
Abstract
Introduction
The objective of this work was to assess the efficacy and safety of risankizumab in psoriatic arthritis (PsA) over 76 weeks.
Methods
In this double-blind, dose-ranging phase 2 study, adults with active PsA were randomized 2:2:2:1:2 to risankizumab 150 mg at weeks 0, 4, 8, 12, and 16 (arm 1), 150 mg at weeks 0, 4, and 16 (arm 2), 150 mg at weeks 0 and 12 (arm 3), 75 mg at week 0 (arm 4), or placebo (arm 5). Patients completing week 24 could receive risankizumab 150 mg in a 52-week open-label extension study. Efficacy assessments included American College of Rheumatology (ACR) responses, Psoriasis Area Severity Index (PASI) responses, minimal disease activity (MDA), and 28-joint Disease Activity Score based on C-reactive protein (DAS28[CRP]).
Results
Of 185 randomized patients, 173 (93.5%) completed week 16 and 145 (78.4%) entered the open-label extension. Significantly more patients in each risankizumab arm achieved ACR20 at week 16 versus placebo (primary endpoint: pooled arms 1 + 2 [59.5%] versus placebo [35.7%]; treatment difference [90% CI] 24.0 [9.3, 38.7]; P = 0.007). Similarly, significantly more patients in most risankizumab arms achieved ACR20/50/70, PASI75/90/100, MDA, and greater improvements in DAS28(CRP) versus placebo at week 16. These benefits of risankizumab were maintained long term. Treatment-emergent adverse events were comparable across treatment arms. Risankizumab 150 mg was well tolerated over 76 weeks.
Conclusions
Risankizumab improved joint and skin symptoms versus placebo in patients with active PsA over 16 weeks; improvements were sustained long term. Risankizumab was well tolerated over the long term with no new safety findings.
Trial Registration Numbers
NCT02719171 and NCT02986373.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
An unmet need for safe and effective long-term therapeutics remains for psoriatic arthritis, which affects an estimated 0.3–1% of the overall population and 6–30% of patients with psoriasis. |
Risankizumab, a humanized immunoglobulin G1 monoclonal antibody that inhibits interleukin-23 by binding to its p19 subunit, has been approved for the treatment of moderate-to-severe plaque psoriasis and more recently active psoriatic arthritis. |
What was learned from the study? |
In this phase 2 study and its long-term extension, patients with active psoriatic arthritis receiving risankizumab demonstrated significant improvement in joint and skin symptoms versus placebo over 16 weeks, and improvements were maintained over the long term among patients who enrolled in the open-label extension study. |
Overall, the study findings demonstrated that risankizumab was well tolerated, reduced the signs and symptoms of psoriasis and arthritis, and inhibited the progression of joint damage over the long term in patients with active psoriatic arthritis. |
Findings from this study supported the recent approvals of risankizumab for the treatment of active psoriatic arthritis in the United States and the European Union. |
Introduction
Psoriatic arthritis (PsA) is a chronic immune-mediated inflammatory disease characterized by signs and symptoms of arthritis, peripheral synovitis, enthesitis, dactylitis, or spondylitis with skin and/or nail involvement (psoriasis) [1, 2]. PsA is estimated to affect 0.3–1% of the overall population and 6–30% of patients with psoriasis [2, 3]. Treatments for PsA include conventional systemic disease-modified antirheumatic drugs (DMARDs), such as methotrexate, targeted synthetic DMARDs, such as tofacitinib and apremilast, and biologic DMARDs, such as tumor necrosis factor (TNF), interleukin (IL)-17, IL-23, and IL-12/23 inhibitors [2, 4]. Although there have been significant strides in the development of therapeutic options for PsA, an unmet need for safe and effective long-term therapeutics remains.
Interleukin-23, a key regulator of multiple effector cytokines, has been implicated in the pathogenesis of psoriatic skin lesions, synovitis, enthesitis, and bone erosion, making IL-23 inhibitors candidates for the treatment of PsA [4,5,6,7]. Risankizumab, a humanized immunoglobulin G1 monoclonal antibody that inhibits IL-23 by binding to its p19 subunit, has been approved for the treatment of moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy [8,9,10]. At the time of this research, risankizumab was under investigation as a treatment for other immune-mediated inflammatory diseases including PsA, atopic dermatitis, hidradenitis suppurativa, Crohn’s disease, and ulcerative colitis [11,12,13,14]. Since this trial was conducted, risankizumab was approved for active PsA in the United States and European Union based on pivotal phase 3 trials [15, 16].
