
Abstract
Purpose of Review
From invertebrates to vertebrates, the ability to sense nutrient availability is critical for survival. Complex organisms have evolved numerous signaling pathways to sense nutrients and dietary fluctuations, which influence many cellular processes. Although both overabundance and extreme depletion of nutrients can lead to deleterious effects, dietary restriction without malnutrition can increase lifespan and promote overall health in many model organisms. In this review, we focus on age-dependent changes in stem cell metabolism and dietary interventions used to modulate stem cell function in aging.
Recent Findings
Over the last half-century, seminal studies have illustrated that dietary restriction confers beneficial effects on longevity in many model organisms. Many researchers have now turned to dissecting the molecular mechanisms by which these diets affect aging at the cellular level. One subpopulation of cells of particular interest are adult stem cells, the most regenerative cells of the body. It is generally accepted that the regenerative capacity of stem cells declines with age, and while the metabolic requirements of each vary across tissues, the ability of dietary interventions to influence stem cell function is striking.
Summary
In this review, we will focus primarily on how metabolism plays a role in adult stem cell homeostasis with respect to aging, with particular emphasis on intestinal stem cells while also touching on hematopoietic, skeletal muscle, and neural stem cells. We will also discuss key metabolic signaling pathways influenced by both dietary restriction and the aging process, and will examine their role in improving tissue homeostasis and lifespan. Understanding the mechanisms behind the metabolic needs of stem cells will help bridge the divide between a basic science interpretation of stem cell function and a whole-organism view of nutrition, thereby providing insight into potential dietary or therapeutic interventions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the most accessible means of influencing lifespan has involved modulating caloric or dietary intake. Many early studies examining the effects of diet on age have focused on the abilities of caloric restriction, generally considered a 20–40% reduction in calorie intake on a whole, or restriction of specific dietary components such as protein or carbohydrates, to increase lifespan [1,2,3,4]. Diet can profoundly affect the onset of age-associated pathologies, such as cardiovascular disease, neurological impairment, immune dysfunction, metabolic syndrome, diabetes, and cancer [5, 6]. As human lifespan has increased globally over the last several decades [7], interest in improving healthspan, the time in life when an individual has limited chronic diseases, has also increased. Since then, regulation of diet by restriction of specific nutrients, such as carbohydrates, amino acids, or fats, as well as timing of nutrient intake, has garnered much interest due to such interventions modulating tissue growth, regeneration, and repair at the molecular level.
In many tissues, healthy state is maintained by populations of stem cells, whose metabolic requirements are varied and complex, and which often lose their self-renewal capacity with age [8,9,10,11,12]. Numerous studies in both vertebrates and invertebrates have shown that dietary restriction and manipulation can be either directly sensed by stem cells, or indirectly by the surrounding niche, and can subsequently dictate stem cell renewal, differentiation, and overall tissue homeostasis. Improving stem cell function through dietary interventions, or dietary mimetics, is an attractive target for extending both healthspan and possibly human lifespan. In this review, we will primarily focus on metabolic dependencies and regulation within intestinal stem cells, but we will also discuss several other populations of somatic stem cells, including hematopoietic, skeletal muscle, and neural stem cells, highlighting the effects of cellular and systemic age-dependent metabolic changes with respect to tissue regeneration. Finally, we will review how dietary interventions influence stem cell self-renewal and tissue regeneration.
Age-Associated Changes in Somatic Stem Cells
Somatic stem cells are a unique population of undifferentiated cells that have the ability to self-renew and give rise to progenitors that subsequently replenish specialized cells within tissues. The rate at which adult somatic stem cells can proliferate and contribute to tissue regeneration and homeostasis can vary between tissues and with age. Perhaps the best studied and most defined adult stem cell population is hematopoietic stem cells (HSCs). HSCs are located in the complex niche microenvironment of the bone marrow [13] and have the ability to give rise to lineage-committed progenitors that replenish all specialized blood cell types throughout an organism’s life [14]. Seminal discoveries in the late 1950s and 1960s showed that the bone marrow contained a population of cells capable of repopulating the hematopoietic system and giving rise to myeloid and lymphoid lineages [15, 16]. Since then, sophisticated lineage tracing techniques, coupled with irradiation and transplantation experiments, have provided insight on long-lived cells within tissues that retain DNA labels and serve as reservoirs of adult somatic stem cells [17•]. In humans, with age, there is a shift towards increased myeloid cell production, and HSCs transplanted from aged mice into young, irradiated recipients show similar myeloid bias and reduced lymphopoieses [18].
In the 1970s, several influential discoveries were also made in epidermal stem cell biology, where long-lived epidermal cells could be maintained in culture over long periods of time and were shown to express specific keratin markers [19, 20]. Later, it was shown that epidermal cells can be expanded through culturing and engrafted successfully onto burn victims [21, 22]. Subsequently, label-retaining hair follicle stem cells (HFSCs) were also discovered and shown to contribute to hair follicle regeneration. With aging, HFSC numbers decline and exhibit age-dependent transcriptional changes in mice [23, 24••].
In the mammalian gastrointestinal tract, crypt base columnar cells were discovered in the 1970s and were later determined to be highly proliferative, Lgr5 high expressing (Lgr5+) intestinal stem cells (ISCs) that are present both in the small and large intestines. ISCs either self-renew or differentiate into progenitor cells, which themselves undergo several rounds of proliferation before differentiating into one of several intestinal cell types: secretory Paneth cells (the only differentiated cell type that travels downwards into the crypt), Goblet cells, enteroendocrine cells, Tuft cells, or the absorptive enterocytes which make up the majority of the intestinal lining [25]. In addition, the mammalian intestine also harbors a quiescent, “reserve” pool of stem cells at the +4 position in the crypt. These more slowly cycling stem cells have been shown to express a differential repertoire of stem cell markers like Bmi, Tert, and Sox9 compared to the fast cycling, Lgr5+ cells [26, 27] and can also withstand radiation induced DNA damage better than high cycling Lgr5+ cells [28]. Even so, the number of functionally dividing ISCs per crypt is relatively low and reported to be around 5–7 cells each [29]. In Drosophila, which has been an excellent model organism to study the effects of diet on aging, intestinal stem cells express high levels of Escargot and give rise to enterocytes, enteroendocrine cells (EEs), and copper cells in the gastric region [30•]. The majority of Drosophila ISCs reside in the midgut and are primarily quiescent, unless the epithelium is challenged by injury or infection. With age, there is an increase of inflammation and gut dysbiosis, leading to hyperproliferation of ISCs, reduced tissue integrity, and barrier dysfunction [31]. Interestingly, gut intestinal health in Drosophila dictates overall organismal longevity, and compromised tissue homeostasis leads to barrier dysfunction and shortened lifespan [32].
It has been more than 20 years since Potten and colleagues showed that the aged mammalian intestine is less regenerative and has more apoptotic cells following irradiation [33, 34]. With the advent of transgenic lines that allow lineage tracing experiments, as well as ex vivo organoid cultures that can compare the function of stem cells from animals of different ages, there have been a plethora of recent studies characterizing morphological and functional age-associated changes in the mammalian gut. Nalapareddy et al. showed age-dependent diminished crypt numbers and slower proliferation rates in mice, which they largely attributed to diminished Wnt3a expression with aging [35••]. Supplementing organoid cultures of aged mice with exogenous Wnt3a overcame this age-associated deficit. Moorefield et al. also reported diminished organoid formation from aged animals, but an increase in cell numbers and proliferation of Sox9-eGFP-expressing reserve stem cells [36••]. With aging, Mihaylova and colleagues observed diminished proliferation in stem and progenitor cells, as well as diminished capacity for self-renewal in aged guts following irradiation [37••]. Similar to the previous two studies, they also reported lower frequency of formation of organoids from aged animals compared to 3- to 4-month-old mice [38••], attributing some of these changes to decreased fatty acid oxidation (FAO) in the crypts of aged animals. Dietary and pharmacological induction of FAO improved the function of aged ISCs. Pentinmikko and colleagues reported decline in stem cell function and showed that Notum, a secreted Wnt inhibitor that is specifically expressed in Paneth cells, is produced at higher levels in aged Paneth cells compared to young ones [39••]. They further showed that pharmacological inhibition of Notum in vivo improved clonogenicity of Lgr5+ stem cells and protected aged epithelium from DNA-damaging Fluorouracil (5FU) treatment. Igarashi and colleagues showed that supplementation of aged animals with NAD+ rejuvenates aged intestinal stem cells and restores stem cell numbers. In addition, aged crypts and stem cells had significant reduction of SIRT1 protein levels, and treatment with the NAD+ precursor nicotinamide riboside (NR) improved the recovery of DSS-treated aged mice [40]. Together, these studies suggest that there are age-dependent changes in ISCs numbers and function due to intrinsic and systemic metabolic changes, as well as key regulators of stemness such as Wnts. DNA methylation analysis on patient-derived organoids also recently revealed regional differences in epigenetic alterations with age, and importantly, these regional differences were maintained in organoid cultures. Epigenetic clock calculations using DNA methylation showed that aging of small intestinal crypts slows down with midlife [41••]. Interestingly, slower proliferation was observed and recently reported by Tomasetti et al. in human esophagus, duodenum, and colon, where they hypothesize it may be a possible protective mechanism to decelerate cancer incidence with age [42].
