desorption from the electrocatalysts. To increase the selectivity of the H2O2 production, the sizes and exposed planes of the used electrocatalysts should be further optimized. The employment of the 2e− WOR process should be paid more attention as a more effective pathway for the H2O2 production where no gas phase is involved. As confirmed from theoretical calculations and experimental operations, metal oxides (e.g., BiVO4, SnO2 and ZnO) tend to generate H2O2 via the 2e− WOR approach, while others (e.g., IrO2, PtO2 and RhO2) are more favorable for the 4e− WOR approach to produce O2. In this context, it is highly necessary to further study the mechanisms of the H2O2 formation via the 2e− WOR approach. Once advanced metal oxide catalysts are found, electrocatalytic H2O2 production on these metal oxide electrocatalysts can be further enhanced. It is worth mentioning that BDD presented good ability to produce H2O2 at high potentials. However, it only exhibited low Faraday efficiency. To utilize diamond for industrial H2O2 production at high current densities, it is urgently needed to improve its catalytic ability by the formation of its nanostructures or the introduction other catalytic centers (e.g., defects, dopants, other catalysts).
To sum up, recent advances in the design and synthesis of photo/electro-catalysts have improved their activity, selectivity and stability for photo/electro-catalytic H2O2 production in past years. To further improve performance of photo/electro-catalytic H2O2 production, the design of advanced catalysts with various functions is still highly necessary. For photocatalysts, two-channel reaction pathways for generating H2O2 with high efficiency are highly pursued. As for electrocatalysts, their high activity with low cost and strong durability is the key factor moving forward. From a practical application perspective, further understanding the reaction kinetics using theoretical and experimental (especially in situ and operando techniques) as well as the design of new reactors for the H2O2 production at an industrial scale must be conducted in future.
References
R.J. Lewis, G.J. Hutchings, Recent advances in the direct synthesis of H2O2. ChemCatChem 11, 298–308 (2019). https://doi.org/10.1002/cctc.201801435
J. Gao, B. Liu, Progress of electrochemical hydrogen peroxide synthesis over single atom catalysts. ACS Mater. Lett. 2, 1008–1024 (2020). https://doi.org/10.1021/acsmaterialslett.0c00189
A. Torres-Pinto, M.J. Sampaio, C.G. Silva, J.L. Faria, A.M.T. Silva, Recent strategies for hydrogen peroxide production by metal-free carbon nitride photocatalysts. Catalysts 9, 990 (2019). https://doi.org/10.3390/catal9120990
S.C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber et al., Electrochemical synthesis of hydrogen peroxide from water and oxygen. Nat. Rev. Chem. 3, 442–458 (2019). https://doi.org/10.1038/s41570-019-0110-6
S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida et al., Enabling direct H2O2 production through rational electrocatalyst design. Nat. Mater. 12, 1137–1143 (2013). https://doi.org/10.1038/NMAT3795
J. Chen, Q. Ma, X. Zheng, Y. Fang, J. Wang et al., Kinetically restrained oxygen reduction to hydrogen peroxide with nearly 100% selectivity. Nat. Commun. 13, 2808 (2022). https://doi.org/10.1038/s41467-022-30411-7
J. Wu, Y. Han, Y. Bai, X. Wang, Y. Zhou et al., The electron transport regulation in carbon dots/In2O3 electrocatalyst enable 100% selectivity for oxygen reduction to hydrogen peroxide. Adv. Funct. Mater. 32, 2203647 (2022). https://doi.org/10.1002/adfm.202203647
J.M. Campos-Martin, G. Blanco-Brieva, J.L.G. Fierro, Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962–6984 (2006). https://doi.org/10.1002/anie.200503779
S.-I. Yamazaki, Z. Siroma, H. Senoh, T. Loroi, N. Fujiwara et al., A fuel cell with selective electrocatalysts using hydrogen peroxide as both an electron acceptor and a fuel. J. Power Sources 178, 20–25 (2008). https://doi.org/10.1016/j.jpowsour.2007.12.013
S.A.M. Shaegh, N. Nam-Trung, S.M.M. Ehteshami, S.H. Chan, A membraneless hydrogen peroxide fuel cell using prussian blue as cathode material. Energy Environ. Sci. 5, 8225–8228 (2012). https://doi.org/10.1039/c2ee21806b
Y. Liu, P. Vijayakumar, Q. Liu, T. Sakthivel, F. Chen et al., Shining light on anion-mixed nanocatalysts for efficient water electrolysis: fundamentals, progress, and perspectives. Nano-Micro Lett. 14, 43 (2022). https://doi.org/10.1007/s40820-021-00785-2
S. Kato, J. Jung, T. Suenobu, S. Fukuzumi, Production of hydrogen peroxide as a sustainable solar fuel from water and dioxygen. Energy Environ. Sci. 6, 3756–3764 (2013). https://doi.org/10.1039/c3ee42815j
Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Tanaka et al., Carbon nitride-aromatic diimide-graphene nanohybrids: metal-free photocatalysts for solar-to-hydrogen peroxide energy conversion with 0.2% efficiency. J. Am. Chem. Soc. 138, 10019–10025 (2016). https://doi.org/10.1021/jacs.6b05806
S. Gogoi, N. Karak, Solar-driven hydrogen peroxide production using polymer-supported carbon dots as heterogeneous catalyst. Nano-Micro Lett. 9, 40 (2017). https://doi.org/10.1007/s40820-017-0143-7
K. Mase, M. Yoneda, Y. Yamada, S. Fukuzumi, Seawater usable for production and consumption of hydrogen peroxide as a solar fuel. Nat. Commun. 7, 11470 (2016). https://doi.org/10.1038/ncomms11470
X. Zeng, Y. Liu, X. Hu, X. Zhang, Photoredox catalysis over semiconductors for light-driven hydrogen peroxide production. Green Chem. 23, 1466–1494 (2021). https://doi.org/10.1039/d0gc04236f
Z. Yao, J. Zhao, R.J. Bunting, C. Zhao, P. Hu et al., Quantitative insights into the reaction mechanism for the direct synthesis of H2O2 over transition metals: coverage-dependent microkinetic modeling. ACS Catal. 11, 1202–1221 (2021). https://doi.org/10.1021/acscatal.0c04125
S. Siahrostami, S.J. Villegas, A.H.B. Mostaghimi, S. Back, A.B. Farimani et al., A review on challenges and successes in atomic-scale design of catalysts for electrochemical synthesis of hydrogen heroxide. ACS Catal. 10, 7495–7511 (2020). https://doi.org/10.1021/acscatal.0c01641
G.-H. Han, S.-H. Lee, S.-Y. Hwang, K.-Y. Lee, Advanced development strategy of nano catalyst and DFT calculations for direct synthesis of hydrogen peroxide. Adv. Energy Mater. 11, 2003121 (2021). https://doi.org/10.1002/aenm.202003121
Y. Sun, L. Han, P. Strasser, A comparative perspective of electrochemical and photochemical approaches for catalytic H2O2 production. Chem. Soc. Rev. 49, 6605–6631 (2020). https://doi.org/10.1039/d0cs00458h
C. Xia, Y. Xia, P. Zhu, L. Fan, H. Wang, Direct electrosynthesis of pure aqueous H2O2 solutions up to 20% by weight using a solid electrolyte. Science 366, 226 (2019). https://doi.org/10.1126/science.aay1844
Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu et al., High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials. Nat. Catal. 1, 156–162 (2018). https://doi.org/10.1038/s41929-017-0017-x
S.J. Freakley, Q. He, J.H. Harrhy, L. Lu, D.A. Crole et al., Palladium-tin catalysts for the direct synthesis of H2O2 with high selectivity. Science 351, 965–968 (2016). https://doi.org/10.1126/science.aad5705
J.K. Edwards, S.J. Freakley, A.F. Carley, C.J. Kiely, G.J. Hutchings, Strategies for designing supported gold-palladium bimetallic catalysts for the direct synthesis of hydrogen peroxide. Acc. Chem. Res. 47, 845–854 (2014). https://doi.org/10.1021/ar400177c
J. Garcia-Serna, T. Moreno, P. Biasi, M.J. Cocero, J.-P. Mikkola et al., Engineering in direct synthesis of hydrogen peroxide: targets, reactors and guidelines for operational conditions. Green Chem. 16, 2320–2343 (2014). https://doi.org/10.1039/c3gc41600c
R.B. Rankin, J. Greeley, Trends in selective hydrogen peroxide production on transition metal surfaces from first principles. ACS Catal. 2, 2664–2672 (2012). https://doi.org/10.1021/cs3003337
J.K. Edwards, B. Solsona, A.F. Carley, A.A. Herzing, C.J. Kiely et al., Switching off hydrogen peroxide hydrogenation in the direct synthesis process. Science 323, 1037–1041 (2009). https://doi.org/10.1126/science.1168980
K. Dong, Y. Lei, H. Zhao, J. Liang, P. Ding et al., Noble-metal-free electrocatalysts toward H2O2 production. J. Mater. Chem. A 8, 23123–23141 (2020). https://doi.org/10.1039/d0ta08894c
H. Lu, X. Li, S.A. Monny, Z. Wang, L. Wang, Photo-electrocatalytic hydrogen peroxide production based on transition-metal-oxide semiconductors. Chin. J. Catal. 43, 1204–1215 (2022). https://doi.org/10.1016/S1872-2067(21)64028-7
L.-H. Fu, C. Qi, J. Lin, P. Huang, Catalytic chemistry of glucose oxidase in cancer diagnosis and treatment. Chem. Soc. Rev. 47, 6454–6472 (2018). https://doi.org/10.1039/c7cs00891k