The objective of this phase 2 study and its long-term extension was to evaluate the safety and efficacy of risankizumab in patients with active PsA.
Methods
Study Design
This analysis included data from a multi-country, parallel-design, dose-ranging, randomized, double-blind, placebo-controlled, phase 2 core study (NCT02719171) and its 52-week, long-term, open-label extension (NCT02986373). Patients were randomized 2:2:2:1:2 to different dosing regimens of subcutaneous risankizumab (arm 1: 150 mg at weeks 0, 4, 8, 12, and 16; arm 2: 150 mg at weeks 0, 4, and 16; arm 3: 150 mg at weeks 0 and 12; arm 4: single 75-mg dose at week 0) or placebo (arm 5: placebo at weeks 0, 4, 8, 12, and 16) for 16 weeks and then entered a follow-up period for up to 16 weeks in the core study (Fig. 1). Randomization was stratified by prior TNF inhibitor use and concurrent methotrexate use. Patients who completed the week 24 visit were allowed to enroll into the 52-week open-label extension study to receive risankizumab 150 mg administered every 12 weeks for 36 weeks (Fig. 1). During the extension study, patients received open-label risankizumab 150 mg by subcutaneous injection at weeks 0, 12, 24, and 36 of the open-label extension; follow-up visits occurred at weeks 48 and 52 (overall duration 76 weeks). An additional dose was made available at week 4 for those patients who had not achieved the protocol-defined response of an improvement in tender and swollen joint count of ≥ 20% compared with the baseline of the core study and for whom the investigator believed that the additional dose would be beneficial. Patients who had not attained this protocol-defined response at two consecutive visits at the week 12 visit or thereafter were discontinued from the open-label extension study.

Study design. ACR20 American College of Rheumatology 20 criteria response, mTSS modified Total Sharp Score, OLE open-label extension, RZB risankizumab, s.c. subcutaneous
This study was conducted in accordance with the protocol, International Council on Harmonisation guidelines, local regulations, and the ethical principles of the Declaration of Helsinki. An independent ethics committee or institutional review board at each study site approved all study-related documents (Supplementary Table S1). All patients reviewed and signed an informed consent form before any study procedures were conducted.
Participants
Adult patients (≥ 18 years) who had active PsA (≥ 5 tender joints and ≥ 5 swollen joints) for ≥ 6 months prior to screening, who met the Classification Criteria for Psoriatic Arthritis (CASPAR) at screening, had ≥ 1 psoriatic lesion or documented history of psoriasis, and had inadequate response/intolerance to standard doses of nonsteroidal anti-inflammatory drugs (NSAIDs, ≥ 4 weeks), conventional synthetic DMARDs (≥ 3 months), or ≤ 2 TNF inhibitors were eligible for the core study. Patients who received methotrexate therapy for ≥ 3 months (not exceeding 25 mg per week) with a stable dose for ≥ 4 weeks, a stable dose of oral corticosteroids for ≥ 2 weeks (not exceeding equivalent of 10 mg of prednisone per day), or a stable dose of NSAIDs/acetaminophen/paracetamol for ≥ 2 weeks were allowed to enroll.
Exclusion criteria included patients with major chronic inflammatory or connective tissue disease other than PsA (e.g., rheumatoid arthritis, systemic lupus erythematosus, ankylosing spondylitis, Lyme disease, gout) or fibromyalgia or patients who received any therapeutic agent directly targeting IL-12/23, IL-23, or IL-17. Patients with prior use of > 2 TNF inhibitors or any cell-depleting therapies (e.g., rituximab) or use of TNF inhibitors within 12 weeks (etanercept within 8 weeks), leflunomide without cholestyramine washout within 8 weeks or with washout within 4 weeks, systemic non-biologic medications for PsA or psoriasis and photochemotherapy within 4 weeks, topical psoriasis medications, phototherapy, or opioid analgesics within 2 weeks prior to randomization were also excluded.