Multiple studies have also reported an increase in the number of secretory cells in aged intestines, particularly in the distal end of the intestine, suggesting potential skewing to secretory lineages [35••, 38••, 39••] with age. Gerbert and colleagues recently honed in on these potential region-specific effects in the intestinal tract by performing proteomic analysis on many sequential sections throughout the small intestine (SI) and reported a similar age-dependent increase of Paneth cell markers, reduction of Goblet cell numbers in the distal small intestine, and conversely, an increase in enterocyte markers [43••]. Collectively these studies suggest that there are age-dependent alterations in secretory lineage cells, likely in response to microbial or metabolite changes with age [36••, 43••]. We have further summarized these age-dependent phenotypes in Table 1, as well as interventions used in these studies to rejuvenate and restore the function of aged stem cells.
Other tissues also contain populations of somatic stem cells that show age-dependent functional changes, including the muscle and brain. With aging, there is a significant loss of muscle mass and strength, otherwise known as sarcopenia, which is a determinant of frailty. Although the list of proposed mechanisms for age-dependent muscle decline is extensive, and includes multiple hallmarks of aging, the role of muscle stem cells remains to be fully elucidated. In muscle, muscle stem cells (MuSCs) or satellite cells are the undifferentiated precursors to skeletal muscle cells and are located beneath the basement membrane of myofibers [44]. Fry and colleagues showed that depletion of satellite stem cells affects muscle regenerative capacity, without affecting sarcopenia [45]. In the mammalian brain, neural stem cells (NSCs), undifferentiated neural cells with the ability to differentiate into various glial and neuronal cells, are located within the subventricular zone where they can form neuroblasts that will travel to the olfactory bulb to form periglomerular and granule mature neurons [46]. As with other adult stem cell populations, NCSs lose their neurogenic ability with age, which may have numerous consequences. For a comprehensive review on age-dependent decline in neuronal stem cells, we direct our readers to a wonderful recent review by Negredo et al. [47•]. Some tissues also have the unique ability to respond to severe injury by evoking post mitotic cells to rapidly proliferate and give rise to more committed lineages to replenish injured tissue. This is the case of one regenerative response in liver due to injury. In addition, following severe toxin injury, the liver can also mobilize cholangiocytes which can give rise to mature hepatocytes [48].
Metabolic Regulation of Adult Stem Cells in Aging and Dietary Interventions
Molecular Mechanisms
Numerous studies in invertebrates and vertebrates have previously shown the beneficial effects of caloric restriction (CR) on increasing maximum lifespan in yeast, C. elegans, D. melanogaster, and mice [1, 2, 4, 49]. CR is thought to decrease the incidence of metabolic syndromes, cardiovascular disease, spontaneous cancers, autoimmune diseases, and neurological defects, while also improving insulin sensitivity in mice. The effects of CR on immune function are somewhat varied, but dietary restriction or cyclical fasting has generally been thought to rejuvenate the immune system, at least in the elderly [50, 51•]. Many of the beneficial effects of CR are seen through its influence on stem cell capacity for self-renewal, as discussed later, but early studies in humans have brought into question the feasibility of maintaining such a strict regimen of caloric restriction. Participants often report feelings of hunger and lethargy while on a CR diet, and they begin to exhibit decreased muscle mass and bone mineral density [6, 52]. As a result, many researchers have turned to other forms of dietary restriction, such as short-term or cyclical fasting, as well as restriction of specific components of the diet like amino acids, to study effects on lifespan.
Restricted protein diets have long been studied as a means of lifespan and healthspan extension, and these diets can have systemic effects on insulin signaling and glucose homeostasis. In Drosophila, feeding a low-protein, high-carbohydrate diet by specifically reducing the percentage of casein in dietary yeast leads to prolonged maximum survival [53]. Restriction of methionine in particular leads to an increase in lifespan similar to dietary restriction, but these effects may be more tied to the ratio of amino acids available [54]. However, aged mice (24 months) on a low protein diet begin to lose weight following 2 weeks on the diet [55], lending credence to the argument that protein restriction in the elderly may in fact be harmful. Mice on restricted protein diets have reduced adiposity, decreased blood glucose and insulin levels, and exhibit improved glucose tolerance even when fed an otherwise high-fat, high-sugar “Western” diet [56, 57]. Conversely, excess amino acids in the diet are associated with decreased lifespan and higher mutation rates in yeast [55] and Drosophila [53, 58]. Furthermore, in the study conducted by Solon-Biet et al., a high-protein, low-carbohydrate diet in mice was associated with glucose intolerance while a low-protein, high-carbohydrate diet actually exhibited decreased insulin levels and led to increased lifespan, although the authors do note that the types of carbohydrates consumed could have an influence on insulin sensitivity [58]. Restriction of the branched chain amino acids (BCAAs) leucine, isoleucine, and valine in particular leads to decreased blood glucose levels and improved insulin sensitivity, and extends the lifespan of male mice [59,60,61]. In mice and rats, methionine restriction alone can decrease blood insulin and glucose levels and potentially increase lifespan, and while the results may mimic those of caloric restriction, it is difficult to pin all of these positive effects on longevity on a single amino acid [62,63,••–64].
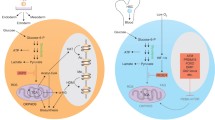
The mammalian target of rapamycin (mTOR) is often the focus of dietary studies like those described previously, as it specifically monitors amino acid availability, energy status, growth factor signaling, and oxygen levels (Fig. 1) [65,66,67]. Two separate complexes may form, mTORC1 and mTORC2, consisting of the serine-threonine kinase mTOR and a host of additional accessory proteins, which together serve as one of the primary means of sensing and responding to nutrient status in cells. When activated in times of nutrient abundance, mTORC1 promotes cell growth by phosphorylating ribosomal and translation initiation factors for protein synthesis, transcription factors associated with lipid synthesis, and coactivators involved with mitochondria biogenesis [65]. Additionally, formation of the autophagosome is inhibited through phosphorylation of the autophagy-initiating complex containing ULK1, Atg13, and FIP200 [68]. Inhibition of mTOR signaling using rapamycin extends the lifespan of yeast, Drosophila, C. elegans, and mice [69,70,71,72], although more recent studies are revealing that prolonged rapamycin treatment may have negative effects on certain tissues despite its positive effects on overall lifespan. It was previously believed that rapamycin only functioned to inhibit mTORC1 assembly, but work by Sarbassov and colleagues showed that prolonged rapamycin treatment also disrupts mTORC2 in multiple cell lines and tumors [73]. Lamming and others went on to show that rapamycin also disrupts mTORC2 in multiple tissues, leading to insulin insensitivity and glucose intolerance, and this mTORC2 disruption has negative effects on lifespan [74,75,76,77,78,79]. Genetic deletion of S6K1, a direct phosphorylation target of mTORC1, also leads to moderately increased lifespan, improved insulin sensitivity, and improved glucose tolerance [80], indicating that a balance must be found to maximize “beneficial” inhibition of mTORC1 and “harmful” disruption of mTORC2.

