
Abstract
Introduction
Baricitinib, a JAK1/JAK2 inhibitor, is approved for treatment of moderate-to-severe rheumatoid arthritis (RA) in China. This single-arm, prospective, multi-center, post-marketing safety study (PMSS) evaluated the safety and effectiveness of baricitinib in Chinese patients.
Methods
This study included adult patients with moderate-to-severe active RA who received baricitinib over periods of approximately 12 and 24 weeks. The primary endpoint was safety, defined as week 12 adverse event (AE)/serious AE incidence. Secondary endpoints were week 24 safety and effectiveness (disease activity score with 28 joints/C-reactive protein [DAS28-CRP] and simplified/Clinical Disease Activity Index [SDAI/CDAI]).
Results
Safety analyses included 667 patients (female, 82.3%; mean age, 53.3 years; mean RA duration, 86.9 months); 106/667 (15.9%) were 65–74 years old and 19/667 (2.8%) were ≥ 75 years old; 87.0% received baricitinib 2 mg QD. Total exposure was 262.1 patient-years (PY). At week 12, AEs had occurred in 214 (32.1%; exposure-adjusted incidence rate [EAIR], 172.5 per 100 PY) patients (serious AEs: 22 [3.3%; EAIR, 15.0]). At week 24, AEs had occurred in 250 (37.5%; EAIR, 125.9) patients (serious AEs: 28 [4.2%; EAIR, 10.9]). Two patients (0.3%) died (of pneumonia and unknown cause); EAIR for death, 0.77. Serious infection occurred in 1.2% of patients (EAIR, 3.1). Hepatotoxicity occurred in 3.4% of patients (EAIR, 9.0). No patients met potential Hy’s law laboratory criteria (alanine/aspartate aminotransferases ≥ 3 × upper limit of normal (ULN) and total bilirubin ≥ 2 × ULN). Malignancy occurred in one patient. No patients experienced venous thromboembolism (VTE) or major adverse cardiovascular events (MACE). At week 24, 52.4%, 27.5%, and 27.6% of patients achieved remission per DAS28-CRP, SDAI, and CDAI, respectively.
Conclusions
This PMSS investigated the safety and effectiveness of baricitinib in clinical practice in China. No VTE/MACE or new safety signals were reported and there was promising effectiveness, supporting the use of baricitinib in Chinese patients with moderate-to-severe active RA.
Trial Registration
EU PAS Register: EUPAS34213.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Targeted inhibitors of Janus kinase (JAK) proteins are recommended for treatment of rheumatoid arthritis (RA) that does not adequately respond to other standard therapy; however, a potentially increased risk of certain adverse events with these therapies has been observed. |
This post-marketing safety study aimed to evaluate the safety and effectiveness of the JAK inhibitor baricitinib in Chinese patients with RA. |
What was learned from this study? |
For Chinese patients who received baricitinib for moderate-to-severe RA as part of routine clinical practice, treatment was generally well tolerated with no new safety signals observed. |
These findings support the use of baricitinib treatment for Chinese patients with moderate-to-severe active RA. |
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by progressive joint damage and considerable morbidity, pain, and reduced health-related quality of life. Worldwide, the prevalence of RA was reported to be 0.46% between 1980 and 2019 [1], and in China was 0.42% in 2013 [2, 3].
Typical treatment for RA involves disease-modifying anti-rheumatic drugs (DMARDs), most commonly the conventional synthetic DMARD (csDMARD), methotrexate. Therapeutic advances have led to the development of increasingly targeted treatments, for example, antibody-based biologic DMARDs (bDMARDs) and targeted synthetic DMARDs (tsDMARDs).
Orally available tsDMARDs that inhibit Janus kinase (JAK) proteins have shown promise. For example, baricitinib (JAK1/2), tofacitinib (JAK1/3), and upadacitinib (JAK1) [4] have demonstrated efficacy in RA treatment [5, 6]. In line with the American College of Rheumatology (ACR), European Alliance of Associations for Rheumatology (EULAR), and Asia-Pacific League of Associations for Rheumatology (APLAR) guidelines [7,8,9], current (2018) Chinese treatment guidelines for patients with moderate-to-severe RA recommend the addition of tsDMARDs or bDMARDs to csDMARD therapy if the treatment target cannot be reached with csDMARDs alone [2]. However, an increased risk of certain adverse events (AEs), including serious infections, major adverse cardiovascular events (MACE), and malignancy, was observed following treatment with the JAK inhibitor tofacitinib compared with TNF inhibitors in a large randomized safety trial [10,11,12,13]. This led to revisions of boxed warnings on US FDA labels of JAK inhibitors and updated EULAR guidelines now recommend that JAK inhibition may be considered, but pertinent risk factors must also be taken into account [8].