Randomization and Blinding
The randomization list was generated using a validated system, involving a pseudorandom number generator to guarantee the reproducibility of the assignments. The randomization list was checked by an independent statistician and used by a third party. An Interactive Response System was used to assign randomization numbers to eligible patients. Patients, investigators, and those involved in trial conduct or analysis remained blinded to the randomized treatment assignments to the week 16 primary analysis database lock. After the last patient completed the 16-week visit, the database was locked for analysis of the primary endpoint and sponsor’s study and project teams were unblinded to treatment and dose assignments. Patients and investigators further remained blinded until after the final database lock.
Endpoints
The primary efficacy endpoint in the core study was American College of Rheumatology (ACR) 20 criteria response at week 16 for pooled arms 1 and 2 versus placebo. Secondary endpoints included ACR20/50/70 responses at week 16 in all dose groups, Psoriasis Area Severity Index (PASI) 90 response at week 16 in patients with psoriasis affecting ≥ 3% of body surface area (BSA) at baseline, change from baseline to week 16 in tender joint count based on 68 joints (TJC68), swollen joint count based on 66 joints (SJC66), Health Assessment Questionnaire Disability Index (HAQ-DI), Short Form Health Survey 36 (SF-36) Physical Component Summary (PCS) and Mental Component Summary (MCS), dactylitis count in patients with dactylitis at baseline, Spondyloarthritis Research Consortium of Canada (SPARCC) enthesitis index in patients with enthesitis at baseline, and modified Nail Psoriasis Severity Index (mNAPSI) in patients with nail psoriasis.
Other endpoints included change from baseline in the 28-joint Disease Activity Score based on C-reactive protein (DAS28[CRP]) and Leeds Enthesitis Index (LEI) to week 16, and proportion of patients achieving Minimal Disease Activity (MDA) defined as meeting 5 of the 7 following criteria: TJC ≤ 1, SJC ≤ 1, PASI ≤ 1 or BSA ≤ 3%, patient assessment of pain (visual analog scale [VAS] 0–100 mm) ≤ 15, patient global activity (VAS) ≤ 20, HAQ-DI ≤ 0.5, and tender entheseal points ≤ 1.
Endpoints in the open-label extension study included ACR20/50/70 and PASI75/90/100 at all timepoints and change from baseline in HAQ-DI, SF-36 PCS, and SF-36 MCS over time. Radiographic progression of structural joint damage was measured using the modified Total Sharp Score (mTSS), which was modified for PsA with the addition of distal interphalangeal hand joints (scored 0–528) [17]. Changes from core study baseline to core study week 24 and to open-label extension weeks 24 and 48 (overall follow-up duration 72 weeks) in mTSS were assessed. Radiographic progression was defined as a change in mTSS of ≥ 0.5. To obtain the total mTSS score, scores for erosions and joint space narrowing in both the hands and feet were added together. Missing joint score imputation was performed for erosion and joint space narrowing. Radiographs from both the core and open-label extension studies were read together centrally by two qualified physicians/radiologists who were blinded to the site number, patient number, treatment allocation in the core study, time sequence of the radiographs, and clinical response.
Long-term safety and tolerability of risankizumab were also assessed. Safety was assessed by reporting of adverse events (AEs), clinical laboratory values, physical evaluation, vital signs, and electrocardiograph. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) coding dictionary version 20.0. Intensity of AEs was assessed by Rheumatology Common Toxicity Criteria (RCTC) version 2.0. All AEs occurring between start of treatment and end of the residual effect period were considered treatment emergent; the residual effect period was defined as 15 weeks after the last trial medication application and included AEs reported through end of study visit. AEs that started before first drug intake and deteriorated under treatment were also considered as treatment emergent. During the open-label extension, a treatment-emergent AE was defined as any AE with an onset date on or after the first dose of study drug and up to 140 days after the last dose of study drug, excluding AEs that occurred outside of 140 days from last dose in the core study but before first dose in the open-label extension.