Nutrient sensing by the mTOR, AMPK, and PPAR signaling pathways. When growth factors and nutrients are abundant, mTORC1 is active and promotes anabolic processes, proliferation and differentiation, while AMPK and PPAR signaling are suppressed. When nutrients become scarce, AMPK is activated and mTORC1 becomes inhibited, resulting in increased catabolic processes and autophagy. At the same time, PPAR mediated fatty acid oxidation and ketogenesis are induced and enhance stem cell function, particularly in intestinal stem cells. With aging, fatty acid oxidation may be limited and mTORC1 activity may be hyperactivated leading to functional decline in stem cells. Interventions such as rapamycin aim to inhibit mTORC1, and treatments such as PPAR agonists or dietary regiments that enhance fatty acid oxidation and ketogenesis may improve age-associated decline in stem cell function, particularly in the gut
Like the mTOR complexes, the AMPK signaling pathway monitors nutrient status and regulates subsequent cell growth and differentiation processes by responding to changes in ATP, ADP, and AMP levels. In times of nutrient depletion, AMP and ADP concentrations rise, and AMPK is activated by phosphorylation of LKB1 at Thr172. This phosphorylation event is made possible through a conformational change induced by AMP/ADP binding at the regulatory γ subunit of AMPK. In turn, AMPK can directly and indirectly inhibit mTOR activity to reduce anabolic processes and promote autophagy [81]. Targets of AMPK include components of the mTOR complex (Raptor), transcription factors and enzymes that regulate lipid and fatty acid metabolism (SREPB1, ACC), and regulators of mitochondrial homeostasis (MFF) [82] among many others [83, 84]. High glucose levels can lead to decreased lifespan in C. elegans via inhibition of AMPK activity [62], whereas increased AMPK activity is correlated with increased lifespan, dependent upon correct balance of mitochondrial fusion and fission dynamics [85]. The beneficial effects of dietary restriction on longevity in C. elegans are in part dependent upon AMPK activity [86, 87]. Conversely, in instances of BCAA restriction, AMPK activity is upregulated through its phosphorylation, in contrast with the decrease in mTOR activity [59]. Several studies of dietary interventions, including metformin treatment or α-ketoglutarate supplementation, have found indirect effects on AMPK function. Such treatments ultimately alter the AMP:ATP ratio of the cell, leading to the activation of AMPK and the inhibition of mTOR [86, 88].
Hematopoietic Stem Cells
Within the stem cell, changes in metabolism are able to direct various cellular phenomena such as senescence, proliferation, stemness, differentiation, apoptosis, and resistance to cancer treatment. HSCs remain in a largely quiescent state, with estimations of less than 10% to be actively dividing. Aging in the hematopoietic system is characterized by loss of self-renewal capacity of HSCs, expansion of HSC numbers, myeloid lineage skewing, and clonal hematopoiesis [89]. On the molecular level, these age-associated perturbations are thought to be largely driven by accumulated mutations and oxidative stress. ROS levels and redox state are important determinants of adult stem cell homeostasis in a number of tissues, and elevated ROS production is necessary for both differentiation and homing of HSCs to the bone marrow niche. Aged HSCs have increased ROS production [90], and a long-standing hypothesis was that age-dependent oxidative damage leads to aberrant phenotypes in HSCs. However, studies using accelerated mouse models of aging have shown that aging phenotypes in HSCs that occur due to mitochondrial dysfunction differ from HSCs from conventionally aged mice [91]. Another feature of aging in the hematopoietic stem cell system is clonal hematopoiesis of indeterminate potential (CHIP), a phenomenon characterized by reduced number of HSC clones that contribute to a large portion of hematopoiesis [89]. Recent studies have shown that with aging, many of the remaining HSC clones harbor mutations for important epigenetic enzymes, such as TET2 and DNMT3A, which leads to an increased risk of age-associated hematopoietic malignancies [92,93,94]. Another recent study showed that Kat6b, a histone modifier that is normally enriched in long-term HSCs, is suppressed in aged HSCs, which contributes to skewed production of myeloid cells [95••].
Hematopoietic stem cells must also maintain a balance of mTOR signaling over time. The activity of this pathway is linked to progenitor proliferation, yet inhibition of its activity with rapamycin in aged HSCs leads to improved stem cell function and renewal [11, 96]. Though once again, recent work is highlighting the different roles that these nutrient sensing pathways play in varying cell populations within a system, and in the niches in which they reside. For example, genetic loss of mTOR activity in the bone marrow microenvironment that informs HSC development can lead to aging phenotypes of both hematopoietic stem cells and progenitors [97]. Autophagy has also been shown to play an important role in HSC homeostasis and recently was shown to be impaired with age, leading to accumulation of active mitochondria and more metabolically active HSCs [98••].
Stem and progenitor cell function has also been shown to be regulated by mitochondrial metabolism and fatty acid oxidation [99]. Dome+ progenitor cells in the lymph glands of Drosophila use FAO as their primary metabolic state, unlike pre-progenitors and intermediate progenitors [100]. Kratchmarov et al. found that while FAO inhibition did not affect total conventional dendritic cell (cDC) or plasmacytoid dendritic cell (pDC) development, it significantly increased the amount of cDC2 cells and decreased cDC1 cells, suggesting that while anabolism drives progenitor division, catabolic mechanisms may push cells into alternative differentiation fates [101]. Similarly, AMPK-a1 deficiency both in vitro and in vivo did not change the cDC/pDC ratio but did change cDC differentiation toward favoring cDC2, suggesting that AMPK-a1 signaling may play a role in dendritic cell progenitor fate [102]. Regarding HSC exhaustion, Ma et al. found that Hes1 deletion upregulated both PPAR targets and FAO-related genes [103]. Inhibition of PPAR improved hematopoietic repopulation of Hes1-deficient HSCs, as did genetic deletion of CPT1A, a rate-limiting enzyme in fatty acid import into the mitochondria, suggesting that Hes1 regulates HSC hematopoiesis through suppression of PPAR-related pathways that otherwise would cause hematopoietic stem cell exhaustion. Caloric restriction has prevented age-associated decline of HSCs in certain mouse genetic backgrounds [104], but it does not prevent age-related decline of hematopoietic stem cell functioning in other studies [105].
Intestinal Stem Cells
Numerous studies in Drosophila have informed how nutrient sensing and aging in the gut can regulate intestinal stem cells, tissue homeostasis, and longevity [106••]. Amino acid restriction leads to inactivation of mTORC1 in Drosophila [107], and further studies show that methionine restriction in particular can lead to lifespan extension, the effects of which are dependent upon mTOR signaling [62]. Early studies in the mammalian intestine have shown that it can respond to prolonged dietary restriction by decreasing villus length, indicating that nutrient availability can have direct consequences on absorptive surface area. More recent studies have shown how dietary restriction and fluctuations in nutrients can be sensed by the mammalian intestinal epithelium and can influence stem cell self-renewal and differentiation, as well as epithelial regeneration [9, 38••, 40, 43••, 108].
We and others have hypothesized that the beneficial effects of caloric restriction may be due to prolonged periods of fasting. To test this hypothesis, we recently asked how short-term, 24 hour fasting affects ISC function and numbers, especially in the context of aging. Interestingly, this short-term fasting led to a significant boost of both young and aged stem cell function and protected the intestine from radiation-induced damage. Unbiased RNAseq analysis of ISCs revealed fasting induced transcriptional upregulation of PPAR targets and subsequent metabolic switch to fatty acid oxidation. Pharmacological or genetic ablation of the rate-limiting enzyme for mitochondrial fatty acid import, CPT1A, blunted the effects of fasting in cultured organoids and reduced stem cell numbers in vivo, but treatment with PPARδ agonist GW501516 increased stem cell numbers, boosted stem cell function, and increased FAO rates in intestinal crypts from aged animals. Collectively, this suggests that FAO promotes stem cell self-renewal and that its disruption compromises ISC function [38••]. Long-term exposure to high fat diet (HFD) also drives a PPARδ-dependent program in intestinal stem cells and boosts stem cell numbers and function, while promoting niche independence of stem cells and increasing beta catenin activity. Importantly, chronic HFD exposure can enhance tumor-initiation capacity in progenitor cells deleted for the tumor suppressor Apc [79].