While data establishing the safety profile and RA-associated effectiveness of baricitinib in clinical trials globally and in predominantly Chinese populations have been previously reported [5, 14,15,16,17,18], real-world data, including patients who may have been excluded from clinical trial populations, are important to fully characterize the safety and effectiveness of baricitinib. These data are important in Chinese patients following the 2019 approval of baricitinib in China and as required by the Chinese National Medical Products Administration (NMPA) according to local regulations. Furthermore, these data are particularly important given the relatively low use of bDMARDs in China, potentially due to their high cost [19, 20].
In this post-marketing safety study (PMSS) conducted from July 2020 to September 2022, we aimed to evaluate the real-world safety and effectiveness of the JAK inhibitor baricitinib for the treatment of Chinese patients with moderate-to-severe active RA.
Methods
Study Design
This was a prospective, observational, multi-center PMSS of baricitinib for the treatment of adult (≥ 18 years old) Chinese patients with moderate-to-severe active RA. All patients received baricitinib as part of routine care; patients who were enrolled in another clinical study or who had contraindications to baricitinib were excluded.
The primary endpoint was safety, as measured by the incidence of AEs/serious AEs at 12 weeks. Safety, effectiveness, and patient-reported outcomes (PROs) of duration of morning joint stiffness (MJS) and pain visual analog scale (VAS) at week 24 were secondary endpoints; a full list of endpoints is provided in the Supplementary Material.
Data were collected at baseline (before first dose) after which patients had two or three subsequent post-baseline visits over the course of approximately 24 weeks of observation. Safety follow-up to monitor for AEs was conducted until 30 days after treatment discontinuation or commencement of a new RA medication, whichever occurred first.
The PMSS was conducted in compliance with applicable laws in China and in accordance with the Chinese Guidelines for Good Clinical Practice and the principles outlined in the Declaration of Helsinki. The study received IRB and ethics approval at each site (Supplementary Material); the central ethics board was the Peking Union Medical College Hospital Ethics Committee (approval number HS-2327). All participants provided written, informed consent prior to participation.
Assessments
Safety
Safety was analyzed in the Safety Analysis Set (SAS), which comprised all patients who received ≥ 1 dose of baricitinib.
AEs were classified using preferred terms (PTs) from the Medical Dictionary for Regulatory Activities (MedDRA) v25.1; severity was graded as mild, moderate, or severe according to the investigator. Serious AEs were defined as per the International Conference on Harmonisation and detailed in the Supplementary Material: they were any AE that resulted in death; initial or prolonged inpatient hospitalization; a life-threatening experience (that is, immediate risk of dying); persistent or significant disability/incapacity; congenital anomaly/birth defect; or may jeopardize the patient or require intervention. AEs of special interest (AESIs) were serious infections, hepatotoxicity, and venous thromboembolism (VTE), which included deep-vein thrombosis (DVT) and pulmonary embolism (PE). Serious infections were defined as serious AEs in the Medical Dictionary for Regulatory Activities (MedDRA) System Organ Class (SOC) of infections and infestations. Hepatotoxicity and VTE data were collected as recorded by the investigators in electronic case report forms (eCRF). MACE included myocardial infarction, cardiovascular death, and stroke. AE relationship to treatment was determined by the practicing investigator.
Laboratory tests (lab samples were collected in an observational capacity only) were continually collected during the whole study period and included routine blood tests, blood biochemistry, coagulation tests, routine urine tests, and serological tests. Definitions of grades/categories used for laboratory tests are provided in the Supplementary Material.
Effectiveness
Effectiveness was analyzed in the Effectiveness Analysis Set (EAS), which comprised all patients in the SAS who had a baseline and ≥ 1 post-baseline effectiveness observation and who had received baricitinib for ≥ 10 weeks.
Disease activity was measured using the Disease Activity Score in 28 Joints based on C-reactive protein (DAS28-CRP), the Simple Disease Activity Index (SDAI), and the Clinical Disease Activity Index (CDAI), each giving composite scores as previously described [21, 22]. Categories of remission and low, moderate, and high disease activity were defined based on thresholds for each scale as detailed in the Supplementary Material.
Patient-reported mean duration of MJS and pain VAS were assessed at weeks 12 and 24, as collected in electronic diaries.
Statistical Analysis and Sample Size Calculations
The Chinese Registry of rhEumatoiD arthrITis (CREDIT) was used to collect data [23].