Statistical Analyses
Sample size was determined on the basis of a one-sided comparison between the average in ACR20 response at week 16 of arm 1 and arm 2 versus placebo. With the assumed week 16 ACR20 response of 38% in the combined arms (arm 1 and arm 2) and 15% in the placebo arm, 40 participants in each arm would provide 85% power to detect a 23% difference in proportion (combination of arm 1 and arm 2 versus placebo) using a one-sided test of 0.05 significance. Although not included in the hypothesis testing strategy, enrollment of 40 participants was planned in arm 3 and 20 participants in arm 4 for pharmacokinetic modeling. The planned total sample size was therefore 180.
In the core study, efficacy was assessed in the Full Analysis Set (FAS), which included all patients who were randomized and received at least 1 dose of assigned study drug. Safety was assessed based on actual treatment in all patients who received at least 1 dose of study drug. In the open-label extension, efficacy and safety were assessed based on all patients who received at least 1 dose of study drug in the extension. In the core study, superiority of risankizumab versus placebo was assessed pooling the two most intense dose regimens (arm 1 and arm 2) at week 16. The difference in proportion of participants who achieved ACR20 between the pooled risankizumab arms 1 and 2 and the placebo arm (arm 5) was tested using the stratified Cochran–Mantel–Haenszel risk difference method stratified by prior TNF inhibitor use and concurrent methotrexate use. Pairwise comparisons of the risankizumab dose groups versus placebo were conducted using the same stratified Cochran–Mantel–Haenszel methods. Continuous secondary endpoints were analyzed using a mixed model repeated measures model (MMRM), which is valid under the missing at random (MAR) assumption. Missing data were imputed for efficacy variables only for up to week 24; data after week 24 were analyzed as observed. Missing values included a missed visit or dropout from the study. Missing data were imputed using non-responder imputation (NRI) for binary variables and last observation carried forward (LOCF) for the continuous variables in the core study. No statistical tests were conducted for the efficacy analyses in the open-label extension; data were summarized as observed and only descriptive statistics were assessed (i.e., patients who did not have an evaluation on a scheduled visit were excluded from the summary for that visit).
Results
The first patient in the core study was enrolled in May 2016 and the last follow-up visit in the extension study was in July 2018. Overall, 185 patients were randomized across 47 sites in 11 countries; 173 (93.5%) patients completed the 24-week core study, and 145 (78.4%) entered the open-label extension at 37 sites across ten countries (Germany did not participate in the extension study; Fig. 2). Baseline characteristics were similar between the groups in the core study; mean age was 49.0 years versus 51.5 years, disease duration since diagnosis was 7.0 years versus 6.7 years, and 42.9 versus 43.4% were female in the placebo versus total risankizumab group (Table 1).

Patient disposition. *Percentages are shown using originally randomized patients (N = 185) as denominator/OLE patients (n = 145) as denominator. OLE open-label extension, RZB risankizumab
Efficacy in Core Study
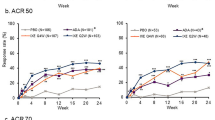
Significantly more patients achieved the primary endpoint of ACR20 in pooled arms 1 and 2 (50/84 [59.5%; 90% CI 50.0–68.6]; treatment difference [90% CI] 24.0 [9.3, 38.7]; P = 0.007) versus placebo at week 16 (Fig. 3a). Similarly, significantly more patients in each of the individual risankizumab arms achieved ACR20 (response range, 57.1–65.0%; each response difference P < 0.05) versus placebo (15/42; 35.7%) at week 16. ACR50 responses were numerically higher in the risankizumab arms but only statistically significant for arm 3 (P ≤ 0.01) versus placebo, and significantly more patients achieved ACR70 in the pooled arms 1 and 2 and individual arms 1, 3, and 4 (P ≤ 0.05) versus placebo at week 16 (Fig. 3a).

Percentage of patients achieving ACR20/50/70 responses at week 16 (a), PASI75/90/100 responses at week 16 among patients with ≥ 3% BSA at baseline (b), and percentage of patients achieving MDA at week 16 among all patients (c). ACR American College of Rheumatology, BSA body surface area, MDA minimal disease activity, NRI non-responder imputation, PASI Psoriasis Area Severity Index, TNF tumor necrosis factor. NRI analyses. The difference in response rates between groups as well as the two-sided P value were calculated using Cochran–Mantel–Haenszel risk difference estimate stratified by prior TNF inhibitor use and concurrent methotrexate use. Statistical comparison versus placebo: ***P ≤ 0.001; **P ≤ 0.01; *P ≤ 0.05
Among patients with psoriasis affecting ≥ 3% of BSA at baseline, significantly more patients achieved PASI75 and PASI90 responses at week 16 versus placebo (all P ≤ 0.01); PASI100 was achieved by significantly more patients in pooled arms 1 and 2 and individual arms 2, 3, and 4 versus placebo at week 16 (all P ≤ 0.05; Fig. 3b).