W. Luo, C. Zhu, S. Su, D. Li, Y. He et al., Self-catalyzed, self-limiting growth of glucose oxidase-mimicking gold nanoparticles. ACS Nano 4, 7451–7458 (2010). https://doi.org/10.1021/nn102592h
J. Fang, T. Sawa, T. Akaike, H. Maeda, Tumor-targeted delivery of polyethylene glycol-conjugated D-amino acid oxidase for antitumor therapy via enzymatic generation of hydrogen peroxide. Cancer Res. 62, 3138–3143 (2002)
A. Parra, E. Casero, F. Pariente, L. Vazquez, E. Lorenzo, Cholesterol oxidase modified gold electrodes as bioanalytical devices. Sens. Actuators B 124, 30–37 (2007). https://doi.org/10.1016/j.snb.2006.11.051
S.C. Perry, S.M. Gateman, J. Sifakis, L. Pollegioni, J. Mauzeroll, Enhancement of the enzymatic biosensor response through targeted electrode surface roughness. J. Electrochem. Soc. 165, G3074–G3079 (2018). https://doi.org/10.1149/2.0121812jes
D. Polcari, S.C. Perry, L. Pollegioni, M. Geissler, J. Mauzeroll et al., Localized detection of D-serine by using an enzymatic amperometric biosensor and scanning electrochemical microscopy. ChemElectroChem 4, 920–926 (2017). https://doi.org/10.1002/celc.201600766
L. Wang, J. Zhang, Y. Zhang, H. Yu, Y. Qu et al., Inorganic metal-oxide photocatalyst for H2O2 production. Small 18, 2104561 (2022). https://doi.org/10.1002/smll.202104561
S. Fukuzumi, Y.-M. Lee, W. Nam, Solar-driven production of hydrogen peroxide from water and dioxygen. Chem. Eur. J. 24, 5016 (2018). https://doi.org/10.1002/chem.201704512
Y. Pang, H. Xie, Y. Sun, M.-M. Titirici, G.-L. Chai, Electrochemical oxygen reduction for H2O2 production: catalysts, pH effects and mechanisms. J. Mater. Chem. A 8, 24996–25016 (2020). https://doi.org/10.1039/d0ta09122g
X. Yang, Y. Zeng, W. Alnoush, Y. Hou, D. Higgins et al., Tuning two-electron oxygen-reduction pathways for H2O2 electrosynthesis via engineering atomically dispersed single metal site catalysts. Adv. Mater. 34, 2107954 (2022). https://doi.org/10.1002/adma.202107954
H.-I. Kim, O.S. Kwon, S. Kim, W. Choi, J.-H. Kim, Harnessing low energy photons (635 nm) for the production of H2O2 using upconversion nanohybrid photocatalysts. Energy Environ. Sci. 9, 1063–1073 (2016). https://doi.org/10.1039/c5ee03115j
Y. Jiang, P. Ni, C. Chen, Y. Lu, P. Yang et al., Selective electrochemical H2O2 production through two-electron oxygen electrochemistry. Adv. Energy Mater. 8, 1801909 (2018). https://doi.org/10.1002/aenm.201801909
Y.-J. Ko, K. Choi, B. Yang, W.H. Lee, J.-Y. Kim et al., A catalyst design for selective electrochemical reactions: direct production of hydrogen peroxide in advanced electrochemical oxidation. J. Mater. Chem. A 8, 9859–9870 (2020). https://doi.org/10.1039/d0ta01869d
L.E.B. Lucchetti, M.O. Almeida, J.M. de Almeida, P.A.S. Autreto, K.M. Honorio et al., Density functional theory studies of oxygen reduction reaction for hydrogen peroxide generation on graphene-based catalysts. J. Electroanal. Chem. 895, 115429 (2021). https://doi.org/10.1016/j.jelechem.2021.115429
L. Cheng, Q. Xiang, Y. Liao, H. Zhang, CdS-based photocatalysts. Energy Environ. Sci. 11, 1362–1391 (2018). https://doi.org/10.1039/c7ee03640j
J. Fu, J. Yu, C. Jiang, B. Cheng, g-C3N4-Based heterostructured photocatalysts. Adv. Energy Mater. 8, 1701503 (2018). https://doi.org/10.1002/aenm.201701503
J. Xiong, J. Di, J. Xia, W. Zhu, H. Li, Surface defect engineering in 2D nanomaterials for photocatalysis. Adv. Funct. Mater. 28, 1801983 (2018). https://doi.org/10.1002/adfm.201801983
H. Hou, X. Zeng, X. Zhang, Production of hydrogen peroxide by photocatalytic processes. Angew. Chem. Int. Ed. 59, 17356–17376 (2020). https://doi.org/10.1002/anie.201911609
Z. Liu, X. Sheng, D. Wang, X. Feng, Efficient hydrogen peroxide generation utilizing photocatalytic oxygen reduction at a triphase interface. Iscience 17, 67 (2019). https://doi.org/10.1016/j.isci.2019.06.023
F. Xue, Y. Si, M. Wang, M. Liu, L. Guo, Toward efficient photocatalytic pure water splitting for simultaneous H2 and H2O2 production. Nano Energy 62, 823–831 (2019). https://doi.org/10.1016/j.nanoen.2019.05.086
A. Izgorodin, E. Izgorodina, D.R. MacFarlane, Low overpotential water oxidation to hydrogen peroxide on a MnOx catalyst. Energy Environ. Sci. 5, 9496–9501 (2012). https://doi.org/10.1039/c2ee21832a
X.J. Shi, Y.R. Zhang, S. Siahrostami, X.L. Zheng, Light-driven BiVO4-C fuel cell with simultaneous production of H2O2. Adv. Energy Mater. 8, 1801158 (2018). https://doi.org/10.1002/aenm.201801158
S. Back, J. Na, Z.W. Ulissi, Efficient discovery of active, selective, and stable catalysts for electrochemical H2O2 synthesis through active motif screening. ACS Catal. 11, 2483–2491 (2021). https://doi.org/10.1021/acscatal.0c05494
J. Zhang, H. Zhang, M.-J. Cheng, Q. Lu, Tailoring the electrochemical production of H2O2: strategies for the rational design of high-performance electrocatalysts. Small 16, 1902845 (2020). https://doi.org/10.1002/smll.201902845
X. Shi, S. Back, T.M. Gill, S. Siahrostami, X. Zheng, Electrochemical synthesis of H2O2 by two-electron water oxidation reaction. Chem 7, 38–63 (2021). https://doi.org/10.1016/j.chempr.2020.09.013
K. Wang, J. Huang, H. Chen, Y. Wang, S. Song, Recent advances in electrochemical 2e oxygen reduction reaction for on-site hydrogen peroxide production and beyond. Chem. Commun. 56, 12109–12121 (2020). https://doi.org/10.1039/d0cc05156j
S. Mavrikis, S.C. Perry, P.K. Leung, L. Wang, C.P. de Leon, Recent advances in electrochemical water oxidation to produce hydrogen peroxide: a mechanistic perspective. ACS Sustainable Chem. Eng. 9, 76–91 (2021). https://doi.org/10.1021/acssuschemeng.0c07263
C. Xia, S. Back, S. Ringe, K. Jiang, F. Chen et al., Confined local oxygen gas promotes electrochemical water oxidation to hydrogen peroxide. Nat. Catal. 3, 125–134 (2020). https://doi.org/10.1038/s41929-019-0402-8
Y. Xue, Y. Wang, Z. Pan, K. Sayama, Electrochemical and photoelectrochemical water oxidation for hydrogen peroxide production. Angew. Chem. Int. Ed. 60, 10469–10480 (2021). https://doi.org/10.1002/anie.202011215
X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin et al., A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 8, 76–80 (2009). https://doi.org/10.1038/NMAT2317
X. Wang, K. Maeda, X. Chen, K. Takanabe, K. Domen et al., Polymer semiconductors for artificial photosynthesis: hydrogen evolution by mesoporous graphitic carbon nitride with visible light. J. Am. Chem. Soc. 131, 1680 (2009). https://doi.org/10.1021/ja809307s
M. Wang, P. Ju, J. Li, Y. Zhao, X. Han et al., Facile synthesis of MoS2/g-C3N4/GO ternary heterojunction with enhanced photocatalytic activity for water splitting. ACS Sustainable Chem. Eng. 5, 7878–7886 (2017). https://doi.org/10.1021/acssuschemeng.7b01386
L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng et al., Doping of graphitic carbon nitride for photocatalysis: a reveiw. Appl. Catal. B 217, 388–406 (2017). https://doi.org/10.1016/j.apcatb.2017.06.003
Y.-X. Ye, C. Wen, J. Pan, J.-W. Wang, Y.-J. Tong et al., Visible-light driven efficient overall H2O2 production on modified graphitic carbon nitride under ambient conditions. Appl. Catal. B 285, 119726 (2021). https://doi.org/10.1016/j.apcatb.2020.119726
G. Dong, L. Zhang, Synthesis and enhanced Cr(VI) photoreduction property of formate anion containing graphitic carbon nitride. J. Phys. Chem. C 117, 4062–4068 (2013). https://doi.org/10.1021/jp3115226
S. Cao, J. Low, J. Yu, M. Jaroniec, Polymeric photocatalysts based on graphitic carbon nitride. Adv. Mater. 27, 2150–2176 (2015). https://doi.org/10.1002/adma.201500033
S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma et al., Exfoliated graphitic carbon nitride nanosheets as efficient catalysts for hydrogen evolution under visible light. Adv. Mater. 25, 2452–2456 (2013). https://doi.org/10.1002/adma.201204453
H. Piao, G. Choi, X. Jin, S.-J. Hwang, Y.J. Song et al., Monolayer graphitic carbon nitride as metal-free catalyst with enhanced performance in photo- and electro-catalysis. Nano-Micro Lett. 14, 55 (2022). https://doi.org/10.1007/s40820-022-00794-9
Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto et al., Highly selective production of hydrogen peroxide on graphitic carbon nitride (g-C3N4) photocatalyst activated by visible light. ACS Catal. 4, 774–780 (2014). https://doi.org/10.1021/cs401208c
Y. Shiraishi, Y. Kofuji, H. Sakamoto, S. Tanaka, S. Ichikawa et al., Effects of surface defects on photocatalytic H2O2 production by mesoporous graphitic carbon nitride under visible light irradiation. ACS Catal. 5, 3058–3066 (2015). https://doi.org/10.1021/acscatal.5b00408
Z. Zhu, H. Pan, M. Murugananthan, J. Gong, Y. Zhang, Visible light-driven photocatalytically active g-C3N4 material for enhanced generation of H2O2. Appl. Catal. B 232, 19–25 (2018). https://doi.org/10.1016/j.apcatb.2018.03.035