The sample size was calculated based on the incidence of serious infection (1.4%) observed in a previous clinical trial (Lilly, data on file). With an assumed incidence of 1.4%, a sample size of 530 patients was estimated to provide adequate precision (95% confidence intervals [CIs] of 0.4–2.4%). Therefore, the final sample was considered to require at least 600 patients, with 667 calculated to be required if assuming a safety follow-up drop-out rate of 10%.
All statistical analyses were exploratory, based on the observational nature of the study, and were conducted using SAS v9.4. There was no imputation for missing data, unless specified otherwise.
Exposure-adjusted incidence rates (EAIRs) were calculated as the number of patients with events divided by overall baricitinib exposure time subjects to events, expressed per 100 patient-years (PY). Patient exposure was censored at the time of the first AE, i.e., (the start date of first AE or the date of cut off (for those without AE) for each patient − the first dose date for each patient + 1)/365.25. 95% CIs for EAIRs were calculated based on a Poisson distribution. Further statistical details can be found in the Supplementary Material.
Results
Patient Disposition and Baseline Characteristics
In total, 699 patients were screened; 667 received baricitinib and were included in the SAS and of those, 514 were included in the EAS (Fig. 1). Overall, 197 patients (29.5%) withdrew from the study, most commonly due to the patient’s decision (15.1%) or loss to follow-up (7.0%).

Patient disposition. AE adverse event, EAS effectiveness analysis set, SAS Safety Analysis Set
At baseline, patients in the SAS had a mean (SD) age of 53.3 (12.5) years (15.9% were 65–74 years old; 2.8% ≥ 75 years old), 82.3% were female, the mean (SD) body mass index (BMI) was 22.3 (3.4) kg/m2, 12.3% were current or former smokers, and 25.2% had prior/concomitant cardiovascular disease (Table 1). All 667 patients had definite RA per ACR/EULAR 2010 criteria, with a mean (SD) diagnosis score of 8.3 (1.5), and a mean duration of RA of 7.2 (8.3) years. Prior RA therapies had been received by 72.0% of patients and included csDMARDs (70.2%), bDMARDs (4.3%), and the tsDMARD tofacitinib (5.2%). Concomitant therapies included csDMARDs (86.8%; most commonly methotrexate [54.3%], leflunomide [35.5%], hydroxychloroquine [24.9%]) and systemic corticosteroids (39.4%).
Safety
The median overall duration of baricitinib exposure was 167 days, the total baricitinib exposure was 262.1 PY. Overall, 580 patients (87.0%) received baricitinib at 2 mg once daily (QD), 53 (7.9%) received baricitinib at 4 mg QD, and 34 (5.1%) received a mixed dosage of 2 and 4 mg QD.
At week 12,214 patients (32.1%) experienced an AE (EAIR, 172.5 per 100 PY). Twenty-two patients (3.3%) experienced a serious AE (EAIR, 15.0 per 100 PY). At week 24, in total, 250 patients (37.5%) experienced an AE; the EAIR for AEs was 125.9 per 100 PY (Table 2). Twenty-eight patients (4.2%) experienced a serious AE; the EAIR for serious AEs was 10.9 per 100 PY. Two patients (0.3%) died; both had multiple concomitant diseases and received the 2-mg dose. One 73-year-old patient (receiving multiple concomitant medications) died of treatment-related pneumonia and a 69-year-old patient died 11 days after their last dose of baricitinib with unknown cause of death, not considered related to baricitinib. The EAIR for death was 0.77 per 100 PY. Twenty-four patients (3.6%) discontinued treatment due to AEs; the EAIR for AEs leading to discontinuation was 9.2 per 100 PY. Treatment-related AEs were reported in 18.0% of patients, with an EAIR of 50.7 per 100 PY.
EAIRs for AEs, serious AEs, and AESIs did not increase with prolonged exposure from 12 to 24 weeks (Table 2). The three most common AEs by PT at 24 weeks were ‘hepatic function abnormal’ (3.3%), upper respiratory tract infection (2.7%), and ‘platelet count increased’ (2.4%; Supplementary Material Table S1). Serious AEs that had occurred in ≥ 2 patients at 24 weeks were pneumonia (four patients; 0.6%), rheumatoid arthritis (three patients; 0.4%), and arthralgia (two patients; 0.3%; Supplementary Material Table S2).