At baseline, 21.4–29.3% and 59.5–70.7% of patients across treatment groups had dactylitis and enthesitis, respectively. Among these patients, those who received risankizumab generally showed numerical but not significant improvements in SPARCC enthesitis index (significant only in arm 4; P ≤ 0.01) and LEI at week 16 compared with patients who received placebo; results were more variable for dactylitis count (Table 2). Improvements in TJC68, SJC66, HAQ-DI, SF-36 PCS, SF-36 MCS, and mNAPSI were also variable. However, significantly more patients achieved MDA across risankizumab arms versus placebo at week 16 (all P ≤ 0.01; Fig. 3c), and significant improvements in DAS28(CRP) were observed in risankizumab pooled arms 1 and 2 and arms 2, 3, and 4 versus placebo at week 16 (P ≤ 0.01; Table 2).
Long-Term Effectiveness
ACR20/50/70 response rates increased throughout the 52-week open-label extension, with 75.2, 43.6, and 24.8% of patients entering the extension, respectively, achieving these responses at week 52 (Supplementary Fig. S1). Similarly, the percentages of patients with PASI75/90/100 responses increased during the 52-week open-label extension with 82.9, 73.2, and 61.0%, respectively, achieving these responses at week 52 (Supplementary Fig. S1). Improvements were also observed in SF-36 PCS, SF-36 MCS, and HAQ-DI at week 52 (Supplementary Fig. S2). No radiographic progression was observed throughout the study (Supplementary Fig. S3).
Safety in Core Study
During the core study, no discernible differences between treatment groups were observed in treatment-emergent AEs (including serious AEs and AEs leading to discontinuation), laboratory abnormalities, and vital signs (Table 3). The most common AEs with risankizumab were viral upper respiratory tract infection (total risankizumab, n = 25 [17.5%] vs. placebo, n = 2 [4.8%]), upper respiratory tract infection (total risankizumab, n = 8 [5.6%] vs. placebo, n = 5 [11.9%]), and headache (total risankizumab, n = 8 [5.6%] vs. placebo, n = 3 [7.1%]). Only a few patients experienced AEs in areas of safety interest, including serious infections, major adverse cardiovascular events (MACE), and malignancies. Two patients reported serious infections (both in risankizumab arm 4). The first patient reported serious urinary tract infection, influenza, and sepsis; the patient also had the only reported serious malignancy (stage IV ovarian cancer) and three serious MACE (acute myocardial infarction [reported as non-ST-elevation myocardial infarction], congestive cardiac failure, and coronary artery occlusion). The patient did not discontinue the study drug. The second patient reported serious gastroenteritis (two separate events) and pyelonephritis; the patient did not discontinue study drug (Table 3). One additional patient experienced an adjudicated MACE (stroke, and the patient did not discontinue study drug).
One serious hypersensitivity event (anaphylactic reaction) was reported in the study (risankizumab arm 1); the event occurred after the first injection of risankizumab in a biologic naïve patient, and the patient discontinued treatment. No deaths or cases of tuberculosis were reported in the study.
Long-Term Safety
Among the patients enrolled into the open-label extension, 73.8% of patients experienced an AE over the course of the core study and open-label extension (Supplementary Table S2). Six patients (4.1%) experienced a serious AE and 5 patients (3.4%) experienced AEs that led to study drug discontinuation. Two patients experienced serious infections (urosepsis in 1 patient and staphylococcal infection, haemophilus pneumonia, and pseudomonal pneumonia in 1 patient) and 3 patients experienced adjudicated MACE (stroke [n = 2] and myocardial infarction [n = 1]). One patient experienced colorectal cancer and one patient fibroadenoma of the breast.