S. Li, G. Dong, R. Hailili, L. Yang, Y. Li et al., Effective photocatalytic H2O2 production under visible light irradiation at g-C3N4 modulated by carbon vacancies. Appl. Catal. B 190, 26–35 (2016). https://doi.org/10.1016/j.apcatb.2016.03.004
Y. You, S. Wang, K. Xiao, T. Ma, Y. Zhang et al., Z-Scheme g-C3N4/Bi4NbO8Cl heterojunction for enhanced photocatalytic hydrogen production. ACS Sustainable Chem. Eng. 6, 16219–16227 (2018). https://doi.org/10.1021/acssuschemeng.8b03075
R. Hao, G. Wang, C. Jiang, H. Tang, Q. Xu, In-situ hydrothermal synthesis of g-C3N4/TiO2 heterojunction photocatalysts with high specific surface area for Rhodamine B degradation. Appl. Surf. Sci. 411, 400–410 (2017). https://doi.org/10.1016/j.apsusc.2017.03.197
W. Liu, C. Song, M. Kou, Y. Wang, Y. Deng et al., Fabrication of ultra-thin g-C3N4 nanoplates for efficient visible-light photocatalytic H2O2 production via two-electron oxygen reduction. Chem. Eng. J. 425, 130615 (2021). https://doi.org/10.1016/j.cej.2021.130615
T. Mahvelati-Shamsabadi, H. Fattahimoghaddam, B.-K. Lee, S. Bae, J. Ryu, Synthesis of hexagonal rosettes of g-C3N4 with boosted charge transfer for the enhanced visible-light photocatalytic hydrogen evolution and hydrogen peroxide production. J. Colloid Interface Sci. 597, 345–360 (2021). https://doi.org/10.1016/j.jcis.2021.04.019
G. Dong, W. Ho, Y. Li, L. Zhang, Facile synthesis of porous graphene-like carbon nitride (C6N9H3) with excellent photocatalytic activity for NO removal. Appl. Catal. B 174, 477–485 (2015). https://doi.org/10.1016/j.apcatb.2015.03.035
Y. Lv, Y. Liu, Y. Zhu, Y. Zhu, Surface oxygen vacancy induced photocatalytic performance enhancement of a BiPO4 nanorod. J. Mater. Chem. A 2, 1174–1182 (2014). https://doi.org/10.1039/c3ta13841k
D. Chen, Z. Wang, T. Ren, H. Ding, W. Yao et al., Influence of defects on the photocatalytic activity of ZnO. J. Phys. Chem. C 118, 15300–15307 (2014). https://doi.org/10.1021/jp5033349
L. Shi, L. Yang, W. Zhou, Y. Liu, L. Yin et al., Photoassisted construction of holey defective g-C3N4 photocatalysts for efficient visible-light-driven H2O2 production. Small 14, 1703142 (2018). https://doi.org/10.1002/smll.201703142
H. Zhao, Q. Jin, M.A. Khan, S. Larter, S. Siahrostami et al., Rational design of carbon nitride for remarkable photocatalytic H2O2 production. Chem. Catalysis 2, 1720–1733 (2022). https://doi.org/10.1016/j.checat.2022.04.015
H. Li, J. Shang, Z. Ai, L. Zhang, Efficient visible light nitrogen fixation with BiOBr nanosheets of oxygen vacancies on the exposed 001 facets. J. Am. Chem. Soc. 137, 6393–6399 (2015). https://doi.org/10.1021/jacs.5b03105
G. Dong, W. Ho, C. Wang, Selective photocatalytic N2 fixation dependent on g-C3N4 induced by nitrogen vacancies. J. Mater. Chem. A 3, 23435–23441 (2015). https://doi.org/10.1039/c5ta06540b
J. Lei, B. Chen, W. Lv, L. Zhou, L. Wang et al., Robust photocatalytic H2O2 production over inverse opal g-C3N4 with carbon vacancy under visible light. ACS Sustainable Chem. Eng. 7, 16467–16473 (2019). https://doi.org/10.1021/acssuschemeng.9b03678
P. Niu, G. Liu, H.-M. Cheng, Nitrogen vacancy-promoted photocatalytic activity of graphitic carbon nitride. J. Phys. Chem. C 116, 11013–11018 (2012). https://doi.org/10.1021/jp301026y
X. Qu, S. Hu, P. Li, Z. Li, H. Wang et al., The effect of embedding N vacancies into g-C3N4 on the photocatalytic H2O2 production ability via H2 plasma treatment. Diamond Relat. Mater. 86, 159–166 (2018). https://doi.org/10.1016/j.diamond.2018.04.027
H. Fattahimoghaddam, T. Mahvelati-Shamsabadi, B.-K. Lee, Enhancement in photocatalytic H2O2 production over g-C3N4 nanostructures: a collaborative approach of nitrogen deficiency and supramolecular precursors. ACS Sustainable Chem. Eng. 9, 4520–4530 (2021). https://doi.org/10.1021/acssuschemeng.0c08884
J. Luo, Y. Liu, C. Fan, L. Tang, S. Yang et al., Direct attack and indirect transfer mechanisms dominated by reactive oxygen species for photocatalytic H2O2 production on g-C3N4 possessing nitrogen vacancies. ACS Catal. 11, 11440–11450 (2021). https://doi.org/10.1021/acscatal.1c03103
X. Meng, L. Liu, S. Ouyang, H. Xu, D. Wang et al., Nanometals for solar-to-chemical energy conversion: from semiconductor-based photocatalysis to plasmon-mediated photocatalysis and photo-thermocatalysis. Adv. Mater. 28, 6781–6803 (2016). https://doi.org/10.1002/adma.201600305
X. Chang, J. Yang, D. Han, B. Zhang, X. Xiang et al., Enhancing light-driven production of hydrogen peroxide by anchoring Au onto C3N4 catalysts. Catalysts 8, 147 (2018). https://doi.org/10.3390/catal8040147
G. Zuo, S. Liu, L. Wang, H. Song, P. Zong et al., Finely dispersed Au nanoparticles on graphitic carbon nitride as highly active photocatalyst for hydrogen peroxide production. Catal. Commun. 123, 69–72 (2019). https://doi.org/10.1016/j.catcom.2019.02.011
J. Cai, J. Huang, S. Wang, J. Iocozzia, Z. Sun et al., Crafting mussel-inspired metal nanoparticle-decorated ultrathin graphitic carbon nitride for the degradation of chemical pollutants and production of chemical resources. Adv. Mater. 31, 1806314 (2019). https://doi.org/10.1002/adma.201806314
Y. Wang, X. Wang, M. Antonietti, Polymeric graphitic carbon nitride as a heterogeneous organocatalyst: from photochemistry to multipurpose catalysis to sustainable chemistry. Angew. Chem. Int. Ed. 51, 68–89 (2012). https://doi.org/10.1002/anie.201101182
G. Liu, P. Niu, L. Yin, H.-M. Cheng, α-Sulfur crystals as a visible-light-active photocatalyst. J. Am. Chem. Soc. 134, 9070–9073 (2012). https://doi.org/10.1021/ja302897b
T. Xiong, W. Cen, Y. Zhang, F. Dong, Bridging the g-C3N4 interlayers for enhanced photocatalysis. ACS Catal. 6, 2462–2472 (2016). https://doi.org/10.1021/acscatal.5b02922
Q. Xu, L. Zhang, J. Yu, S. Wageh, A.A. Al-Ghamdi et al., Direct Z-scheme photocatalysts: principles, synthesis, and applications. Mater. Today 21, 1042–1063 (2018). https://doi.org/10.1016/j.mattod.2018.04.008
Q. Chen, J. Li, L. Cheng, H. Liu, Construction of CdLa2S4/MIL-88A(Fe) heterojunctions for enhanced photocatalytic H2 evolution activity via a direct Z-scheme electron transfer. Chem. Eng. J. 379, 122389 (2020). https://doi.org/10.1016/j.cej.2019.122389
L. Yang, G. Dong, D.L. Jacobs, Y. Wang, L. Zang et al., Two-channel photocatalytic production of H2O2 over g-C3N4 nanosheets modified with perylene imides. J. Catal. 352, 274–281 (2017). https://doi.org/10.1016/j.jcat.2017.05.010
Z. Li, N. Xiong, G. Gu, Fabrication of a full-spectrum-response Cu2(OH)2CO3/g-C3N4 heterojunction catalyst with outstanding photocatalytic H2O2 production performance via a self-sacrificial method. Dalton Trans. 48, 182–189 (2019). https://doi.org/10.1039/c8dt04081h
X. Zhao, Y. You, S. Huang, Y. Wu, Y. Ma et al., Z-scheme photocatalytic production of hydrogen peroxide over Bi4O5Br2/g-C3N4 heterostructure under visible light. Appl. Catal. B 278, 119251 (2020). https://doi.org/10.1016/j.apcatb.2020.119251
D.-L. Long, E. Burkholder, L. Cronin, Polyoxometalate clusters, nanostructures and materials: from self- assembly to designer materials and devices. Chem. Soc. Rev. 36, 105–121 (2007). https://doi.org/10.1039/b502666k
H. Lv, Y.V. Geletii, C. Zhao, J.W. Vickers, G. Zhu et al., Polyoxometalate water oxidation catalysts and the production of green fuel. Chem. Soc. Rev. 41, 7572–7589 (2012). https://doi.org/10.1039/c2cs35292c
S.-S. Wang, G.-Y. Yang, Recent advances in polyoxometalate-catalyzed reactions. Chem. Rev. 115, 4893–4962 (2015). https://doi.org/10.1021/cr500390v
J. Ettedgui, Y. Diskin-Posner, L. Weiner, R. Neumann, Photoreduction of carbon dioxide to carbon monoxide with hydrogen catalyzed by a rhenium(I) phenanthroline-polyoxometalate hybrid complex. J. Am. Chem. Soc. 133, 188–190 (2011). https://doi.org/10.1021/ja1078199
B. Rausch, M.D. Symes, G. Chisholm, L. Cronin, Decoupled catalytic hydrogen evolution from a molecular metal oxide redox mediator in water splitting. Science 345, 1326–1330 (2014). https://doi.org/10.1126/science.1257443
S. Zhao, X. Zhao, H. Zhang, J. Li, Y. Zhu, Covalent combination of polyoxometalate and graphitic carbon nitride for light-driven hydrogen peroxide production. Nano Energy 35, 405–414 (2017). https://doi.org/10.1016/j.nanoen.2017.04.017
S. Zhao, X. Zhao, S. Ouyang, Y. Zhu, Polyoxometalates covalently combined with graphitic carbon nitride for photocatalytic hydrogen peroxide production. Catal. Sci. Technol. 8, 1686–1695 (2018). https://doi.org/10.1039/c8cy00043c
S. Zhao, X. Zhao, Polyoxometalates-derived metal oxides incorporated into graphitic carbon nitride framework for photocatalytic hydrogen peroxide production under visible light. J. Catal. 366, 98–106 (2018). https://doi.org/10.1016/j.jcat.2018.08.003