Serious infections were reported in eight patients (1.2%), most commonly pneumonia (four patients; 0.6%); all were considered related to treatment. Hepatotoxicity was reported in 23 patients (3.4%), most commonly ‘hepatic function abnormal’ (17 patients; 2.5%). Eighteen patients (2.7%) experienced treatment-related hepatotoxicity and one patient experienced a serious AE of hepatotoxicity. No patients had concurrent alanine/aspartate aminotransferase (ALT/AST) ≥ 3 × ULN and serum total bilirubin ≥ 2 × ULN. No AEs of VTE or MACE (myocardial infarction, cardiovascular death, and stroke) were reported. At 24 weeks, herpes zoster infection was reported in seven patients (1.0%); pulmonary tuberculosis in one patient (0.1%); and new primary malignancy (thyroid cancer considered unrelated to treatment) in one patient (0.1%).
Clinically meaningful, treatment-emergent changes in hematological parameters were uncommon (Table 3). No patients experienced changes from baseline that were grade < 3 to ≥ 3 in white blood cell count, hemoglobin, or platelets. Change from baseline in neutrophil count was from grade < 3 to ≥ 3 in 2/503 patients (0.4%) and in lymphocytes, 3/503 patients (0.6%). Thrombocytosis (platelets increase to > 600 × 109/L) was observed in 2/506 patients (0.4%).
Changes in treatment-emergent blood chemistry parameters are summarized in Table 4. ALT levels increased from < 3 × to ≥ 3 × ULN in 6/488 patients (1.2%), and from < 5 × to ≥ 5 × ULN in 1/488 patients (0.2%). AST levels increased from < 3 × to ≥ 3 × ULN in 4/462 patients (0.9%). Alkaline phosphatase (ALP) increased from < 1.5 × to ≥ 1.5 × ULN in 4/313 patients (1.3%). No patients had clinically meaningful increases in total bilirubin (< 2 × to ≥ 2 × ULN), creatinine (grade < 3 to ≥ 3), or creatine kinase (grade < 3 to ≥ 3).
Effectiveness
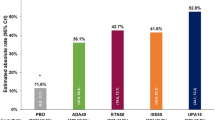
At 24 weeks, the proportion of patients who achieved remission per DAS28-CRP, SDAI, and CDAI was 52.4, 27.5, and 27.6%, respectively (Fig. 2). The least squares (LS) mean change from baseline to week 24 (95% CI) in DAS28-CRP, SDAI, and CDAI was − 1.9 (− 2.1 to − 1.8), − 19.3 (− 20.5 to − 18.1), and − 17.7 (− 18.9 to − 16.6), respectively (Supplementary Material Table S3).

DAS28-CRP, SDAI, and CDAI measures of effectiveness at 24 weeks (EAS). Patients with unknown effectiveness scores were not included; the denominator for each percentage is indicated at the top of the bars. Percentages may not sum to 100% due to rounding. CDAI Clinical Disease Activity Index, DAS28-CRP disease activity score 28-C-reactive protein, EAS effectiveness analysis set, SDAI simple disease activity index
At 24 weeks, patient-reported mean duration of MJS and pain VAS scores decreased from baseline, with mean (SD) changes from baseline of − 28.3 (47.0) and − 3.0 (2.5), respectively (Supplementary Material Table S4).
Discussion
In this real-world, observational study of baricitinib for Chinese patients with moderate-to-severe active RA, the safety profile was generally consistent with previous reports from clinical trials [17, 24, 25] and no new safety signals were observed. Baricitinib treatment demonstrated effectiveness in the treatment of Chinese patients with RA, with notable proportions of patients able to achieve remission per DAS28-CRP, SDAI, and CDAI scales.
Notably, use of prior bDMARDs was lower in our study (4.9%) than in the Japanese PMSS (71%) [26], reflecting the difference in bDMARD access and approvals between China and global populations [19]. Indeed, the majority of patients in our study (87.0%) received the 2-mg dose of baricitinib, and not the 4-mg dose used in previous clinical trials of baricitinib and the Japanese PMSS [25, 26]. As this PMSS was conducted from 2020 onwards, prescribing physicians followed Chinese recommendations for dosage at the time, which required patients to initiate baricitinib at 2 mg QD as the only approved dosage. However, the study protocol was amended in March 2021 to reflect the additional approval of the 4-mg dose in China, which is indicated for patients with an inadequate response to the 2-mg dose for at least 3 months or to TNF inhibitors.