Cheng and colleagues recently showed that intestinal stem cells are highly enriched for HMGCS2, the rate-limiting enzyme in ketone body production, and that deletion of HMGCS2 in the intestine decreases beta-hydroxybutyrate (BOHB) levels in crypts, reduces stemness, and skews differentiation to a secretory lineage phenotype [109••]. Conversely, a high-fat ketogenic diet can enhance stem cell function in an HMGCS2-dependent fashion, where high levels of BOHB in ISCs act as endogenous HDAC inhibitors to regulate intestinal stem cell function and differentiation through Notch signaling [109••]. Gebert and colleagues also recently showed that dietary restriction followed by refeeding modulates HMGCS2 levels in the gut and influences secretory lineage differentiation [43••]. Meanwhile, ISC proliferation can also be regulated by cholesterol metabolism, where increased cholesterol biosynthesis can lead to increased organoid growth or ISC proliferation in vivo [110]. Another group has shown that a fasting-mimicking diet can modulate the microbiota and partially reverse inflammatory markers in dextran sulfate sodium (DSS) -induced colitis [111], and future studies in this area of research will shed more light on how diet influences the microbiome and its metabolic repertoire.
Even within the small intestine, there appears to be metabolic variability among stem cells in the intestinal crypt. Stine et al. found that both PRDM16 and PPAR expressions were highest in the duodenal crypts and decreased distally, suggesting duodenal stem cells rely more on fatty acid oxidation [112]. This was further supported by the finding that PRDM16 KO duodenal crypts contain lower numbers of proliferating cells and increased apoptotic cells in the transit-amplifying zone, suggesting that PRDM16 helps regulate epithelial cell renewal through FAO promotion [112]. Recently, it was also shown that loss of HNF4 leads to lower levels of FAO-related gene expression, and that knockout of HNF4a and HNF4g leads to increases in apoptosis, especially in the intestinal crypt base, indicating that HNF4 is necessary for stem cell survival. This was further supported through the supplementation of HNF4 DKO organoids with acetate, similar to levels present in the colonic lumen, which not only rescued the organoids from a spheroid morphology but also restored Lgr5-GFP-positive stem cells, showing that acetyl-CoA deficiency from the HNF4-related FAO defects could be limiting the renewal of stem cells in the intestinal crypt [113]. Interestingly, pharmacological inhibition of pyruvate metabolism or loss of mitochondrial pyruvate carrier (MPC) expression in Lgr5+ stem cells leads to increased stem cell proliferation, and a similar hyperproliferative phenotype was observed with the loss of Drosophila MPC [114]. Along the same lines, inhibition of MPC1 in hair follicle stem cells can induce activation and proliferation by promoting metabolism of pyruvate to lactate by lactate dehydrogenase [115].
With regards to mTOR signaling, in the aging mouse intestinal crypt, Pentinmikko et al. found that old Paneth cells exhibit increased mTORC1 activity, as evidenced by increased phosphorylation of the S6 ribosomal protein [39••]. Conversely, old intestinal stem cells show decreased pS6 levels compared to their younger counterparts, but do not maintain their capacity for self-renewal with age [38••, 40]. Indeed, current models point towards inverse trends in mTORC1 activation in Paneth cells and ISCs under the same treatment conditions [39••, 116], due to the fact that nutrient availability can be sensed by the niche and communicated to ISCs through paracrine secreted signals [9]. Caloric restriction increases stem cell numbers by suppressing mTORC1 signaling in niche Paneth cells, leading to increased levels of Bst1, an enzyme that converts NAD+ to cyclic ADP ribose (cADPR) to induce stem cell renewal [9]. Simultaneously, mTORC1 inhibition in ISCs due to CR promotes stem cell renewal and resistance to radiation-induced damage [117]. The interdependent relationship between ISCs and Paneth cells is further illustrated by the work of Rodríguez-Colman and colleagues, who showed that ISCs depend heavily on lactate produced by nearby Paneth cells to fuel their own mitochondrial oxidative phosphorylation, which leads to differentiation of ISCs [118]. As a result, the role of Paneth cells in regulating ISC function has garnered much interest from a metabolic perspective in recent years, and it is becoming more apparent that treatments such as rapamycin can have varied effects on different cell types even within the same niche, and thus varied ability to affect cellular aging.
Current studies are examining combination treatments that target pathways in addition to mTOR such as the Sirtuins, in order to account for these mixed responses across cell types and tissues. It is worth noting, too, that while rapamycin treatment alone can induce boosts in stem cell function and self-renewal that are generally seen with CR [9], treatment of crypts from calorie-restricted mice with rapamycin blocks these positive effects [40]. Thus, the influence of caloric restriction on intestinal aging is mediated in part by mTOR signaling, but is more complicated than simple mTOR inactivation.
In a similar fashion, AMPK phosphorylation is elevated in the intestinal crypts of calorie-restricted mice, and in the intestinal stem cells in particular [116]. As with mTOR signaling, the CR-enhanced self-renewal capacity of ISCs is blocked by inhibition of AMPK signaling in the ISCs, indicating that the beneficial effects of caloric restriction in ISCs are mediated through the activity of AMPK [116]. In intestinal stem cells as in hematopoietic stem cells, AMPK and mTOR together coordinate stem cell renewal, senescence, and even disease initiation with age, but the complexities of the relationship between fluctuating activities of each pathway are still being determined [119].
Other Somatic Stem Cells
Like intestinal stem cells, neural stem cells exhibit marked decline in function with age and become less able to counteract neuron loss or damage with regeneration over time [47•]. Proliferative neural stem cells seem to turn to fatty acid oxidation when faced with mitochondrial dysfunction, shown through an increase in carnitine metabolites and the inability of palmitate to sustain cell viability without carnitine [120], suggesting that cells with mitochondrial dysfunction become more dependent on FAO to sustain cell proliferation. Yang et al. discovered that physiological levels of short-chain fatty acids, such as acetate, butyrate, and propionate, increased the growth rate of human neural progenitor cells that were derived from a human embryonic stem cell line, as well as encouraged the cells to undergo mitosis with no effect on apoptosis [121]. This suggests that short-chain fatty acids could regulate development of the neural system in early stages [121]. While treatment of mice with the gut hormone acyl-ghrelin did not increase proliferation in the subventricular zone, nor does it affect the number of adult-born neurons or the rate of neuronal differentiation, the increase of endogenous acyl-ghrelin through calorie restriction does increase the activation of new adult-born cells [122].
Knobloch et al. found that compared to proliferating neural stem and progenitor cells (NSPCs), quiescent NSPCs have higher expression levels of CPT1A. Inducing quiescence upregulates Spot14, a negative regulator of malonyl-CoA, suggesting that high levels of Spot14 along with low levels of malonyl-CoA help promote fatty acid oxidation. A massive die-off of quiescent NSPCs occurred after etomoxir treatment, while treatment of proliferative NSPCs led to a reduction of proliferation, indicating that FAO is much more crucial to quiescent NSPCs than proliferative ones, further supported through the upregulated expression of PPARα and its targets in quiescent NSPCs [123]. As an organism ages, neural stem cells also show a decline in mTOR signaling, which is believed to play a role in differentiation of stem and progenitor populations in the brain [96]. It has been found that induction of mTOR signaling promotes proliferation of NSCs in old mice, and in fact, that rapamycin treatment of young mice can generate NSCs with a more aged profile [124]. This provides another example that while rapamycin treatment appears to extend organismal lifespan on the whole, its tissue and cell specific effects must be considered in detail before it is more universally applied as an aging intervention. On the other hand, AMPK signaling is also implicated in the decline of neurological function with age, and its activity is elevated in old NSCs compared to young [125], yet some AMPK activity may be necessary for maintenance of autophagy and prevention of neurological disease [126].