Discussion
This phase 2, dose-finding study and its long-term extension assessed the use of risankizumab for the treatment of patients with active PsA. The results demonstrated that risankizumab was effective in treating the symptoms of active PsA versus placebo over 16 weeks, with sustained effectiveness over the long term with open-label risankizumab treatment. The safety profile was favorable and overall, these findings support the use of risankizumab for the treatment of PsA.
Patients with PsA present with symptoms of both psoriasis and arthritis, and thus therapeutic interventions are needed that treat both the joint and skin components of the disease. The findings of this study demonstrated significant efficacy of risankizumab versus placebo over 16 weeks in treating both the arthritic and psoriatic manifestations of PsA; a significantly greater proportion of patients achieved the primary endpoint of ACR20 response at week 16 in the pooled risankizumab arms 1 and 2 versus placebo as well as ACR20/50/70 responses across most risankizumab dose groups. Among patients with psoriasis affecting ≥ 3% of BSA at baseline, significantly more patients achieved PASI75/90/100 responses across dose groups (except PASI100 in arm 1) at week 16 versus placebo. Of note, the ACR50 and ACR70 responses were greatest in the risankizumab 150 mg at weeks 0 and 12 dose group (arm 3), while the PASI responses were more variable across dose groups at week 16.
The effects of risankizumab in treating arthritic manifestations and the skin manifestations of the disease appeared to be sustained with long-term treatment, as shown by improvements in ACR responses (75% of patients were ACR20 responders and over 40% were ACR50 responders at week 52 of the open-label extension) and PASI responses (≥ 60% of patients achieved PASI75/90/100 responses at week 52 of the open-label extension) in patients entering the open-label extension.
Improvements in other endpoints assessing arthritic manifestations or quality of life were more variable; however, improvements in disease activity were significant versus placebo for all risankizumab arms when assessed by MDA and for all but one risankizumab dose group when assessed by DAS28(CRP) at week 16.
Most patients with PsA experience progressive joint damage over time, evident as bone loss, erosions, and joint space narrowing on radiographs [18, 19]. Treatment with biologic DMARDs has been shown to inhibit progression of structural damage in clinical studies of PsA compared with placebo [20, 21]. In this study, no radiographic progression was observed with risankizumab treatment over 72 weeks’ duration; however, larger studies are needed to confirm these observations.
Overall, the safety profile was comparable for the risankizumab treatment groups and the placebo group in the core study; similar proportions of patients reported AEs, severe AEs, serious AEs, and AEs leading to discontinuation. Serious infections, MACE, and malignancies were reported in only a few patients receiving risankizumab (≤ 2 each).
Risankizumab 150 mg was safe and well tolerated in patients receiving up to 76 weeks of treatment. No deaths were reported and the incidence of serious AEs and AEs leading to discontinuation was low. Overall, no new safety signals were noted, and safety of risankizumab was consistent with previous psoriasis studies [9].
Limitations of the trial include small patient populations in each arm, which limit the precision of the outcomes and the accuracy of translation into phase 3 results, and the use of the 2:2:2:1:2 randomization scheme that included the lowest number of patients in the low-dose arm. Furthermore, the study was not designed to test difference across treatment arms (there was no control for type I error); however, efficacy against placebo was demonstrated. The strengths of the study include that it assessed key domains of PsA, a heterogeneous disease, and confirmed the efficacy of IL-23 inhibitors.
Conclusions
Patients with active PsA receiving risankizumab demonstrated significant improvement in joint and skin symptoms versus placebo over 16 weeks. Furthermore, the improvements were maintained over the long term among patients who enrolled in the open-label extension study. Overall, the findings of this study demonstrated that risankizumab was well tolerated, reduced the signs and symptoms of psoriasis and arthritis, and inhibited the progression of joint damage over the long term in patients with active PsA. These results supported further evaluation of risankizumab for the treatment of PsA in a phase 3 program.
References
Coates LC, Helliwell PS. Psoriatic arthritis: state of the art review. Clin Med (Lond). 2017;17:65–70.
Ogdie A, Coates LC, Gladman DD. Treatment guidelines in psoriatic arthritis. Rheumatology (Oxford). 2020;59:i37–46.
Gladman DD, Antoni C, Mease P, Clegg DO, Nash P. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis. 2005;64(Suppl 2):ii14–7.