With appropriate caution for cross-trial comparisons, baseline characteristics were generally comparable between our study and previous clinical trials or post-marketing studies [17, 24,25,26]. The mean age in our study was 53.2 years, slightly older than the 48.0 years reported in Chinese patients in RA-BALANCE [24], but younger than the 64 years reported in a 24-week Japanese baricitinib PMSS for RA [26]. The mean BMI was 22.3 kg/m2 in our study similar to the Japanese PMSS, but lower than that of patients enrolled in clinical trials (mean BMI 27.7 kg/m2) [27]. Finally, in our study, baseline DAS28-CRP, SDAI, and CDAI remission rates were 12.7, 4.8, and 4.7%, respectively, similar to the Japanese PMSS, which reported baseline remission rates of 12.6, 3.1, and 2.9% for each scale, respectively [26].
In our study, AEs were reported in 37.5% of patients at 24 weeks (EAIR [95% CI], 125.9 [110.8–142.5] per 100 PY), which is higher than in the Japanese PMSS (26.9%; incidence rate [IR], 99.7 [95.1–104.2] per 100 PY) [26]; however, the incidence of AEs did not increase from 12 to 24 weeks (EAIR [95% CI], 172.5 [150.2–197.3] per 100 PY). Serious AE incidences were similar to those reported in the 24-week Japanese PMSS [26].
Two patients died in this study; both had multiple concomitant diseases and received the 2-mg dose of baricitinib. One 73-year-old patient died of treatment-related pneumonia (receiving multiple concomitant medications, including the immunomodulatory leflunomide, methotrexate, and Tripterygium wilfordii) and one 69-year-old patient died of an unknown cause, not considered related to treatment. The EAIR for death in this study of 0.77 (95% CI, 0.1–2.8) per 100 PY was similar to global data: 0.0 [95% CI, not available–1.9] and 0.6 [95% CI, 0.1–1.8] per 100 PY in the 2- and 4-mg dose groups to week 24, respectively [25], and the incidence rate of 0.56 (95% CI, 0.45–0.70) per 100 PY in a long-term extension study [17]. The EAIR for death in this study is also comparable to the Japanese PMSS (0.85 [0.46–1.24] per 100 PY) [26].
In our study, a total of eight patients (1.2%) had serious infections, which was similar to the proportion reported for Chinese patients in the RA-BALANCE study (0.9%) and in the Japanese PMSS (1.9%) [24, 26]. The EAIR for serious infections in our study was similar to the incidence rate reported in both the 2-mg and 4-mg cohorts in the 3-year integrated analysis of clinical trials to week 24 [25].
The incidence of hepatotoxicity was slightly higher in this study than the EAIR reported in the Japanese 24-week PMSS (7.15 per 100 PY) [26] and the EAIR for abnormal hepatic function (6.5 per 100 PY) in a Japanese integrated analysis of nine clinical trials of baricitinib in RA with a median of 1.6 years on treatment [28]. However, the incidences of ALT and AST changes in our study were comparable to previous reports [26, 28] and no patients met laboratory criteria for Hy’s law [29], suggesting that hepatocellular injury was unlikely to occur with baricitinib treatment.
In the phase 3b/4 ORAL Surveillance study, which included patients with RA who were ≥ 50 years old with at least one additional cardiovascular risk factor, increased risks of MACE, malignancy, and serious infections were observed in patients who received tofacitinib compared with TNF inhibitors after a median follow-up of 4.0 years [10, 11]. In an observational study using propensity-matched data from multiple databases in the US, Europe, and Japan, there was a statistically significant increase in the incidence of VTE with baricitinib compared with TNF inhibition, but not with MACE or serious infection [30]. However, no VTE or MACE occurred among patients who received baricitinib in the two Japanese databases, though these patients were relatively few in number (n = 384) [30]. In the Japanese baricitinib PMSS, the IR of malignancy (including lymphoma) was 0.91 per 100 PY, IR of MACE was 0.38 per 100 PY, and IR of VTE was 0.38 per 100 PY [26]. These were generally similar to the IRs in the global integrated safety analysis:, IR, 0.9 per 100 PY, IR, 0.5 per 100 PY, and IR, 0.5 per 100 PY for malignancy, MACE, and deep vein thrombosis events of special interest, respectively, in all patients who received baricitinib [17]. In a Korean real-world study, JAK inhibition did not increase the risk of MACE or cancer compared with TNF inhibition in patients with RA [31]. In our study, no MACE or VTE were observed and malignancy (deemed unrelated to baricitinib by the investigator) was observed in one patient. However, the exposure (24-week observation period) and the sample size of our study limits the ability to observe AEs with a long latency time. In addition, it should be noted that patients in our study had a relatively low mean baseline BMI of 22.3 kg/m2, as well as low proportions of patients who had ever smoked (12.3%), which may have been protective against MACE and VTE events. Together, there were differences in those previous studies, it is crucial to assess the long-term safety profile of baricitinib, and further studies are required.