In muscle, mTOR signaling is associated with muscle size, where its increased activity can contribute to hypertrophy but can also lead to muscle degeneration as a result of oxidative damage [127]. Consequently, age-associated muscle loss may be correlated with hyperactive mTOR activity in old muscle, and in rats sarcopenia can be prevented by treatment with rapamycin [127,123,124,130]. In contrast, the ability of skeletal muscle to activate AMPK appears to decrease with age in rats, and may lead to several age-associated impairments in mitochondrial function and lipid metabolism [83]. When treating satellite cells both in vivo and ex vivo with metformin, an AMPK agonist, Pax7 downregulation and the terminal differentiation of satellite cells are delayed, which is consistent with the inhibition of mTOR and reduction in RPS6 phosphorylation that induce a quiescent metabolic state [131]. Importantly, muscle satellite cells that maintain a more quiescent phenotype following metformin treatment have the potential to counteract age-associated muscle decline [131]. For both injured and non-injured control mice, there were significantly more satellite cells 1 week after injury in calorie-restricted mice than the control mice at 6 months of age; however, at 12 months of age, there were significantly fewer satellite cells in the calorie-restricted male mice than the control mice [132]. Treatment with metformin or administration of a calorie-restricted diet may induce AMPK activity in muscle and help to maintain the function of muscle satellite cells while preventing age-associated senescence [131, 133].
Satellite cells isolated from irradiated mice are more likely to have incomplete oxidation of 14C-palmitate and reduced levels of insulin-stimulated glucose uptake, regardless of whether mice were on a regular chow or high fat diet [134]. After observing impairments in the lipid profiles and increases in insulin resistance in childhood cancer survivors who had radiotherapy and chemotherapy, this, along with the experiments done in mice, suggests a metabolic reprogramming of satellite cells due to irradiation that could cause long-term metabolic dysfunction [134]. When using fetal and perinatal satellite cells along with post-injury myogenic cells, mitochondrial FAO genes had little change in gene expression while peroxisomal FAO genes were upregulated in activated cells. Satellite cells injured with notexin had significantly higher gene expression related to peroxisomal FAO than quiescent cells, which was not seen with mitochondrial FAO genes, suggesting different uses for the two FAO pathways in satellite cells. Culturing satellite cells with the peroxisomal FAO inhibitor, thioridazine, caused increases in levels of myogenin, while etomoxir inhibition of mitochondrial FAO did not, suggesting that inhibition of peroxisomal FAO induces differentiation in myogenic cells [135]. After culturing bovine satellite cells for 24 h with linoleic or oleic acid, expression levels significantly increased for genes such as PPARα, PPARγ, ACOX, LPL, FABP4, and CPT1, suggesting that unsaturated fatty acids like linoleic or oleic acids could promote FAO in muscle satellite cells [136].
Conclusion
Somatic stem cells integrate critical environmental inputs that inform decisions on self-renewal, differentiation, and subsequent tissue turnover. Aging is a risk factor for many diseases, and recent studies are starting to uncover the molecular mechanisms of how environmental factors, such as diet, can influence stem cell behavior over time. With aging, many adult stem cell populations accumulate damage and become impaired in their function, which leads to inefficient tissue repair and may predispose to age-associated diseases such as cancer. Although numerous studies have shown that dietary restriction confers beneficial effects on overall organismal lifespan, we are just starting to uncover the complexities of metabolic dependencies in stem cells and how the availability of specific nutrients are sensed within a heterogeneous population of stem, progenitor, and niche cells and communicated between each other. The advent of new single cell technologies has already begun to enhance our ability to resolve the complex metabolic heterogeneity and interactions that exist in certain niches, such as in the gut crypt and bone marrow.
Recent studies using single cells technologies have also revealed that seemingly uniform, terminally differentiated cells in the liver and intestinal villus have specific transcriptional metabolic profiles that are driving cell function. These differences among hepatocytes and enterocytes were largely influenced by location, proximity to nutrients, and oxygen supply within their respective tissues [137, 138]. In the upcoming years, as multiple single cell -omic technologies advance and integrate, we will begin to see more advanced tissue maps of transcriptional, proteomic, and metabolomic signatures of stem and progenitor cells and their corresponding niches, both under homeostatic conditions as well as during aging and other pathological states. These types of studies will also likely layer dietary patterns with other environmental factors to expand our understanding of how nutrients and systemic metabolism impact tissue homeostasis. Finally, the constant improvement and engineering of primary 3D organoid cultures will allow us to more precisely examine intrinsic and extrinsic age-associated changes, as well as measure the activity of metabolic pathways, in response to defined nutrient conditions. We will be able to incorporate and study other signals in these systems, such as cytokines, hormones, and microbial metabolites. All of these advances will conceivably lead to better strategies and therapies for tissue repair with age, while carefully avoiding interventions that may accelerate age-dependent diseases such as cancer.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Weindruch R. The retardation of aging by caloric restriction: Studies in rodents and primates. Toxicol Pathol. 1996;24:742–5.
Mahoney LB, Denny CA, Seyfried TN. Caloric restriction in C57BL/6J mice mimics therapeutic fasting in humans. Lipids Health Dis. 2006;5:1–11.
Taormina G, Mirisola MG. Calorie restriction in mammals and simple model organisms. Biomed Res Int. 2014;2014:1–10.
Lee GD, Wilson MA, Zhu M, Wolkow CA, de Cabo R, Ingram DK, et al. Dietary deprivation extends lifespan in Caenorhabditis elegans. Aging Cell. 2006;5:515–24.
Huffman DM, Schafer MJ, LeBrasseur NK. Energetic interventions for healthspan and resiliency with aging. Exp Gerontol Elsevier Inc. 2016;86:73–83.
Barzilai N, Huffman DM, Muzumdar RH, Bartke A. The critical role of metabolic pathways in aging. Diabetes. 2012;61:1315–22.
World health statistics 2020: monitoring health for the SDGs, sustainable development goals. Geneva: World Health Organization; 2020. Licence: CC BY-NC-SA 3.0 IGO.
Mihaylova MM, Sabatini DM, Yilmaz ÖH. Dietary and metabolic control of stem cell function in physiology and cancer. Cell Stem Cell. 2014;14:292–305.
Yilmaz ÖH, Katajisto P, Lamming DW, Gultekin Y, Bauer-Rowe KE, Sengupta S, Birsoy K, Dursun A, Yilmaz VO, Selig M, Nielsen GP, Mino-Kenudson M, Zukerberg LR, et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. 2012; 486: 4905.
Cheng CW, Adams GB, Perin L, Wei M, Zhou X, Lam BS, et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell- based regeneration and reverse immunosuppression. Cell Stem Cell Elsevier Inc. 2014;14:810–23.
Chen C, Liu Y, Liu Y, Zheng P. MTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009;2:ra75.
Ghosh-Choudhary S, Liu J, Finkel T. Metabolic Regulation of Cell Fate and Function. Trends Cell Biol. Elsevier Ltd; 2020; 30: 20112.
Dexter TM, Allen TD, Lajtha LG. Conditions controlling the proliferation of haemopoietic stem cells in vitro. Symp Soc Dev Biol. 1978:149–60.
Bryder D, Rossi DJ, Weissman IL. Hematopoietic stem cells: the paradigmatic tissue-specific stem cell. Am J Pathol American Society for Investigative Pathology. 2006;169:338–46.
Becker AJ, McCulloch EA, Till JE. Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature. 1963;197:452–4.
Morrison S, Uchida N, Weissman I. The biology of hematopoietic stem cells. Annu Rev Cell Dev Biol. 1995;11:35–71.
Post Y, Clevers H. Defining adult stem cell function at its simplest: the ability to replace lost cells through mitosis. Cell Stem Cell Elsevier Inc. 2019;25:174–83. This review discusses updated definitions and characteristics of stemness and propose a revised definition of stem cell function – “the ability to replace lost tissue through cell division.
Dykstra B, De Haan G. Hematopoietic stem cell aging and self-renewal. Cell Tissue Res. 2008;331:91–101.
Rheinwald JG, Green H. Formation of a keratinizing epithelium in culture by a cloned cell line derived from a teratoma. Cell. 1975;6:317–30.
Fuchs E, Green H. The expression of keratin genes in epidermis and cultured epidermal cells. Cell. 1978;15:887–97.