S. Zhao, X. Zhao, Insights into the role of singlet oxygen in the photocatalytic hydrogen peroxide production over polyoxometalates-derived metal oxides incorporated into graphitic carbon nitride framework. Appl. Catal. B 250, 408–418 (2019). https://doi.org/10.1016/j.apcatb.2019.02.031
S. Hu, X. Qu, P. Li, F. Wang, Q. Li et al., Photocatalytic oxygen reduction to hydrogen peroxide over copper doped graphitic carbon nitride hollow microsphere: the effect of Cu(I)-N active sites. Chem. Eng. J. 334, 410–418 (2018). https://doi.org/10.1016/j.cej.2017.10.016
X. Qu, S. Hu, J. Bai, P. Li, G. Lu et al., Synthesis of band gap-tunable alkali metal modified graphitic carbon nitride with outstanding photocatalytic H2O2 production ability via molten salt method. J. Mater. Sci. Technol. 34, 1932–1938 (2018). https://doi.org/10.1016/j.jmst.2018.04.019
Y. Chen, X. Yan, J. Xu, L. Wang, K+, Ni and carbon co-modification promoted two-electron O2 reduction for photocatalytic H2O2 production by crystalline carbon nitride. J. Mater. Chem. A 9, 24056–24063 (2021). https://doi.org/10.1039/d1ta06960h
G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin et al., Iodine modified carbon nitride semiconductors as visible light photocatalysts for hydrogen evolution. Adv. Mater. 26, 805–809 (2014). https://doi.org/10.1002/adma.201303611
Z.-A. Lan, G. Zhang, X. Wang, A facile synthesis of Br-modified g-C3N4 semiconductors for photoredox water splitting. Appl. Catal. B 192, 116–125 (2016). https://doi.org/10.1016/j.apcatb.2016.03.062
C. Zhang, J. Bai, L. Ma, Y. Lv, F. Wang et al., Synthesis of halogen doped graphite carbon nitride nanorods with outstanding photocatalytic H2O2 production ability via saturated NH4X (X = Cl, Br) solution-hydrothermal post-treatment. Diamond Relat. Mater. 87, 215–222 (2018). https://doi.org/10.1016/j.diamond.2018.06.013
S. Kim, G.-H. Moon, H. Kim, Y. Mun, P. Zhang et al., Selective charge transfer to dioxygen on KPF6-modified carbon nitride for photocatalytic synthesis of H2O2 under visible light. J. Catal. 357, 51–58 (2018). https://doi.org/10.1016/j.jcat.2017.10.002
L. Zhou, J. Lei, F. Wang, L. Wang, M.R. Hoffmann et al., Carbon nitride nanotubes with in-situ grafted hydroxyl groups for highly efficient spontaneous H2O2 production. Appl. Catal. B 288, 119993 (2021). https://doi.org/10.1016/j.apcatb.2021.119993
Y. Zheng, Y. Luo, Q. Ruan, J. Yu, X. Guo et al., Plasma-Tuned nitrogen vacancy graphitic carbon nitride sphere for efficient photocatalytic H2O2 production. J. Colloid Interface Sci. 609, 75–85 (2022). https://doi.org/10.1016/j.jcis.2021.12.006
Y. Shao, J. Hu, T. Yang, X. Yang, J. Qu et al., Significantly enhanced photocatalytic in-situ H2O2 production and consumption activities for efficient sterilization by ZnIn2S4/g-C3N4 heterojunction. Carbon 190, 337–347 (2022). https://doi.org/10.1016/j.carbon.2022.01.019
J. Xiong, X. Li, J. Huang, X. Gao, Z. Chen et al., CN/rGO@BPQDs high-low junctions with stretching spatial charge separation ability for photocatalytic degradation and H2O2 production. Appl. Catal. B 266, 118602 (2020). https://doi.org/10.1016/j.apcatb.2020.118602
Y. Kofuji, S. Ohkita, Y. Shiraishi, H. Sakamoto, S. Tanaka et al., Graphitic carbon nitride doped with biphenyl diimide: efficient photocatalyst for hydrogen peroxide production from water and molecular oxygen by sunlight. ACS Catal. 6, 7021–7029 (2016). https://doi.org/10.1021/acscatal.6b02367
Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan et al., Efficient visible-light-driven selective oxygen reduction to hydrogen peroxide by oxygen-enriched graphitic carbon nitride polymers. Energy Environ. Sci. 11, 2581–2589 (2018). https://doi.org/10.1039/c8ee01316k
H.-I. Kim, Y. Choi, S. Hu, W. Choi, J.-H. Kim, Photocatalytic hydrogen peroxide production by anthraquinone-augmented polymeric carbon nitride. Appl. Catal. B 229, 121–129 (2018). https://doi.org/10.1016/j.apcatb.2018.01.060
S. Zhao, T. Guo, X. Li, T. Xu, B. Yang et al., Carbon nanotubes covalent combined with graphitic carbon nitride for photocatalytic hydrogen peroxide production under visible light. Appl. Catal. B 224, 725–732 (2018). https://doi.org/10.1016/j.apcatb.2017.11.005
C. Feng, L. Tang, Y. Deng, J. Wang, Y. Liu et al., A novel sulfur-assisted annealing method of g-C3N4 nanosheet compensates for the loss of light absorption with further promoted charge transfer for photocatalytic production of H2 and H2O2. Appl. Catal. B 281, 119539 (2021). https://doi.org/10.1016/j.apcatb.2020.119539
N. Lu, N. Liu, Y. Hui, K. Shang, N. Jiang et al., Characterization of highly effective plasma-treated g-C3N4 and application to the photocatalytic H2O2 production. Chemosphere 241, 124927 (2020). https://doi.org/10.1016/j.chemosphere.2019.124927
Y. Li, W. Ho, K. Lv, B. Zhu, S.C. Lee, Carbon vacancy-induced enhancement of the visible light-driven photocatalytic oxidation of NO over g-C3N4 nanosheets. Appl. Surf. Sci. 430, 380–389 (2018). https://doi.org/10.1016/j.apsusc.2017.06.054
Y. Zhao, Y. Liu, Z. Wang, Y. Ma, Y. Zhou et al., Carbon nitride assisted 2D conductive metal-organic frameworks composite photocatalyst for efficient visible light-driven H2O2 production. Appl. Catal. B 289, 120035 (2021). https://doi.org/10.1016/j.apcatb.2021.120035
A. Fujishima, K. Honda, Electrochemical photolysis of water at a semiconductor electrode. Nature 238, 37 (1972). https://doi.org/10.1038/238037a0
A. Fujishima, T.N. Rao, D.A. Tryk, Titanium dioxide photocatalysis. J. Photochem. Photobiol. C 1, 1–21 (2000). https://doi.org/10.1016/S1389-5567(00)00002-2
H. Goto, Y. Hanada, T. Ohno, M. Matsumura, Quantitative analysis of superoxide ion and hydrogen peroxide produced from molecular oxygen on photoirradiated TiO2 particles. J. Catal. 225, 223–229 (2004). https://doi.org/10.1016/j.jcat.2004.04.001
T. Baran, S. Wojtyla, A. Minguzzi, S. Rondinini, A. Vertova, Achieving efficient H2O2 production by a visible-light absorbing, highly stable photosensitized TiO2. Appl. Catal. B 244, 303–312 (2019). https://doi.org/10.1016/j.apcatb.2018.11.044
B.O. Burek, D.W. Bahnemann, J.Z. Bloh, Modeling and optimization of the photocatalytic reduction of molecular oxygen to hydrogen peroxide over titanium dioxide. ACS Catal. 9, 25–37 (2019). https://doi.org/10.1021/acscatal.8b03638
M. Teranishi, S.-I. Naya, H. Tada, In-situ liquid phase synthesis of hydrogen peroxide from molecular oxygen using gold nanoparticle-loaded titanium(IV) dioxide photocatalyst. J. Am. Chem. Soc. 132, 7850 (2010). https://doi.org/10.1021/ja102651g
X.Z. Li, C.C. Chen, J.C. Zhao, Mechanism of photodecomposition of H2O2 on TiO2 surfaces under visible light irradiation. Langmuir 17, 4118–4122 (2001). https://doi.org/10.1021/la010035s
M. Teranishi, R. Hoshino, S.-I. Naya, H. Tada, Gold-nanoparticle-loaded carbonate-modified titanium(IV) oxide surface: visible-light-driven formation of hydrogen peroxide from oxygen. Angew. Chem. Int. Ed. 55, 12773–12777 (2016). https://doi.org/10.1002/anie.201606734
M. Teranishi, S.-I. Naya, H. Tada, Temperature- and pH-dependence of hydrogen peroxide formation from molecular oxygen by gold nanoparticle-loaded titanium(IV) oxide photocatalyst. J. Phys. Chem. C 120, 1083–1088 (2016). https://doi.org/10.1021/acs.jpcc.5b10626
R. Miah, T. Ohsaka, Cathodic detection of H2O2 using iodide-modified gold electrode in alkaline media. Anal. Chem. 78, 1200–1205 (2006). https://doi.org/10.1021/ac0515935
T. Hirakawa, K. Yawata, Y. Nosaka, Photocatalytic reactivity for O2·- and OH· radical formation in anatase and rutile TiO2 suspension as the effect of H2O2 addition. Appl. Catal. A 325, 105–111 (2007). https://doi.org/10.1016/j.apcata.2007.03.015
D. Tsukamoto, A. Shiro, Y. Shiraishi, Y. Sugano, S. Ichikawa et al., Photocatalytic H2O2 production from ethanol/O2 system using TiO2 loaded with Au-Ag bimetallic alloy nanoparticles. ACS Catal. 2, 599–603 (2012). https://doi.org/10.1021/cs2006873
E.M. Goliaei, N. Seriani, Structure and electronic properties of small silver-gold clusters on titania photocatalysts for H2O2 production: an investigation with density functional theory. J. Phys. Chem. C 123, 2855–2863 (2019). https://doi.org/10.1021/acs.jpcc.8b09300
X.T. Zheng, A. Ananthanarayanan, K.Q. Luo, P. Chen, Glowing graphene quantum dots and carbon dots: properties, syntheses, and biological applications. Small 11, 1620–1636 (2015). https://doi.org/10.1002/smll.201402648
Q. Mei, K. Zhang, G. Guan, B. Liu, S. Wang et al., Highly efficient photoluminescent graphene oxide with tunable surface properties. Chem. Commun. 46, 7319–7321 (2010). https://doi.org/10.1039/c0cc02374d
L. Zheng, H. Su, J. Zhang, L.S. Walekar, H.V. Molamahmood et al., Highly selective photocatalytic production of H2O2 on sulfur and nitrogen for co-doped graphene quantum dots tuned TiO2. Appl. Catal. B 239, 475–484 (2018). https://doi.org/10.1016/j.apcatb.2018.08.031