Changes from baseline in hematological or other laboratory parameters were observed in our study; however, clinically meaningful events (grade ≥ 3 change) were uncommon, consistent with a pooled analysis of eight clinical trials of baricitinib in which hematological changes were evaluated for up to 128 weeks [32]. In that pooled analysis, grade ≥ 3 neutrophil count decreases were seen in < 1% of patients who received baricitinib [32]; grade ≥ 3 decreased lymphocyte count was seen in 1.3% with baricitinib 2 mg and 0.8% with baricitinib 4 mg [32]; and drug discontinuation due to hematological abnormalities occurred in a low proportion (< 1%) of patients [32]. This is consistent with our study, wherein no discontinuations due to hematological abnormalities occurred.
While our study was designed to assess the real-world safety of baricitinib, it is notable that 52.4% of patients achieved DAS28-CRP remission at 24 weeks. This proportion is comparable to the Japanese PMSS (61.3%) [26], which is higher than in clinical trials [16, 18]. In our study, the LS mean change from baseline to week 24 for DAS28-CRP, SDAI, and CDAI was − 1.9, − 19.3, and − 17.7, respectively. In the Spanish ORBIT-RA real-world study of patients with refractory RA, the mean (SD) change from baseline in SDAI and CDAI at 6 months was − 13.4 (11.6) and − 13.0 (11.4), respectively [33]. However, caution is warranted when interpreting any comparisons due to differences in patient populations and methodological differences between the studies. For example, patients in our study who received baricitinib for < 10 weeks were excluded from the effectiveness analysis, while patients in clinical trials who received rescue therapy or discontinued were defined as non-responders [13, 16].
In our study, there was some discrepancy between the DAS28-CRP and SDAI/CDAI measures of effectiveness. This may be expected, as previous studies, including an observational study using the Chinese CREDIT database with 30,501 patients, have found that that the proportion of discordance was lowest between SDAI and CDAI (8.7%) and highest between DAS28-CRP and CDAI (32.8%) [34]. This suggests that despite high correlation between indices, they are not interchangeable [34]. Clinical trials have also found differences: in the RA-BEAM trial at week 24, 34% of patients who received baricitinib achieved DAS28-CRP remission, compared with 16% by SDAI/CDAI [5].
Patients reported reductions from baseline in both mean scores of duration of MJS and pain VAS in our study. While this observational study was not designed for statistical comparison of PROs, these results are in line with those reported in analyses of clinical trials wherein statistically significant improvements in PROs with baricitinib treatment were reported compared with placebo [5, 18, 35].
Limitations of this study include its single-arm, observational design, precluding robust conclusions regarding the safety or effectiveness of baricitinib treatment for RA in Chinese patients. Additionally, the 24-week maximum observation period limits safety conclusions drawn from these data for patients who receive baricitinib for longer than 24 weeks. However, EAIRs for AEs, treatment-related AEs, serious AEs, AEs leading to discontinuation, and AESIs did not increase from 12 to 24 weeks of observation, suggesting that a longer duration of baricitinib treatment does not lead to an increased risk of AEs. Taken together with previous studies of baricitinib treatment [5, 14,15,16,17,18], including long-term safety data, the results of this study do not suggest an increased risk of AEs with 24 weeks of baricitinib treatment. In our study, the majority of patients (86.8%) received concomitant csDMARDs. This is in line with the approval of baricitinib in China, where the approval of baricitinib is in combination with csDMARDs. However, due to the observational nature of this study which was designed to provide real-world data on safety and effectiveness of baricitinib, data regarding baricitinib monotherapy or other concomitant treatment patterns are not available.
Conclusions
In this real-world, observational study of baricitinib treatment in Chinese patients with moderate-to-severe active RA, baricitinib was generally well tolerated and the safety profile was consistent with previous reports, with no new safety signals. Baricitinib demonstrated promising effectiveness, with over half of evaluable patients achieving DAS28-CRP remission at 24 weeks. These encouraging safety and effectiveness data support the use of baricitinib for Chinese patients with RA.
Data Availability
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
References
Almutairi KB, et al. The prevalence of rheumatoid arthritis: a systematic review of population-based studies. J Rheumatol. 2021;48(5):669–76.
Tian X, et al. 2018 Chinese guidelines for the diagnosis and treatment of rheumatoid arthritis. Rheumatol Immunol Res. 2021;2(1):1–14.