O’Connor NE, Mulliken JB, Banks-Schlegel S, Kehinde O, Green H. Grafting of burns with cultured epithelium prepared from autologous epidermal cells. Lancet. 1981;317:75–8.
Green H. Cultured cells for the Treatment of Disease. Sci Am. 1991;265:96–102.
Keyes BE, Segal JP, Heller E, Lien WH, Chang CY, Guo X, Oristian DS, Zheng D, Fuchs E. Nfatc1 orchestrates aging in hair follicle stem cells. Proc Natl Acad Sci U S A. 2013; 110: E49509.
Ge Y, Miao Y, Gur-Cohen S, Gomez N, Yang H, Nikolova M, et al. The aging skin microenvironment dictates stem cell behavior. Proc Natl Acad Sci U S A. 2020;117:5339–50. This recent study showed age-dependent transcriptional changes in extra cellular matrix remodeling genes in HFSC and age-dependent decline in hair regeneration following wounding.
van der Flier LG, Clevers H. Stem Cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–60.
Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915–20.
Montgomery RK, Carlone DL, Richmond CA, Farilla L, Kranendonk MEG, Henderson DE, et al. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci U S A. 2011;108:179–84.
Basak O, Born M, Korving J, Beumer J, Elst S, Es JH, et al. Mapping early fate determination in L gr5 + crypt stem cells using a novel K i67- RFP allele. EMBO J. 2014;33:2057–68.
Kozar S, Morrissey E, Nicholson AM, van der Heijden M, Zecchini HI, Kemp R, et al. Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell. 2013;13:626–33.
Rodriguez-Fernandez IA, Tauc HM. Jasper H. Hallmarks of aging Drosophila intestinal stem cells. Mech Ageing Dev Elsevier. 2020;190:111285. This recent, comprehensive review discusses many age-dependent alterations in Drosophila intestinal stem cells and their consequences.
Biteau B, Hochmuth CE, Jasper H. Maintaining tissue homeostasis: dynamic control of somatic stem cell activity. Cell Stem Cell Elsevier Inc. 2011;9:402–11.
Rera M, Clark RI, Walker DW. Intestinal barrier dysfunction links metabolic and inflammatory markers of aging to death in Drosophila. Proc Natl Acad Sci U S A. 2012;109:21528–33.
Martin K, Potten CS, Roberts SA, Kirkwood TBL. Altered stem cell regeneration in irradiated intestinal crypts of senescent mice. J Cell Sci. 1998;111:2297–303.
Martin K, Kirkwood TBL, Potten CS. Age changes in stem cells of murine small intestinal crypts. Exp Cell Res. 1998;241:316–23.
Nalapareddy K, Nattamai KJ, Kumar RS, Karns R, Wikenheiser-Brokamp KA, Sampson LL, et al. Canonical Wnt Signaling ameliorates aging of intestinal stem cells. Cell Rep ElsevierCompany. 2017;18:2608–21. Its imporance is discussed in Table 1.
Moorefield EC, Andres SF, Blue RE, Van Landeghem L, Mah AT, Santoro MA, et al. Aging effects on intestinal homeostasis associated with expansion and dysfunction of intestinal epithelial stem cells. Aging (Albany NY). 2017;9:1898–915. Its imporance is discussed in Table 1.
Mihaylova MM, Cheng CW, Cao AQ, Tripathi S, Mana MD, Bauer-Rowe KE, Abu-Remaileh M, Clavain L, Erdemir A, Lewis CA, Freinkman E, Dickey AS, La Spada AR, et al. Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell. 2018; 22: 769-778.e4. Its imporance is discussed in Table 1.
Mihaylova MM, Cheng CW, Cao AQ, Tripathi S, Mana MD, Bauer-Rowe KE, Abu-Remaileh M, Clavain L, Erdemir A, Lewis CA, Freinkman E, Dickey AS, La Spada AR, et al. Fasting Activates fatty acid oxidation to enhance intestinal stem cell function during homeostasis and aging. Cell Stem Cell. 2018; 22: 769-778.e4. Its imporance is discussed in Table 1.
Pentinmikko N, Iqbal S, Mana M, Andersson S, Cognetta AB, Suciu RM, et al. Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nature Springer US. 2019;571:398–402. Its imporance is discussed in Table 1.
Igarashi M, Miura M, Williams E, Jaksch F, Kadowaki T, Yamauchi T, et al. NAD+ supplementation rejuvenates aged gut adult stem cells. Aging Cell. 2019;18:1–10.
Lewis SK, Nachun D, Martin MG, Horvath S, Coppola G, Jones DL. DNA Methylation analysis validates organoids as a viable model for studying human intestinal aging. Cmgh Elsevier Inc. 2020;9:527–41. This study used methylation analysis and the human epigenetic clock to show that there are region specific differences in the aged human GI tract. Importantly, organoids derived from defined regions maintained segmental differences in age associated methylation patterns.
Tomasetti C, Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science (80- ). 2015; 347: 78–81.
Gebert N, Cheng CW, Kirkpatrick JM, Di Fraia D, Yun J, Schädel P, et al. Region-Specific proteome changes of the intestinal epithelium during aging and dietary restriction. Cell Rep. 2020;31:107565. This study implemented region specific, proteomic analysis of the small intestines in young and aged mice and mapped age dependent changes, including alterations in metabolic pathways and response to dietary interventions.
Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–5.
Fry CS, Lee JD, Mula J, Kirby TJ, Jackson JR, Liu F, et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat Med Nature Publishing Group. 2015;21:76–80.
Alvarez-Buylla A, Herrera DG, Wichterle H. The subventricular zone: source of neuronal precursors for brain repair. Prog Brain Res. 2000;127:1–11.
Navarro Negredo P, Yeo RW, Brunet A. Aging and rejuvenation of neural stem cells and their niches. Cell Stem Cell Elsevier Inc. 2020;27:202–23. This recent, comprehensive review covers many aspects and consequences of aging on neuronal stem cells.
Raven A, Lu WY, Man TY, Ferreira-Gonzalez S, O’Duibhir E, Dwyer BJ, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature Nature Publishing Group. 2017;547:350–4.
Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell Elsevier Inc. 2015;161:106–18.
Buono R, Longo VD. Starvation, Stress resistance, and cancer. Trends Endocrinol Metab Elsevier Ltd. 2018;29:271–80.
Longo VD, Di Tano M, Mattson MP, Guidi N. Intermittent and periodic fasting, longevity and disease. Nat Aging Springer US. 2021;1:47–59. This very recent review describes different dietary restriction strategies, as well as their implementation and outcomes in both human and rodent studies.
Libert S, Guarente L. Metabolic and neuropsychiatric effects of calorie restriction and sirtuins. Annu Rev Physiol. 2013;75:669–84.
Min KJ, Tatar M. Restriction of amino acids extends lifespan in Drosophila melanogaster. Mech Ageing Dev. 2006;127:643–6.
Grandison RC, Piper MDW, Partridge L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature. Nature Publishing Group; 2009; 462: 1061-1064.
Levine ME, Suarez JA, Brandhorst S, Balasubramanian P, Cheng CW, Madia F, et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab Elsevier Inc. 2014;19:407–17.
Fontana L, Cummings NE, Arriola Apelo SI, Neuman JC, Kasza I, Schmidt BA, et al. Decreased Consumption of branched-chain amino acids improves metabolic health. Cell Rep. 2016;16:520–30.
Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J Physiol. 2018;596:623–45.
Solon-Biet SM, Mitchell SJ, Coogan SCP, Cogger VC, Gokarn R, McMahon AC, et al. Dietary Protein to carbohydrate ratio and caloric restriction: comparing metabolic outcomes in mice. Cell Rep The Authors. 2015;11:1529–34.
Xiao F, Yu J, Guo Y, Deng J, Li K, Du Y, et al. Effects of individual branched-chain amino acids deprivation on insulin sensitivity and glucose metabolism in mice. Metabolism Elsevier Inc. 2014;63:841–50.
Richardson NE, Konon EN, Schuster HS, Mitchell AT, Boyle C, Rodgers AC, et al. Lifelong restriction of dietary branched-chain amino acids has sex-specific benefits for frailty and life span in mice. Nat Aging Springer US. 2021;1:73–86. This very recent study showed that restricting dietary BCAAs increased survival in two progeroid mouse models. The authors further showed significant lifespan extension of wild type male, but not female mice, when mice are placed on lifelong low BCAA diet.