Gossec L, Baraliakos X, Kerschbaumer A, et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann Rheum Dis. 2020;79:700–12.
Boutet MA, Nerviani A, Gallo Afflitto G, Pitzalis C. Role of the IL-23/IL-17 axis in psoriasis and psoriatic arthritis: the clinical importance of its divergence in skin and joints. Int J Mol Sci. 2018;19:530.
Deodhar A, Helliwell PS, Boehncke WH, et al. Guselkumab in patients with active psoriatic arthritis who were biologic-naive or had previously received TNFalpha inhibitor treatment (DISCOVER-1): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet. 2020;395:1115–25.
Mease PJ, Rahman P, Gottlieb AB, et al. Guselkumab in biologic-naive patients with active psoriatic arthritis (DISCOVER-2): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet. 2020;395:1126–36.
SKYRIZI™ (risankizumab-rzaa) Prescribing Information. AbbVie Inc., North Chicago, IL. 2022.
Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392:650–61.
Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551–60.
Blair HA. Risankizumab: a review in moderate to severe plaque psoriasis. Drugs. 2020;80:1235–45.
Clinical study: a study of the efficacy and safety of risankizumab in participants with Crohn’s disease. 2020 [cited July 7, 2022]; Available from https://clinicaltrials.gov/ct2/show/NCT03105102?term=risankizumab&recrs=ab&draw=2&rank=6.
McDonald J, Maliyar K, Gooderham MJ. Risankizumab, an IL-23p19 inhibitor for psoriasis: a review of the current literature. Skin Therapy Lett. 2020;25:1–4.
Clinical study: a study to evaluate risankizumab in adult and adolescent subjects with moderate to severe atopic dermatitis. 2020 [cited July 7, 2022]. Available from https://clinicaltrials.gov/ct2/show/NCT03706040?term=risankizumab&recrs=d&draw=2&rank=6.
Kristensen LE, Keiserman M, Papp K, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 1 trial. Ann Rheum Dis. 2022;81:225–31.
Ostor A, Van den Bosch F, Papp K, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 2 trial. Ann Rheum Dis. 2022;81:351–8.
van der Heijde D, Sharp J, Wassenberg S, Gladman DD. Psoriatic arthritis imaging: a review of scoring methods. Ann Rheum Dis. 2005;64(Suppl 2):ii61–4.
Kane D, Stafford L, Bresnihan B, FitzGerald O. A prospective, clinical and radiological study of early psoriatic arthritis: an early synovitis clinic experience. Rheumatology (Oxford). 2003;42:1460–8.
Ritchlin CT, Colbert RA, Gladman DD. Psoriatic arthritis. N Engl J Med. 2017;376:2095–6.
Mease P, van der Heijde D, Landewe R, et al. Secukinumab improves active psoriatic arthritis symptoms and inhibits radiographic progression: primary results from the randomised, double-blind, phase III FUTURE 5 study. Ann Rheum Dis. 2018;77:890–7.
Landewe R, Ritchlin CT, Aletaha D, et al. Inhibition of radiographic progression in psoriatic arthritis by adalimumab independent of the control of clinical disease activity. Rheumatology (Oxford). 2019;58:1025–33.
Acknowledgements
Funding
AbbVie and Boehringer Ingelheim funded the core study (NCT02719171) and AbbVie funded the extension study (NCT02986373). No honoraria or payments were made for authorship. AbbVie funded the journal’s Rapid Service Fee.