Jin S, et al. Chinese Registry of rheumatoid arthritis (CREDIT): II. Prevalence and risk factors of major comorbidities in Chinese patients with rheumatoid arthritis. Arthritis Res Ther. 2017;19(1):251.
Smolen JS, et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet. 2019;393(10188):2303–11.
Taylor PC, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. 2017;376(7):652–62.
Lee EB, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med. 2014;370(25):2377–86.
Fraenkel L, et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res (Hoboken). 2021;73(7):924–39.
Smolen JS, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann Rheum Dis. 2023;82:3–18.
Lau CS, et al. 2018 update of the APLAR recommendations for treatment of rheumatoid arthritis. Int J Rheum Dis. 2019;22(3):357–75.
Balanescu AR, et al. Infections in patients with rheumatoid arthritis receiving tofacitinib versus tumour necrosis factor inhibitors: results from the open-label, randomised controlled ORAL Surveillance trial. Ann Rheum Dis. 2022;81(11):1491–503.
Ytterberg SR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386(4):316–26.
Curtis JR, et al. Malignancy risk with tofacitinib versus TNF inhibitors in rheumatoid arthritis: results from the open-label, randomised controlled ORAL Surveillance trial. Ann Rheum Dis. 2023;82(3):331–43.
FDA. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions; 2021 [cited 15 Dec 2022]. https://www.fda.gov/media/151936/download.
Fleischmann R, et al. Baricitinib, methotrexate, or combination in patients with rheumatoid arthritis and no or limited prior disease-modifying antirheumatic drug treatment. Arthritis Rheumatol. 2017;69(3):506–17.
Fleischmann R, et al. Efficacy and safety of long-term baricitinib with and without methotrexate for the treatment of rheumatoid arthritis: experience with baricitinib monotherapy continuation or after switching from methotrexate monotherapy or baricitinib plus methotrexate. Arthritis Care Res (Hoboken). 2020;72(8):1112–21.
Dougados M, et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA-BUILD study. Ann Rheum Dis. 2017;76(1):88–95.
Taylor PC, et al. Safety of baricitinib for the treatment of rheumatoid arthritis over a median of 4.6 and up to 9.3 years of treatment: final results from long-term extension study and integrated database. Ann Rheum Dis. 2022;81(3):335–43.
Li Z, et al. Baricitinib in patients with rheumatoid arthritis with inadequate response to methotrexate: results from a phase 3 study. Clin Exp Rheumatol. 2020;38(4):732–41.
An Y, et al. The usage of biological DMARDs and clinical remission of rheumatoid arthritis in China: a real-world large scale study. Clin Rheumatol. 2017;36(1):35–43.
Tan C, et al. Sequences of biological treatments for patients with moderate-to-severe rheumatoid arthritis in the era of treat-to-target in China: a cost-effectiveness analysis. Clin Rheumatol. 2022;41(1):63–73.
Wells G, et al. Validation of the 28-joint Disease Activity Score (DAS28) and European League Against Rheumatism response criteria based on C-reactive protein against disease progression in patients with rheumatoid arthritis, and comparison with the DAS28 based on erythrocyte sedimentation rate. Ann Rheum Dis. 2009;68(6):954–60.
Aletaha D, Smolen J. The Simplified Disease Activity Index (SDAI) and the Clinical Disease Activity Index (CDAI): a review of their usefulness and validity in rheumatoid arthritis. Clin Exp Rheumatol. 2005;23(5 Suppl 39):S100–8.
Yu C, et al. Chinese registry of rheumatoid arthritis (CREDIT): I. Introduction and prevalence of remission in Chinese patients with rheumatoid arthritis. Clin Exp Rheumatol. 2018;36(5):836–40.
Yang Y, et al. Efficacy and safety of baricitinib in Chinese rheumatoid arthritis patients and the subgroup analyses: results from study RA-BALANCE. Rheumatol Ther. 2020;7(4):851–66.
Genovese MC, et al. Safety profile of baricitinib for the treatment of rheumatoid arthritis over a median of 3 years of treatment: an updated integrated safety analysis. Lancet Rheumatol. 2020;2(6):e347–57.
Takagi M, et al. Safety and effectiveness of baricitinib for rheumatoid arthritis in Japanese clinical practice: 24-week results of all-case post-marketing surveillance. Mod Rheumatol. 2023;33:647–56.
Taylor PC, et al. Cardiovascular safety during treatment with baricitinib in rheumatoid arthritis. Arthritis Rheumatol. 2019;71(7):1042–55.