L. Zheng, J. Zhang, Y.H. Hu, M. Long, Enhanced photocatalytic production of H2O2 by nafion coatings on S, N-codoped graphene-quantum-dots-modified TiO2. J. Phys. Chem. C 123, 13693–13701 (2019). https://doi.org/10.1021/acs.jpcc.9b02311
R.X. Cai, Y. Kubota, A. Fujishima, Effect of copper ions on the formation of hydrogen peroxide from photocatalytic titanium dioxide particles. J. Catal. 219, 214–218 (2003). https://doi.org/10.1016/S0021-9517(03)00197-0
V. Maurino, C. Minero, E. Pelizzetti, G. Mariella, A. Arbezzano et al., Influence of Zn (II) adsorption on the photocatalytic activity and the production of H2O2 over irradiated TiO2. Res. Chem. Intermed. 33, 319–332 (2007). https://doi.org/10.1163/156856707779238711
V. Maurino, C. Minero, G. Mariella, E. Pelizzetti, Sustained production of H2O2 on irradiated TiO2 - fluoride systems. Chem. Commun. (2005). https://doi.org/10.1039/b418789j
Y. Shiraishi, S. Kanazawa, D. Tsukamoto, A. Shiro, Y. Sugano et al., Selective hydrogen peroxide formation by titanium dioxide photocatalysis with benzylic alcohols and molecular oxygen in water. ACS Catal. 3, 2222–2227 (2013). https://doi.org/10.1021/cs400511q
G. Zuo, B. Li, Z. Guo, L. Wang, F. Yang et al., Efficient photocatalytic hydrogen peroxide production over TiO2 passivated by SnO2. Catalysts 9, 623 (2019). https://doi.org/10.3390/catal9070623
K. Awa, S.-I. Naya, M. Fujishima, H. Tada, A three-component plasmonic photocatalyst consisting of gold nanoparticle and TiO2-SnO2 nanohybrid with heteroepitaxial junction: hydrogen peroxide synthesis. J. Phys. Chem. C 124, 7797–7802 (2020). https://doi.org/10.1021/acs.jpcc.9b11875
M. Nagamitsu, K. Awa, H. Tada, Hydrogen peroxide synthesis from water and oxygen using a three-component nanohybrid photocatalyst consisting of Au particle-loaded rutile TiO2 and RuO2 with a heteroepitaxial junction. Chem. Commun. 56, 8190–8193 (2020). https://doi.org/10.1039/d0cc03327h
C. Chu, D. Huang, Q. Zhu, E. Stavitski, J.A. Spies et al., Electronic tuning of metal nanoparticles for highly efficient photocatalytic hydrogen peroxide production. ACS Catal. 9, 626–631 (2019). https://doi.org/10.1021/acscatal.8b03738
R. Ma, L. Wang, H. Wang, Z. Liu, M. Xing et al., Solid acids accelerate the photocatalytic hydrogen peroxide synthesis over a hybrid catalyst of Titania nanotube with carbon dot. Appl. Catal. B 244, 594–603 (2019). https://doi.org/10.1016/j.apcatb.2018.11.087
X. Wu, X. Zhang, S. Zhao, Y. Gong, R. Djellabi et al., Highly-efficient photocatalytic hydrogen peroxide production over polyoxometalates covalently immobilized onto titanium dioxide. Appl. Catal. A 591, 117271 (2020). https://doi.org/10.1016/j.apcata.2019.117271
G.-H. Moon, W. Kim, A.D. Bokare, N.-E. Sung, W. Choi, Solar production of H2O2 on reduced graphene oxide-TiO2 hybrid photocatalysts consisting of earth-abundant elements only. Energy Environ. Sci. 7, 4023–4028 (2014). https://doi.org/10.1039/c4ee02757d
R. He, D. Xu, B. Cheng, J. Yu, W. Ho, Review on nanoscale Bi-based photocatalysts. Nanoscale Horiz. 3, 464–504 (2018). https://doi.org/10.1039/c8nh00062j
L. Huang, Z. Duan, Y. Song, Q. Li, L. Chen, BiVO4 microplates with oxygen vacancies decorated with metallic Cu and Bi nanoparticles for CO2 photoreduction. ACS Appl. Nano Mater. 4, 3576–3585 (2021). https://doi.org/10.1021/acsanm.1c00115
J.H. Kim, J.S. Lee, Elaborately modified BiVO4 photoanodes for solar water splitting. Adv. Mater. 31, 1806938 (2019). https://doi.org/10.1002/adma.201806938
W. Zhao, Y. Feng, H. Huang, P. Zhou, J. Li et al., A novel Z-scheme Ag3VO4/BiVO4 heterojunction photocatalyst: study on the excellent photocatalytic performance and photocatalytic mechanism. Appl. Catal. B 245, 448–458 (2019). https://doi.org/10.1016/j.apcatb.2019.01.001
T. Hirakawa, Y. Nosaka, Selective production of superoxide ions and hydrogen peroxide over nitrogen- and sulfur-doped TiO2 photocatalysts with visible light in aqueous suspension systems. J. Phys. Chem. C 112, 15818–15823 (2008). https://doi.org/10.1021/jp8055015
M.A. Henderson, A surface science perspective on TiO2 photocatalysis. Surf. Sci. Rep. 66, 185–297 (2011). https://doi.org/10.1016/j.surfrep.2011.01.001
A. Kudo, K. Omori, H. Kato, A novel aqueous process for preparation of crystal form-controlled and highly crystalline BiVO4 powder from layered vanadates at room temperature and its photocatalytic and photophysical properties. J. Am. Chem. Soc. 121, 11459–11467 (1999). https://doi.org/10.1021/ja992541y
H. Hirakawa, S. Shiota, Y. Shiraishi, H. Sakamoto, S. Ichikawa et al., Au nanoparticles supported on BiVO4: effective inorganic photocatalysts for H2O2 production from water and O2 under visible light. ACS Catal. 6, 4976–4982 (2016). https://doi.org/10.1021/acscatal.6b01187
V. Kumar, S. O’Donnell, B. Zoellner, J. Martinez, G. Wang et al., Interfacing plasmonic nanoparticles with ferroelectrics for hot-carrier-driven photocatalysis: impact of schottky barrier height. ACS Appl. Energy Mater. 2, 7690–7699 (2019). https://doi.org/10.1021/acsaem.9b01682
X. Chen, R.T. Pekarek, J. Gu, A. Zakutayev, K.E. Hurst et al., Transient evolution of the built-in field at junctions of GaAs. ACS Appl. Mater. Interfaces 12, 40339–40346 (2020). https://doi.org/10.1021/acsami.0c11474
K. Wang, M. Wang, J. Yu, D. Liao, H. Shi et al., BiVO4 microparticles decorated with Cu @Au core-shell nanostructures for photocatalytic H2O2 production. ACS Appl. Nano Mater. 4, 13158–13166 (2021). https://doi.org/10.1021/acsanm.1c02688
M. Teranishi, T. Kunimoto, S.-I. Naya, H. Kobayashi, H. Tada, Visible-light-driven hydrogen peroxide synthesis by a hybrid photocatalyst consisting of bismuth vanadate and bis(hexafluoroacetylacetonato)copper (II) complex. J. Phys. Chem. C 124, 3715–3721 (2020). https://doi.org/10.1021/acs.jpcc.9b11568
Q. Li, X. Li, S. Wageh, A.A. Al-Ghamdi, J. Yu, CdS/graphene nanocomposite photocatalysts. Adv. Energy Mater. 5, 1500010 (2015). https://doi.org/10.1002/aenm.201500010
A. Kudo, Y. Miseki, Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 38, 253–278 (2009). https://doi.org/10.1039/b800489g
Y. Xu, M.A.A. Schoonen, The absolute energy positions of conduction and valence bands of selected semiconducting minerals. Am. Mineral. 85, 543–556 (2000). https://doi.org/10.2138/am-2000-0416
Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang et al., Highly efficient visible-light-driven photocatalytic hydrogen production of CdS-cluster-decorated graphene nanosheets. J. Am. Chem. Soc. 133, 10878–10884 (2011). https://doi.org/10.1021/ja2025454
S. Thakur, T. Kshetri, N.H. Kim, J.H. Lee, Sunlight-driven sustainable production of hydrogen peroxide using a CdS-graphene hybrid photocatalyst. J. Catal. 345, 78–86 (2017). https://doi.org/10.1016/j.jcat.2016.10.028
D. Stone, Y. Ben-Shahar, N. Waiskopf, U. Banin, The metal type governs photocatalytic reactive oxygen species formation by semiconductor-metal hybrid nanoparticles. ChemCatChem 10, 5119–5123 (2018). https://doi.org/10.1002/cctc.201801306
J.H. Lee, H. Cho, S.O. Park, J.M. Hwang, Y. Hong et al., High performance H2O2 production achieved by sulfur-doped carbon on CdS photocatalyst via inhibiting reverse H2O2 decomposition. Appl. Catal. B 284, 119690 (2021). https://doi.org/10.1016/j.apcatb.2020.119690
J. Lee, O.K. Farha, J. Roberts, K.A. Scheidt, S.T. Nguyen et al., Metal-organic framework materials as catalysts. Chem. Soc. Rev. 38, 1450–1459 (2009). https://doi.org/10.1039/b807080f
H. Wang, X. Yuan, Y. Wu, G. Zeng, X. Chen et al., Synthesis and applications of novel graphitic carbon nitride/metal-organic frameworks mesoporous photocatalyst for dyes removal. Appl. Catal. B 174, 445–454 (2015). https://doi.org/10.1016/j.apcatb.2015.03.037
R. Li, W. Zhang, K. Zhou, Metal-organic-framework-based catalysts for photoreduction of CO2. Adv. Mater. 30, 1705512 (2018). https://doi.org/10.1002/adma.201705512
J. Qiu, X. Zhang, Y. Feng, X. Zhang, H. Wang et al., Modified metal-organic frameworks as photocatalysts. Appl. Catal. B 231, 317–342 (2018). https://doi.org/10.1016/j.apcatb.2018.03.039
J. Guo, Y. Wan, Y. Zhu, M. Zhao, Z. Tang et al., Advanced photocatalysts based on metal nanoparticle/metal-organic framework composites. Nano Res. 14, 2037–2052 (2021). https://doi.org/10.1007/s12274-020-3182-1
Y. Isaka, Y. Kondo, Y. Kawase, Y. Kuwahara, K. Mori et al., Photocatalytic production of hydrogen peroxide through selective two-electron reduction of dioxygen utilizing amine-functionalized MIL-125 deposited with nickel oxide nanoparticles. Chem. Commun. 54, 9270–9273 (2018). https://doi.org/10.1039/c8cc02679c
Y. Isaka, Y. Kawase, Y. Kuwahara, K. Mori, H. Yamashita et al., Two-phase system utilizing hydrophobic metal-organic frameworks (MOFs) for photocatalytic synthesis of hydrogen peroxide. Angew. Chem. Int. Ed. 58, 5402–5406 (2019). https://doi.org/10.1002/anie.201901961