Xiao F, Huang Z, Li H, Yu J, Wang C, Chen S, et al. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes. 2011;60:746–56.
Lee D, Hwang W, Artan M, Jeong DE, Lee SJ. Effects of nutritional components on aging. Aging Cell. 2015;14:8–16.
Miller RA, Buehner G, Chang Y, Harper JM, Sigler R, Smith-Wheelock M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell. 2005;4:119–25.
Orentreich N, Matias JR, DeFelice A, Zimmerman JA. Low Methionine ingestion by rats extends life span. J Nutr. 1993;123:269–74.
Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–94.
Cummings NE, Lamming DW. Regulation of metabolic health and aging by nutrient-sensitive signaling pathways. Mol Cell Endocrinol. 2017;455:13–22.
Saxton RA, Sabatini DM. mTOR Signaling in growth, metabolism, and disease. Cell Elsevier Inc. 2017;168:960–76.
Laplante M, Sabatini DM. MTOR signaling in growth control and disease. Cell Elsevier. 2012;149:274–93.
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature Nature Publishing Group. 2009;460:392–5.
Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by Modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–90.
Powers RW III, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–84.
Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, et al. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab Elsevier Inc. 2012;15:713–24.
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged Rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68.
Apelo SIA, Lin A, Brinkman JA, Meyer E, Morrison M, Tomasiewicz JL, et al. Ovariectomy uncouples lifespan from metabolic health and reveals a sexhormone-dependent role of hepatic mtorc2 in aging. Elife. 2020;9:1–17.
Chellappa K, Brinkman JA, Mukherjee S, Morrison M, Alotaibi MI, Carbajal KA, et al. Hypothalamic mTORC2 is essential for metabolic health and longevity. Aging Cell. 2019;18:1–21.
Lamming DW, Ye L, Katajisto P. Rapamycin-Induced Insulin Resistance Is Mediated by mTORC2 Loss and Uncoupled from Longevity. Science 2012; 335: 1638-1643.
Lamming DW, Ye L, Katajisto P. Rapamycin-induced insulin resistance is mediated by mTORC2 Loss and uncoupled from longevity. Science. 2012;(80):335.
Schreiber KH, Arriola Apelo SI, Yu D, Brinkman JA, Velarde MC, Syed FA, et al. A novel rapamycin analog is highly selective for mTORC1 in vivo. Nat Commun Springer US. 2019;10:1–12.
Yu D, Tomasiewicz JL, Yang SE, Miller BR, Wakai MH, Sherman DS, et al. Calorie-restriction-induced insulin sensitivity is mediated by adipose mTORC2 and not required for lifespan extension. Cell Rep ElsevierCompany. 2019;29:236–248.e3.
Selman C, Tullet JMA, Wieser D, Irvine E, Lingard SJ, Choudhury AI, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;461(80):140–4.
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol Nature Publishing Group. 2011;13:1016–23.
Toyama EQ, Herzig S, Courchet J, Lewis Jr. TL, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress 2016; 351: 275–82.
Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, et al. Aging-associated reductions in AMP-Activated Protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–6.
Cantó C, Auwerx J. Calorie restriction: Is AMPK a key sensor and effector? Physiology. 2011;26:214–24.
Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K, et al. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab Elsevier Inc. 2017;26:884–896.e5.
Slack C, Foley A, Partridge L. Activation of AMPK by the putative dietary restriction mimetic metformin is insufficient to extend lifespan in Drosophila. PLoS One. 2012;7:1–7.
Greer EL, Brunet A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell. 2009;8:113–27.
Su Y, Wang T, Wu N, Li D, Fan X, Xu Z, et al. Alpha-ketoglutarate extends Drosophila lifespan by inhibiting mTOR and activating AMPK. Aging (Albany NY). 2019;11:4183–97.
Verovskaya EV, Dellorusso PV, Passegué E. Losing sense of self and surroundings: hematopoietic stem cell aging and leukemic transformation. Trends Mol Med. 2019;25:494–515.
Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–51.
Norddahl GL, Pronk CJ, Wahlestedt M, Sten G, Nygren JM, Ugale A, et al. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell Elsevier Inc. 2011;8:499–510.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med Nature Publishing Group. 2014;20:1472–8.
Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal Hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87.
Khokhar ES, Borikar S, Eudy E, Stearns T, Young K, Trowbridge JJ. Aging-associated decrease in the histone acetyltransferase KAT6B is linked to altered hematopoietic stem cell differentiation. Exp Hematol Elsevier Inc. 2020;82:43–52.e4. In this study identified histone acetyltransferase KAT6B as a novel epigenetic regulator of hematopoietic differentiation in an RNAi screen and also showed its expression is significantly decreased with aging.
Meng D, Frank AR, Jewell JL. mTOR signaling in stem and progenitor cells. Development. 2018;145:dev152595.
Ramalingam P, Poulos MG, Gutkin MC, Katsnelson L, Freire AG, Lazzari E, Butler JM. Endothelial mTOR maintains hematopoiesis during aging. J Exp Med. 2020; 217: e20191212.
Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature Nature Publishing Group. 2017;543:205–10. This study provides mechanistic insight of why aged hematopoietic stem cells have elevated autophagy levels and further showed that loss of autophagy leads to accumulation of mitochondria and activated metabolic state, which accelerates myeloid differentiation.
Luo H, Mu WC, Karki R, Chiang HH, Mohrin M, Shin JJ, et al. Mitochondrial Stress-initiated aberrant activation of the NLRP3 inflammasome regulates the functional deterioration of hematopoietic stem cell aging. Cell Rep ElsevierCompany. 2019;26:945–954.e4.
Tiwari SK, Toshniwal AG, Mandal S, Mandal L. Fatty acid b-oxidation is required for the differentiation of larval hematopoietic progenitors in drosophila. Elife. 2020;9:1–35.
Kratchmarov R, Viragova S, Kim MJ, Rothman NJ, Liu K, Reizis B, Reiner SL. Metabolic control of cell fate bifurcations in a hematopoietic progenitor population. Immunol Cell Biol. 2018; 96: 863–71.
Kratchmarov R, Viragova S, Kim MJ, Rothman NJ, Liu K, Reizis B, et al. Metabolic control of cell fate bifurcations in a hematopoietic progenitor population. Immunol Cell Biol. 2018;96:863–71.
Ma Z, Xu J, Wu L, Wang J, Lin Q, Chowdhury FA, et al. Hes1 deficiency causes hematopoietic stem cell exhaustion. Stem Cells. 2020;38:756–68.
Ertl RP, Chen J, Astle CM, Duffy TM, Harrison DE. Effects of dietary restriction on hematopoietic stem-cell aging are genetically regulated. Blood. 2008;111:1709–16.
Lazare S, Ausema A, Reijne AC, van Dijk G, van Os R, de Haan G. Lifelong dietary intervention does not affect hematopoietic stem cell function. Exp Hematol ISEH - International Society for Experimental Hematology. 2017;53:26–30.
Rodriguez-Fernandez IA, Qi Y, Jasper H. Loss of a proteostatic checkpoint in intestinal stem cells contributes to age-related epithelial dysfunction. Nat Commun Springer US. 2019;10:1–15. In this study the authors describe a “proteostatic checkpoint” in Drosophila ISCs, which is compromised in aged flies and can be restored with an Nrf2 activator. Treatment with the Nrf2 activator reduced age dependent protein aggregation and restored inhibition of ISC proliferation.
Tsokanos F, Albert M, Demetriades C, Spirohn K, Boutros M, Teleman AA. eIF 4A inactivates TORC 1 in response to amino acid starvation. EMBO J. 2016;35:1058–76.
Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong SJ, et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature Nature Publishing Group. 2016;531:53–8.
Cheng CW, Biton M, Haber AL, Gunduz N, Eng G, Gaynor LT, et al. Ketone Body signaling mediates intestinal stem cell homeostasis and adaptation to diet. Cell. 2019;178:1115–1131.e15. This recent study provided evidence that the rate limiting enzyme in ketogenesis, HMGCS2, is highly expressed in ISCs. Dietary inputs regulate endogenous production of ketones in ISCs, which subsequently dictates stemness and differentiation.
Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martín MG, et al. Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell. 2018;22:206–220.e4.
Rangan P, Choi I, Wei M, Navarrete G, Guen E, Brandhorst S, et al. Fasting-Mimicking Diet Modulates Microbiota and promotes intestinal regeneration to reduce inflammatory bowel disease pathology. Cell Rep ElsevierCompany. 2019;26:2704–2719.e6.
Stine RR, Sakers AP, TeSlaa T, Kissig M, Stine ZE, Kwon CW, et al. PRDM16 maintains Homeostasis of the intestinal epithelium by controlling region-specific metabolism. Cell Stem Cell Elsevier Inc. 2019;25:830–845.e8.
Chen L, Vasoya RP, Toke NH, Parthasarathy A, Luo S, Chiles E, Flores J, Gao N, Bonder EM, Su X, Verzi MP. HNF4 Regulates Fatty Acid Oxidation and Is Required for Renewal of Intestinal Stem Cells in Mice. Gastroenterology 2020; 158: 98599.
Schell JC, Wisidagama DR, Bensard C, Zhao H, Wei P, Tanner J, et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat Cell Biol. 2017;19:1027–36.
Flores A, Schell J, Krall AS, Jelinek D, Miranda M, Grigorian M, et al. Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat Cell Biol. 2017;19:1017–26.
Igarashi M, Guarente L. mTORC1 and SIRT1 Cooperate to foster expansion of gut adult stem cells during calorie restriction. Cell Elsevier Inc. 2016;166:436–50.
Yousefi M, Nakauka-Ddamba A, Berry CT, Li N, Schoenberger J, Simeonov KP, Cedeno RJ, Yu Z, Lengner CJ. Calorie Restriction Governs Intestinal Epithelial Regeneration through Cell-Autonomous Regulation of mTORC1 in Reserve Stem Cells. Stem Cell Reports. ElsevierCompany.; 2018; 10: 703–11.
Rodríguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, Pras-Raves M, et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature. 2017;543:424–7.
Ghosh J, Kapur R. Regulation of hematopoietic stem cell self-renewal and leukemia maintenance by the PI3K-mTORC1 pathway. Curr Stem Cell Rep Current Stem Cell Reports. 2016;2:368–78.
Audano M, Pedretti S, Crestani M, Caruso D, De Fabiani E, Mitro N. Mitochondrial dysfunction increases fatty acid β-oxidation and translates into impaired neuroblast maturation. FEBS Lett. 2019;593:3173–89.
Ratcliff M, Rees D, McGrady S, Buntwal L, Hornsby AKE, Bayliss J, et al. Calorie restriction activates new adult born olfactory-bulb neurones in a ghrelin-dependent manner but acyl-ghrelin does not enhance subventricular zone neurogenesis. J Neuroendocrinol. 2019;31:1–11.
Knobloch M, Pilz GA, Ghesquière B, Kovacs WJ, Wegleiter T, Moore DL, et al. A Fatty acid oxidation-dependent metabolic shift regulates adult neural stem cell activity. Cell Rep. 2017;20:2144–55. This study showed that quiescent NSPCs require high levels of FAO and inhibition of FAO through Malonyl-CoA treatment was sufficient to increase proliferation of NSPCs.
Yang LL, Millischer V, Rodin S, MacFabe DF, Villaescusa JC, Lavebratt C. Enteric short-chain fatty acids promote proliferation of human neural progenitor cells. J Neurochem. 2020; 154: 63546.
Romine J, Gao X, Xu XM, So KF, Chen J. The proliferation of amplifying neural progenitor cells is impaired in the aging brain and restored by the mTOR pathway activation. Neurobiol Aging Elsevier Inc. 2015;36:1716–26.
Wang BZ, Yang JJ, Zhang H, Smith CA, Jin K. AMPK signaling regulates the age-related decline of hippocampal neurogenesis. Aging Dis. 2019;10:1058–74.
Gao J, Perera G, Bhadbhade M, Halliday GM, Dzamko N. Autophagy activation promotes clearance of α-synuclein inclusions in fibril-seeded human neural cells. J Biol Chem. 2019;294:14241–56.
Tang H, Inoki K, Brooks SV, Okazawa H, Lee M, Wang J, et al. mTORC1 underlies age-related muscle fiber damage and loss by inducing oxidative stress and catabolism. Aging Cell. 2019;18:1–20.
Baar EL, Carbajal KA, Ong IM, Lamming DW. Sex- and tissue-specific changes in mTOR signaling with age in C57BL/6J mice. Aging Cell. 2016;15:155–66.
Houtkooper RH, Argmann C, Houten SM, Cant́o C, Jeninga EH, Andreux Ṕeńelope A., Thomas C, Doenlen R, Schoonjans K, Auwerx J. The metabolic footprint of aging in mice. Sci Rep. 2011; 1: 1-11.
Joseph GA, Wang S, Zhou W, Kimble G, Tse H, Eash J, et al. Partial inhibition of mTORC1 in Aged rats counteracts the decline in muscle mass and reverses molecular signaling associated with sarcopenia. Mol Cell Biol. 2019;39:1–16.
Pavlidou T, Marinkovic M, Rosina M, Fuoco C, Vumbaca S, Gargioli C, et al. Metformin delays satellite cell activation and maintains quiescence. Stem Cells Int. 2019;2019:1–19.
Boldrin L, Ross JA, Whitmore C, Doreste B, Beaver C, Eddaoudi A, et al. The effect of calorie restriction on mouse skeletal muscle is sex, strain and time-dependent. Sci Rep. 2017;7:1–16.
White JP, Billin AN, Campbell ME, Russell AJ, Huffman KM, Kraus WE. The AMPK/p27Kip1 axis regulates autophagy/apoptosis decisions in aged skeletal muscle stem cells. Stem Cell Rep ElsevierCompany. 2018;11:425–39.
Amorim NML, Kee A, Coster ACF, Lucas C, Bould S, Daniel S, et al. Irradiation impairs mitochondrial function and skeletal muscle oxidative capacity: significance for metabolic complications in cancer survivors. Metabolism Elsevier Inc. 2020;103:154025.
Pala F, Di Girolamo D, Mella S, Yennek S, Chatre L, Ricchetti M, et al. Distinct metabolic states govern skeletal muscle stem cell fates during prenatal and postnatal myogenesis. J Cell Sci. 2018;131:jcs212977.
Belal SA, Sivakumar AS, Kang DR, Cho S, Choe HS, Shim KS. Modulatory effect of linoleic and oleic acid on cell proliferation and lipid metabolism gene expressions in primary bovine satellite cells. Animal Cells Syst (Seoul) Taylor & Francis. 2018;22:324–33.
Halpern KB, Shenhav R, Matcovitch-Natan O, Tóth B, Lemze D, Golan M, et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature. 2017;542:1–5.
Moor AE, Harnik Y, Ben-Moshe S, Massasa EE, Rozenberg M, Eilam R, et al. Spatial reconstruction of single enterocytes uncovers broad zonation along the intestinal villus axis. Cell Elsevier Inc. 2018;175:1156–1167.e15.
Acknowledgements
We thank D.W.Lamming, C.W. Cheng and members of the Mihaylova lab for critical reading and comments; in particular M. Festing, J. Sledziona and K. Akkaya. We apologize for being unable to discuss many primary studies due to space limitations. This work is supported in part by NIH (K99/R00 AG054760) and the Glenn Foundation for Medical Research and AFAR Grants for Junior Faculty. A.K. is supported in part by the CMBP NIH Pre Doctoral Training Grant (T32 GM 86252-8). Figures created with BioRender.com. B.S. was funded in part by the Foods for Health Post-Doctoral Fellowship. The authors declare no competing interests.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Metabolism and Stem Cells
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Keller, A., Temple, T., Sayanjali, B. et al. Metabolic Regulation of Stem Cells in Aging. Curr Stem Cell Rep 7, 72–84 (2021). https://doi.org/10.1007/s40778-021-00186-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-021-00186-6