Harigai M, et al. Safety profile of baricitinib in Japanese patients with active rheumatoid arthritis with over 1.6 years median time in treatment: an integrated analysis of Phases 2 and 3 trials. Mod Rheumatol. 2020;30(1):36–43.
FDA. Drug-induced liver injury: premarketing clinical evaluation; 2009 [cited 23 Jan 2023]. https://www.fda.gov/media/116737/download.
Salinas CA, et al. Evaluation of VTE, MACE, and serious infections among patients with RA treated with baricitinib compared to TNFi: a multi-database study of patients in routine care using disease registries and claims databases. Rheumatol Ther. 2023;10(1):201–23.
Min HK, et al. Risk of cancer, cardiovascular disease, thromboembolism, and mortality in patients with rheumatoid arthritis receiving Janus kinase inhibitors: a real-world retrospective observational study using Korean health insurance data. Epidemiol Health. 2023;45: e2023045.
Kay J, et al. Changes in selected haematological parameters associated with JAK1/JAK2 inhibition observed in patients with rheumatoid arthritis treated with baricitinib. RMD Open. 2020;6(3): e001370.
Hernandez-Cruz B, et al. Real-world treatment patterns and clinical outcomes of baricitinib in rheumatoid arthritis patients in Spain: results of a multicenter, observational study in routine clinical practice (the ORBIT-RA Study). Rheumatol Ther. 2022;9(2):589–608.
Song X, et al. Chinese registry of rheumatoid arthritis: IV. Correlation and consistency of rheumatoid arthritis disease activity indices in China. Chin Med J (Engl). 2021;134(12):1465–70.
Yang Y, et al. Patient-reported outcomes from a randomized, double-blind, placebo controlled, phase III study of baricitinib versus placebo in patients with moderately to severely active rheumatoid arthritis and an inadequate response to methotrexate therapy: results from the RA-BALANCE study. Ther Adv Musculoskelet Dis. 2021;13: 175972X0211006964.
Acknowledgements
We thank the patients and their families for making this trial possible, and the investigators and clinical study teams who participated. The authors would like to thank Yu Dong, PhD, Rong Chen, PhD, and Jinnan Li, MPH at Eli Lilly and Company for review and critical suggestions for improvement.
Investigators
For a list of all study investigators, see Supplementary Material.
Medical Writing/Editorial Assistance
The authors would like to thank Adam Gill, MRes, and Jake Burrell, PhD, at Rude Health Consulting, for medical writing and editorial support, which was funded by Eli Lilly and Company, China.
Funding
This study was funded by Eli Lilly and Company, Suzhou, China, who also paid the journal’s publication fee.
Author information
Authors and Affiliations
Contributions
In addition to the below individual contributions, all authors provided critical revision of the manuscript and approval of the final version for publication. Study conception and design: Wu Chan-yuan, Zeng Xiao-feng. Data acquisition: Wu Chan-yuan, Wang Qian, Shi Jian, Zhang Xiu-ying, Du Rong, Gu Jie-ruo, Liu Qi-huan, Li Meng-tao, Zeng Xiao-feng. Data interpretation: Wu Chan-yuan, Wang Qian, Yu Jiao, Xu Jia-wei, Zhang Yan-jie, Zhu Hao, Li Meng-tao, Zeng Xiao-feng.
Corresponding author
Ethics declarations
Conflict of Interest
Yu Jiao, Xu Jia-wei, Zhang Yan-jie, and Zhu Hao are employees of Eli Lilly and Company, Suzhou, China. Wu Chan-yuan, Wang Qian, Shi Jian, Zhang Xiu-ying, Du Rong, Gu Jie-ruo, Liu Qi-huan, Li Meng-tao and Zeng Xiao-feng have no conflicts of interest to declare.
Ethical Approval
The PMSS was conducted in compliance with applicable laws in China and in accordance with the Chinese Guidelines for Good Clinical Practice and the principles outlined in the Declaration of Helsinki. The study received IRB and ethics approval at each site (Supplementary Material); the central ethics board was the Peking Union Medical College Hospital Ethics Committee (approval number HS-2327). All participants provided written, informed consent prior to participation.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wu, Cy., Wang, Q., Shi, J. et al. Safety and Effectiveness of Baricitinib in Chinese Patients with Moderate-to-Severe Rheumatoid Arthritis: 24-Week Results from a Post-Marketing Safety Study. Rheumatol Ther 10, 1609–1622 (2023). https://doi.org/10.1007/s40744-023-00596-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-023-00596-4