Y. Kawase, Y. Isaka, Y. Kuwahara, K. Mori, H. Yamashita et al., Ti cluster-alkylated hydrophobic MOFs for photocatalytic production of hydrogen peroxide in two-phase systems. Chem. Commun. 55, 6743–6746 (2019). https://doi.org/10.1039/c9cc02380a
X. Chen, Y. Kuwahara, K. Mori, C. Louis, H. Yamashita, A hydrophobic titanium doped zirconium-based metal organic framework for photocatalytic hydrogen peroxide production in a two-phase system. J. Mater. Chem. A 8, 1904–1910 (2020). https://doi.org/10.1039/c9ta11120d
J.-D. Xiao, L. Han, J. Luo, S.-H. Yu, H.-L. Jiang et al., Integration of plasmonic effects and schottky junctions into metal-organic framework composites: steering charge flow for enhanced visible-light photocatalysis. Angew. Chem. Int. Ed. 57, 1103–1107 (2018). https://doi.org/10.1002/anie.201711725
C. Liu, Q. Sun, L. Lin, J. Wang, C. Zhang et al., Ternary MOF-on-MOF heterostructures with controllable architectural and compositional complexity via multiple selective assembly. Nat. Commun. 11, 4971 (2020). https://doi.org/10.1038/s41467-020-18776-z
J. Ran, J. Qu, H. Zhang, T. Wen, H. Wang et al., 2D metal organic framework nanosheet: a universal platform promoting highly efficient visible-light-induced hydrogen production. Adv. Energy Mater. 9, 1803402 (2019). https://doi.org/10.1002/aenm.201803402
C. Liu, T. Bao, L. Yuan, C. Zhang, J. Wang et al., Semiconducting MOF@ZnS heterostructures for photocatalytic hydrogen peroxide production: heterojunction coverage matters. Adv. Funct. Mater. 32, 2111404 (2022). https://doi.org/10.1002/adfm.202111404
S. Chen, T. Luo, X. Li, K. Chen, J. Fu et al., Identification of the highly active Co-N4 coordination motif for selective oxygen reduction to hydrogen peroxide. J. Am. Chem. Soc. 144, 14505–14516 (2022). https://doi.org/10.1021/jacs.2c0119414505
X. Li, S. Tang, S. Dou, H.J. Fan, T.S. Choksi et al., Molecule confined isolated metal sites enable the electrocatalytic synthesis of hydrogen peroxide. Adv. Mater. 34, 2104891 (2022). https://doi.org/10.1002/adma.202104891
J. Wu, M. Hou, Z. Chen, W. Hao, X. Pan et al., Composition engineering of amorphous nickel boride nanoarchitectures enabling highly efficient electrosynthesis of hydrogen peroxide. Adv. Mater. 34, 2202995 (2022). https://doi.org/10.1002/adma.202202995
P.T. Smith, Y. Kim, B.P. Benke, K. Kim, C.J. Chang, Supramolecular tuning enables selective oxygen reduction catalyzed by cobalt porphyrins for direct electrosynthesis of hydrogen peroxide. Angew. Chem. Int. Ed. 59, 4902–4907 (2020). https://doi.org/10.1002/anie.201916131
J.Y. Zhang, C.A. Xia, H.F. Wang, C. Tang, Recent advances in electrocatalytic oxygen reduction for on-site hydrogen peroxide synthesis in acidic media. J. Energy Chem. 67, 432–450 (2022). https://doi.org/10.1016/j.jechem.2021.10.013
S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvio, V. Colic et al., Toward the decentralized electrochemical production of H2O2: a focus on the catalysis. ACS Catal. 8, 4064–4081 (2018). https://doi.org/10.1021/acscatal.8b00217
A.W.E. Chan, R. Hoffmann, W. Ho, Theoretical aspects of photoinitiated chemisorption, dissociation, and desorption of oxygen on platinum (111). Langmuir 8, 1111–1119 (1992). https://doi.org/10.1021/la00040a017
X. Song, N. Li, H. Zhang, H. Wang, L. Wang et al., Promotion of hydrogen peroxide production on graphene-supported atomically dispersed platinum: effects of size on oxygen reduction reaction pathway. J. Power Sources 435, 226771 (2019). https://doi.org/10.1016/j.jpowsour.2019.226771
S. Yang, J. Kim, Y.J. Tak, A. Soon, H. Lee, Single-atom catalyst of platinum supported on titanium nitride for selective electrochemical reactions. Angew. Chem. Int. Ed. 55, 2058–2062 (2016). https://doi.org/10.1002/anie.201509241
C.H. Choi, M. Kim, H.C. Kwon, S.J. Cho, S. Yun et al., Tuning selectivity of electrochemical reactions by atomically dispersed platinum catalyst. Nat. Commun. 7, 10922 (2016). https://doi.org/10.1038/ncomms10922
S. Yang, Y.J. Tak, J. Kim, A. Soon, H. Lee, Support effects in single-atom platinum catalysts for electrochemical oxygen reduction. ACS Catal. 7, 1301–1307 (2017). https://doi.org/10.1021/acscatal.6b02899
R.-Q. Zhang, T.-H. Lee, B.-D. Yu, C. Stampfl, A. Soon, The role of titanium nitride supports for single-atom platinum-based catalysts in fuel cell technology. Phys. Chem. Chem. Phys. 14, 16552–16557 (2012). https://doi.org/10.1039/c2cp41392b
C.H. Choi, H.C. Kwon, S. Yook, H. Shin, H. Kim et al., Hydrogen peroxide synthesis via enhanced two-electron oxygen reduction pathway on carbon-coated Pt surface. J. Phys. Chem. C 118, 30063–30070 (2014). https://doi.org/10.1021/jp5113894
Q. Chang, P. Zhang, A.H.B. Mostaghimi, X. Zhao, S.R. Denny et al., Promoting H2O2 production via 2-electron oxygen reduction by coordinating partially oxidized Pd with defect carbon. Nat. Commun. 11, 2178 (2020). https://doi.org/10.1038/s41467-020-15843-3
M. Ledendecker, E. Pizzutilo, G. Malta, G.V. Fortunato, K.J.J. Mayrhofer et al., Isolated Pd sites as selective catalysts for electrochemical and direct hydrogen peroxide synthesis. ACS Catal. 10, 5928–5938 (2020). https://doi.org/10.1021/acscatal.0c01305
Y.L. Wang, S. Gurses, N. Felyey, A. Boubnov, S.S. Mao et al., In-situ deposition of Pd during oxygen reduction yields highly selective and active electrocatalysts for direct H2O2 production. ACS Catal. 9, 8453–8463 (2019). https://doi.org/10.1021/acscatal.9b01758
D. Mei, Z.D. He, Y.L. Zheng, D.C. Jiang, Y.-X. Chen et al., Mechanistic and kinetic implications on the ORR on an Au (100) electrode: pH, temperature and H-D kinetic isotope effects. Phys. Chem. Chem. Phys. 16, 13762–13773 (2014). https://doi.org/10.1039/c4cp00257a
Y. Lu, Y. Jiang, X. Gao, W. Chen, Charge state-dependent catalytic activity of [Au25(SC12H25)18] nanoclusters for the two-electron reduction of dioxygen to hydrogen peroxide. Chem. Commun. 50, 8464–8467 (2014). https://doi.org/10.1039/c4cc01841a
H. Tsunoyama, N. Ichikuni, H. Sakurai, T. Tsukuda, Effect of electronic structures of Au clusters stabilized by poly (N-vinyl-2-pyrrolidone) on aerobic oxidation catalysis. J. Am. Chem. Soc. 131, 7086–7093 (2009). https://doi.org/10.1021/ja810045y
S. Siahrostami, A. Verdaguer-Casdevall, M. Karamad, I. Chorkendorff, I. Stephens et al., Activity and selectivity for O2 Reduction to H2O2 on transition metal surfaces. ECS Trans. 58, 53–62 (2013). https://doi.org/10.1149/05802.0053ecst
J.S. Jirkovsky, I. Panas, E. Ahlberg, M. Halasa, S. Romani et al., Single atom hot-spots at Au-Pd nanoalloys for electrocatalytic H2O2 production. J. Am. Chem. Soc. 133, 19432–19441 (2011). https://doi.org/10.1021/ja206477z
A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida et al., Trends in the electrochemical synthesis of H2O2: enhancing activity and selectivity by electrocatalytic site engineering. Nano Lett. 14, 1603–1608 (2014). https://doi.org/10.1021/nl500037x
E. Pizzutilo, O. Kasian, C.H. Choi, S. Cherevko, G.J. Hutchings et al., Electrocatalytic synthesis of hydrogen peroxide on Au-Pd nanoparticles: from fundamentals to continuous production. Chem. Phys. Lett. 683, 436–442 (2017). https://doi.org/10.1016/j.cplett.2017.01.071
Y. Dong, J. Su, S. Zhou, M. Wang, S. Huang et al., Carbon-based dots for the electrochemical production of hydrogen peroxide. Chem. Commun. 56, 7609–7612 (2020). https://doi.org/10.1039/c9cc09987e
Y.-L. Wang, S.-S. Li, X.-H. Yang, G.-Y. Xu, Z.-C. Zhu et al., One minute from pristine carbon to an electrocatalyst for hydrogen peroxide production. J. Mater. Chem. A 7, 21329–21337 (2019). https://doi.org/10.1039/c9ta04788c
V. Colic, S. Yang, Z. Revay, I.E.L. Stephens, I. Chorkendorff, Carbon catalysts for electrochemical hydrogen peroxide production in acidic media. Electrochim. Acta 272, 192–202 (2018). https://doi.org/10.1016/j.electacta.2018.03.170
Y. Bu, Y. Wang, G.-F. Han, Y. Zhao, X. Ge et al., Carbon-based electrocatalysts for efficient hydrogen peroxide production. Adv. Mater. 33, 2103266 (2021). https://doi.org/10.1002/adma.202103266
Y. Liu, X. Quan, X. Fan, H. Wang, S. Chen, High-yield electrosynthesis of hydrogen peroxide from oxygen reduction by hierarchically porous carbon. Angew. Chem. Int. Ed. 54, 6837–6841 (2015). https://doi.org/10.1002/anie.201502396
L. Tao, Q. Wang, S. Dou, Z. Ma, J. Huo et al., Edge-rich and dopant-free graphene as a highly efficient metal-free electrocatalyst for the oxygen reduction reaction. Chem. Commun. 52, 2764–2767 (2016). https://doi.org/10.1039/c5cc09173j
S. Chen, Z. Chen, S. Siahrostami, T.R. Kim, D. Nordlund et al., Defective carbon-based materials for the electrochemical synthesis of hydrogen peroxide. ACS Sustainable Chem. Eng. 6, 311–317 (2018). https://doi.org/10.1021/acssuschemeng.7b02517
Y.J. Sa, J.H. Kim, S.H. Joo, Active edge-site-rich carbon nanocatalysts with enhanced electron transfer for efficient electrochemical hydrogen peroxide production. Angew. Chem. Int. Ed. 58, 1100–1105 (2019). https://doi.org/10.1002/anie.201812435
M. Yeddala, P. Thakur, A. Anugraha, T.N. Narayanan, Electrochemically derived functionalized graphene for bulk production of hydrogen peroxide. Beilstein J. Nanotechnol. 11, 432–442 (2020). https://doi.org/10.3762/bjnano.11.34
X. Xu, D. Kong, J. Liang, Y. Gao, Q. Yang et al., Bottom-up construction of microporous catalyst with identical active sites for efficient hydrogen peroxide production. Carbon 171, 931–937 (2021). https://doi.org/10.1016/j.carbon.2020.09.080
H.W. Kim, M.B. Ross, N. Kornienko, L. Zhang, J.H. Guo et al., Efficient hydrogen peroxide generation using reduced graphene oxide-based oxygen reduction electrocatalysts. Nat. Catal. 1, 282–290 (2018). https://doi.org/10.1038/s41929-018-0044-2
W. Wang, Y. Hu, Y. Liu, Z. Zheng, S. Chen, Self-powered and highly efficient production of H2O2 through a Zn-air battery with oxygenated carbon electrocatalyst. ACS Appl. Mater. Interfaces 10, 31855–31859 (2018). https://doi.org/10.1021/acsami.8b11703
H. Zhang, Y. Li, Y. Zhao, G. Li, F. Zhang, Carbon black oxidized by air calcination for enhanced H2O2 generation and effective organics degradation. ACS Appl. Mater. Interfaces 11, 27846–27853 (2019). https://doi.org/10.1021/acsami.9b07765
J. Zhu, X. Xiao, K. Zheng, F. Li, G. Ma et al., KOH-treated reduced graphene oxide: 100% selectivity for H2O2 electroproduction. Carbon 153, 6–11 (2019). https://doi.org/10.1016/j.carbon.2019.07.009
G.-F. Han, F. Li, W. Zou, M. Karamad, J.-P. Jeon et al., Building and identifying highly active oxygenated groups in carbon materials for oxygen reduction to H2O2. Nat. Commun. 11, 2209 (2020). https://doi.org/10.1038/s41467-020-15782-z
Y. Xue, X. Du, W. Cai, Z. Sun, Y. Zhang et al., Ramie biomass derived nitrogen-doped activated carbon for efficient electrocatalytic production of hydrogen peroxide. J. Electrochem. Soc. 165, E171–E176 (2018). https://doi.org/10.1149/2.0681805jes
Y.-H. Lee, F. Li, K.-H. Chang, C.-C. Hu, T. Ohsaka, Novel synthesis of N-doped porous carbons from collagen for electrocatalytic production of H2O2. Appl. Catal. B 126, 208–214 (2012). https://doi.org/10.1016/j.apcatb.2012.06.031
J. Zhang, G. Zhang, S. Jin, Y. Zhou, Q. Ji et al., Graphitic N in nitrogen-doped carbon promotes hydrogen peroxide synthesis from electrocatalytic oxygen reduction. Carbon 163, 154–161 (2020). https://doi.org/10.1016/j.carbon.2020.02.084
L. Han, Y. Sun, S. Li, C. Cheng, C.E. Halbig et al., In-plane carbon lattice-defect regulating electrochemical oxygen reduction to hydrogen peroxide production over nitrogen-doped graphene. ACS Catal. 9, 1283–1288 (2019). https://doi.org/10.1021/acscatal.8b03734
V. Perazzolo, C. Durante, R. Pilot, A. Paduano, J. Zheng et al., Nitrogen and sulfur doped mesoporous carbon as metal-free electrocatalysts for the in-situ production of hydrogen peroxide. Carbon 95, 949–963 (2015). https://doi.org/10.1016/j.carbon.2015.09.002
D.-W. Wang, D. Su, Heterogeneous nanocarbon materials for oxygen reduction reaction. Energy Environ. Sci. 7, 576–591 (2014). https://doi.org/10.1039/c3ee43463j
D. Iglesias, A. Giuliani, M. Melchionna, S. Marchesan, A. Criado et al., N-doped graphitized carbon nanohorns as a forefront electrocatalyst in highly selective O2 reduction to H2O2. Chem 4, 106–123 (2018). https://doi.org/10.1016/j.chempr.2017.10.013
K. Gong, F. Du, Z. Xia, M. Durstock, L. Dai, Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 323, 760–764 (2009). https://doi.org/10.1126/science.1168049
X. Liu, L. Dai, Carbon-based metal-free catalysts. Nat. Rev. Mater. 1, 16064 (2016). https://doi.org/10.1038/natrevmats.2016.64
L. Dai, Y. Xue, L. Qu, H.-J. Choi, J.-B. Baek, Metal-free catalysts for oxygen reduction reaction. Chem. Rev. 115, 4823–4892 (2015). https://doi.org/10.1021/cr5003563
T.-P. Fellinger, F. Hasche, P. Strasser, M. Antonietti, Mesoporous nitrogen-doped carbon for the electrocatalytic synthesis of hydrogen peroxide. J. Am. Chem. Soc. 134, 4072–4075 (2012). https://doi.org/10.1021/ja300038p
J. Park, Y. Nabae, T. Hayakawa, M.-A. Kakimoto, Highly selective two-electron oxygen reduction catalyzed by mesoporous nitrogen-doped carbon. ACS Catal. 4, 3749–3754 (2014). https://doi.org/10.1021/cs5008206
Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. Dresp et al., Efficient electrochemical hydrogen peroxide production from molecular oxygen on nitrogen-doped mesoporous carbon catalysts. ACS Catal. 8, 2844–2856 (2018). https://doi.org/10.1021/acscatal.7b03464
J. Li, C. Shi, A. Bao, J. Jia, Development of boron-doped mesoporous carbon materials for use in CO2 capture and electrochemical generation of H2O2. ACS Omega 6, 8438–8446 (2021). https://doi.org/10.1021/acsomega.1c00197
G. Chen, J. Liu, Q. Li, P. Guan, X. Yu et al., A direct H2O2 production based on hollow porous carbon sphere-sulfur nanocrystal composites by confinement effect as oxygen reduction electrocatalysts. Nano Res. 12, 2614–2622 (2019). https://doi.org/10.1007/s12274-019-2496-3
S. Chen, Z. Chen, S. Siahrostami, D. Higgins, D. Nordlund et al., Designing boron nitride islands in carbon materials for efficient electrochemical synthesis of hydrogen peroxide. J. Am. Chem. Soc. 140, 7851–7859 (2018). https://doi.org/10.1021/jacs.8b02798
X.-H. Yan, Z. Meng, J. Liu, T. Xue, Sodium ferric EDTA-derived Fe-N-C material for selectively electrocatalytic synthesis of hydrogen peroxide. Mater. Lett. 217, 171–173 (2018). https://doi.org/10.1016/j.matlet.2018.01.044
A. Byeon, J. Cho, J.G.M. Kim, K.H. Chae, H.-Y. Park et al., High-yield electrochemical hydrogen peroxide production from an enhanced two-electron oxygen reduction pathway by mesoporous nitrogen-doped carbon and manganese hybrid electrocatalysts. Nanoscale Horiz. 5, 832–838 (2020). https://doi.org/10.1039/c9nh00783k
M. Suk, M.W. Chung, M.H. Han, H.-S. Oh, C.H. Choi et al., Selective H2O2 production on surface-oxidized metal-nitrogen-carbon electrocatalysts. Catal. Today 359, 99–105 (2021). https://doi.org/10.1016/j.cattod.2019.05.034
B. You, N. Jiang, M. Sheng, W.S. Drisdell, J. Yano et al., Bimetal-organic framework self-adjusted synthesis of support-free nonprecious electrocatalysts for efficient oxygen reduction. ACS Catal. 5, 7068–7076 (2015). https://doi.org/10.1021/acscatal.5b02325
J. Gao, H.B. Yang, X. Huang, S.-F. Hung, W. Cai et al., Enabling direct H2O2 production in acidic media through rational design of transition metal single atom catalyst. Chem 6, 658–674 (2020). https://doi.org/10.1016/j.chempr.2019.12.008
Y. Sun, L. Silvioli, N.R. Sahraie, W. Ju, J. Li et al., Activity-selectivity trends in the electrochemical production of hydrogen peroxide over single-site metal-nitrogen-carbon catalysts. J. Am. Chem. Soc. 141, 12372–12381 (2019). https://doi.org/10.1021/jacs.9b05576
E. Jung, H. Shin, B.-H. Lee, V. Efremov, S. Lee et al., Atomic-level tuning of Co-N-C catalyst for high-performance electrochemical H2O2 production. Nat. Mater. (2020). https://doi.org/10.1038/s41563-019-0571-5
M. Jin, W. Liu, J. Sun, X. Wang, S. Zhang et al., Highly dispersed Ag clusters for active and stable hydrogen peroxide production. Nano Res. 15, 5842–5847 (2022). https://doi.org/10.1007/s12274-022-4208-7
Z. Zheng, Y.H. Ng, D.-W. Wang, R. Amal, Epitaxial growth of Au-Pt-Ni nanorods for direct high selectivity H2O2 production. Adv. Mater. 28, 9949–9955 (2016). https://doi.org/10.1002/adma.201603662
S. Chen, T. Luo, K. Chen, Y. Lin, J. Fu et al., Chemical identification of catalytically active sites on oxygen-doped carbon nanosheet to decipher the high activity for electro-synthesis hydrogen peroxide. Angew. Chem. Int. Ed. 60, 16607–16614 (2021). https://doi.org/10.1002/anie.202104480
Z. Deng, X. Wang, Mechanism investigation of enhanced electrochemical H2O2 production performance on oxygen-rich hollow porous carbon spheres. Nano Res. 15, 4599–4605 (2022). https://doi.org/10.1007/s12274-022-4095-y
Y. Sun, S. Li, Z.P. Jovanov, D. Bernsmeier, P. Strasser et al., structure, activity, and faradaic efficiency of nitrogen-doped porous carbon catalysts for direct electrochemical hydrogen peroxide production. Chemsuschem 11, 3388–3395 (2018). https://doi.org/10.1002/cssc.201801583
Y. Hu, J. Zhang, T. Shen, Z. Li, K. Chen et al., Efficient electrochemical production of H2O2 on hollow N-doped carbon nanospheres with abundant micropores. ACS Appl. Mater. Interfaces 13, 29551–29557 (2021). https://doi.org/10.1021/acsami.1c05353
H. Gong, Z. Wei, Z. Gong, J. Liu, G. Ye et al., Low-coordinated Co-N-C on oxygenated graphene for efficient electrocatalytic H2O2 production. Adv. Funct. Mater. 32, 2106886 (2022). https://doi.org/10.1002/adfm.202106886
P.-T. Thuan-Nguyen, T. Petenzi, C. Ranjan, H.N. Randriamahazaka, J. Ghilane, Microwave assisted synthesis of carbon dots in ionic liquid as metal free catalyst for highly selective production of hydrogen peroxide. Carbon 130, 544–552 (2018). https://doi.org/10.1016/j.carbon.2018.01.070
L. Wang, L. Duan, L. Tong, L. Sun, Visible light-driven water oxidation catalyzed by mononuclear ruthenium complexes. J. Catal. 306, 129–132 (2013). https://doi.org/10.1016/j.jcat.2013.06.023
I. Katsounaros, S. Cherevko, A.R. Zeradjanin, K.J.J. Mayrhofer, Oxygen electrochemistry as a cornerstone for sustainable energy conversion. Angew. Chem. Int. Ed. 53, 102–121 (2014). https://doi.org/10.1002/anie.201306588
Z.-P. Wu, X.F. Lu, S.-Q. Zang, X.W. Lou, Non-noble-metal-based electrocatalysts toward the oxygen evolution reaction. Adv. Funct. Mater. 30, 1910274 (2020). https://doi.org/10.1002/adfm.201910274
V. Viswanathan, H.A. Hansen, J.K. Norskov, Selective electrochemical generation of hydrogen peroxide from water oxidation. J. Phys. Chem. Lett. 6, 4224–4228 (2015). https://doi.org/10.1021/acs.jpclett.5b02178
S. Siahrostami, G.-L. Li, V. Viswanathan, J.K. Norskov, One- or two-electron water oxidation, hydroxyl radical, or H2O2 evolution. J. Phys. Chem. Lett. 8, 1157–1160 (2017). https://doi.org/10.1021/acs.jpclett.6b02924
X. Shi, S. Siahrostami, G.-L. Li, Y. Zhang, P. Chakthranont et al., Understanding activity trends in electrochemical water oxidation to form hydrogen peroxide. Nat. Commun. 8, 701 (2017). https://doi.org/10.1038/s41467-017-00585-6
A. Nadar, S.S. Gupta, Y. Kar, S. Shetty, A.P. van Bavel et al., Evaluating the reactivity of BiVO4 surfaces for efficient electrocatalytic H2O2 production: a combined experimental and computational study. J. Phys. Chem. C 124, 4152–4161 (2020). https://doi.org/10.1021/acs.jpcc.9b11418
J.H. Baek, T.M. Gill, H. Abroshan, S. Park, X. Shi et al., Selective and efficient Gd-doped BiVO4 photoanode for two-electron water oxidation to H2O2. ACS Energy Lett. 4, 720–728 (2019). https://doi.org/10.1021/acsenergylett.9b00277
L. Li, Z. Hu, J.C. Yu, On-demand synthesis of H2O2 by water oxidation for sustainable resource production and organic pollutant degradation. Angew. Chem. Int. Ed. 59, 20538–20544 (2020). https://doi.org/10.1002/anie.202008031
T. Kang, B. Li, Q. Hao, W. Gao, F. Bin et al., Efficient hydrogen peroxide (H2O2) synthesis by CaSnO3 via two-electron water oxidation reaction. ACS Sustainable Chem. Eng. 8, 15005–15012 (2020). https://doi.org/10.1021/acssuschemeng.0c05449
S.Y. Park, H. Abroshan, X. Shi, H.S. Jung, S. Siahrostami et al., CaSnO3: an electrocatalyst for two-electron water oxidation reaction to form H2O2. ACS Energy Lett. 4, 352–357 (2019). https://doi.org/10.1021/acsenergylett.8b02303
S.R. Kelly, X. Shi, S. Back, L. Vallez, S.Y. Park et al., ZnO as an active and selective catalyst for electrochemical water oxidation to hydrogen peroxide. ACS Catal. 9, 4593–4599 (2019). https://doi.org/10.1021/acscatal.8b04873
J. Li, D. Solanki, Q. Zhu, X. Shen, G. Callander et al., Microstructural origin of selective water oxidation to hydrogen peroxide at low overpotentials: a study on Mn-alloyed TiO2. J. Mater. Chem. A 9, 18498–18505 (2021). https://doi.org/10.1039/d1ta05451a
S.-G. Xue, L. Tang, Y.-K. Tang, C.-X. Li, M.-L. Li et al., Selective electrocatalytic water oxidation to produce H2O2 using a C, N codoped TiO2 electrode in an acidic electrolyte. ACS Appl. Mater. Interfaces 12, 4423–4431 (2020). https://doi.org/10.1021/acsami.9b16937
K. Fuku, Y. Miyase, Y. Miseki, T. Gunji, K. Sayama, Enhanced oxidative hydrogen peroxide production on conducting glass anodes modified with metal oxides. ChemistrySelect 1, 5721–5726 (2016). https://doi.org/10.1002/slct.201601469
Y. Miyase, S. Iguchi, Y. Miseki, T. Gunji, K. Sayama, Electrochemical H2O2 production and accumulation from H2O by composite effect of Al2O3 and BiVO4. J. Electrochem. Soc. 166, H644–H649 (2019). https://doi.org/10.1149/2.0561913jes
F. Kuttassery, S. Mathew, S. Sagawa, S.N. Remello, A. Thomas et al., One electron-initiated two-electron oxidation of water by aluminum porphyrins with earth’s most abundant metal. Chemsuschem 10, 1909–1915 (2017). https://doi.org/10.1002/cssc.201700322
S.N. Remello, F. Kuttassery, S. Mathew, A. Thomas, D. Yamamoto et al., Two-electron oxidation of water to form hydrogen peroxide catalysed by silicon-porphyrins. Sustainable Energy Fuels 2, 1966–1973 (2018). https://doi.org/10.1039/c8se00102b
A. Sebastian, S.N. Remello, F. Kuttassery, S. Mathew, Y. Ohsaki et al., Protolytic behavior of water-soluble zinc (II) porphyrin and the electrocatalytic two-electron water oxidation to form hydrogen peroxide. J. Photochem. Photobiol. A 400, 112619 (2020). https://doi.org/10.1016/j.jphotochem.2020.112619
Y. Ohsaki, A. Thomas, F. Kuttassery, S. Mathew, S.N. Remello et al., Two-electron oxidation of water to form hydrogen peroxide initiated by one-electron oxidation of Tin (IV)-porphyrins. J. Photochem. Photobiol. A 401, 112732 (2020). https://doi.org/10.1016/j.jphotochem.2020.112732
Y. Ando, T. Tanaka, Proposal for a new system for simultaneous production of hydrogen and hydrogen peroxide by water electrolysis. Int. J. Hydrogen Energy 29, 1349–1354 (2004). https://doi.org/10.1016/j.ijhydene.2004.02.001
J.V. Macpherson, A practical guide to using boron doped diamond in electrochemical research. Phys. Chem. Chem. Phys. 17, 2935–2949 (2015). https://doi.org/10.1039/c4cp04022h
B. Marselli, J. Garcia-Gomez, P.A. Michaud, M.A. Rodrigo, C. Comninellis et al., Electrogeneration of hydroxyl radicals on boron-doped diamond electrodes. J. Electrochem. Soc. 150, D79–D83 (2003). https://doi.org/10.1149/1.1553790
P.A. Michaud, M. Panizza, L. Ouattara, T. Diaco, G. Foti et al., Electrochemical oxidation of water on synthetic boron-doped diamond thin film anodes. J. Appl. Electrochem. 33, 151–154 (2003). https://doi.org/10.1023/A:1024084924058
S. Mavrikis, M. Goeltz, S. Rosiwal, L. Wang, C. Ponce de Leon, Boron-doped diamond electrocatalyst for enhanced anodic H2O2 production. ACS Appl. Energy Mater. 3, 3169–3173 (2020). https://doi.org/10.1021/acsaem.0c00093
S. Mavrikis, M. Goeltz, S.C. Perry, F. Bogdan, P.K. Leung et al., Effective hydrogen peroxide production from electrochemical water oxidation. ACS Energy Lett. 6, 2369–2377 (2021). https://doi.org/10.1021/acsenergylett.1c00904
Irkham, A. Fiorani, G. Valenti, N. Kamoshida, F. Paolucci et al., Electrogenerated chemiluminescence by in-situ production of coreactant hydrogen peroxide in carbonate aqueous solution at a boron-doped diamond electrode. J. Am. Chem. Soc. 142, 1518–1525 https://doi.org/10.1021/jacs.9b11842
K. Wenderich, B.A.M. Nieuweweme, G. Mul, B.T. Mei, Selective electrochemical oxidation of H2O to H2O2 using boron-doped diamond: an experimental and techno-economic evaluation. ACS Sustainable Chem. Eng. 9, 7803–7812 (2021). https://doi.org/10.1021/acssuschemeng.1c01244
I. Salmeron, K.V. Plakas, I. Sires, I. Oller, M.I. Maldonado et al., Optimization of electrocatalytic H2O2 production at pilot plant scale for solar-assisted water treatment. Appl. Catal. B 242, 327–336 (2019). https://doi.org/10.1016/j.apcatb.2018.09.045
J. Casado, Towards industrial implementation of Electro-Fenton and derived technologies for wastewater treatment: a review. J. Environ. Chem. Eng. 7, 102823 (2019). https://doi.org/10.1016/j.jece.2018.102823
I. Salmeron, I. Oller, K.V. Plakas, S. Malato, Carbon-based cathodes degradation during electro-Fenton treatment at pilot scale: changes in H2O2 electrogeneration. Chemosphere 275, 129962 (2021). https://doi.org/10.1016/j.chemosphere.2021.129962
Z. Ye, D.R.V. Guelfi, G. Alvarez, F. Alcaide, E. Brillas et al., Enhanced electrocatalytic production of H2O2 at Co-based air-diffusion cathodes for the photoelectro-Fenton treatment of bronopol. Appl. Catal. B 247, 191–199 (2019). https://doi.org/10.1016/j.apcatb.2019.01.029
Acknowledgements
This work was supported by Shanxi Province Science Foundation (20210302124446; 202102070301018), the National Natural Science Joint Foundation (U1710112) and Basic Research Project from the Institute of Coal Chemistry, CAS (SCJC-HN-2022-17).
Funding
Open access funding provided by Shanghai Jiao Tong University.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, Y., Tong, X. & Yang, N. Photocatalytic and Electrocatalytic Generation of Hydrogen Peroxide: Principles, Catalyst Design and Performance. Nano-Micro Lett. 15, 77 (2023). https://doi.org/10.1007/s40820-023-01052-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40820-023-01052-2