Medical Writing, Editorial, and Other Assistance
The authors would like to thank the participants of the study as well as Beate Berner of Boehringer Ingelheim for contributions to the core study. Medical writing support was provided by Maria Hovenden, PhD, and Janet E. Matsuura, PhD, of ICON (Blue Bell, PA, United States) and was funded by AbbVie Inc. AbbVie and Boehringer Ingelheim both contributed to its design and participated in data collection; and participated in data analysis and interpretation of the data and in writing, reviewing, and approving the publication. All authors had access to relevant data and participated in the drafting, review, and approval of this publication.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Philip J. Mease, Stella Aslanyan, Steven J. Padula, Ann Eldred, and Kim A. Papp contributed to the study conception and design. Philip J. Mease, Herbert Kellner, Akimichi Morita, Alan J. Kivitz, Stella Aslanyan, Steven J. Padula, Andrew S. Topp, Ann Eldred, Frank Behrens, and Kim A. Papp contributed to acquisition of data. Substantial contributions to analysis and interpretation of data were provided by all authors, with formal analysis and investigation of the data performed by Andrew S. Topp. The first draft of the manuscript was written by Maria Hovenden and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Disclosures
Philip J. Mease has received research grants, consulting fees, and/or speaker’s fees from AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, Galapagos, Gilead, GlaxoSmithKline, Janssen, Novartis, Pfizer, Sun Pharma, and UCB. Herbert Kellner has received research grants, consulting fees, and/or speaker’s fees from AbbVie, BMS, MSD, Novartis, Pfizer, Roche, and UCB. Akimichi Morita has received research grants, consulting fees, and/or speaker’s fees from AbbVie, Boehringer Ingelheim Japan, Inc., Bristol Myers Squibb, Eli Lilly Japan K.K., Esai, GlaxoSmithKline K. K., Janssen Pharmaceutical K. K., Kyowa Hakko Kirin Co., Ltd, Leo Pharma, Maruho Co., Ltd, Mitsubishi-Tanabe Pharma, Nichi-Iko Pharmaceutical Co., Ltd, Nippon Kayaku Co., Ltd, Novartis, Pfizer Japan Inc, Sun Pharmaceutical Industries, Ltd, Taiho Pharmaceutical Co., Ltd, Torii Pharmaceutical Co., Ltd, and UCB Japan Co., Ltd. Alan J. Kivitz has received consulting fees and/or speaker’s fees from and/or served as an advisor to AbbVie, Amgen, Bendcare, Boehringer Ingelheim, Celgene, ChemoCentryx, ECOR1, Flexion, Genentech, Gilead Sciences Inc, GlaxoSmithKline, Grünenthal, Horizon, Janssen, Lilly, Merck, Novartis, Pfizer, Regeneron, Sanofi, Sanofi-Genzyme, Sun Pharma Advanced Research, and UCB and is a shareholder of Amgen, Gilead Sciences Inc, GlaxoSmithKline, Novartis, Pfizer, and Sanofi. Stella Aslanyan is a full-time salaried employee of Boehringer Ingelheim. Steven J. Padula was a full-time salaried employee of Boehringer Ingelheim at the time of this research. He has since retired from Boehringer Ingelheim. Andrew S. Topp and Ann Eldred are full-time salaried employees of AbbVie and may own stock/options. Frank Behrens has received research grants, consulting fees, and/or speaker’s fees from AbbVie, Amgen, Celgene, Chugai, Eli Lilly, Genzyme, Janssen, Novartis, Pfizer, Roche, Sandoz, and Sanofi. Kim A. Papp has received grants paid to his institution and/or personal honoraria or fees for serving on advisory boards, as a speaker, and as a consultant and grants as an investigator from AbbVie, Akros, Amgen, Anacor (institution only), Arcutis, Astellas, Avillion, Bausch Health, Baxalta (institution only), Baxter, Boehringer Ingelheim, Bristol Myers Squibb, CanFite (institution only), Celgene, Coherus, Dermavent (institution only), Dermira (institution only), Dice Pharmaceuticals (institution only), Dow Pharma (institution only), Eli Lilly, Evelo (institution only), Galapagos (institution only), Galderma, Genentech (institution only), Gilead (institution only), GlaxoSmithKline (institution only), Incyte, Janssen, Kyowa-Hakko Kirin, Leo Pharma, MedImmune (institution only), Meiji Seika Pharma, Merck-Serono, Merck Sharp & Dohme, Mitsubishi Pharma, Moberg Pharma (institution only), Novartis, Pfizer, PRCL Research, Regeneron, Roche, Sanofi-Genzyme, Sun Pharma (institution only), Takeda, and UCB.
Compliance with Ethics Guidelines
This study was conducted in accordance with the protocol, International Council on Harmonisation guidelines, local regulations, and the ethical principles of the Declaration of Helsinki. An independent ethics committee or institutional review board at each study site approved all study-related documents (Supplementary Table S1). All patients reviewed and signed an informed consent form before any study procedures were conducted.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Mease, P.J., Kellner, H., Morita, A. et al. Long-Term Efficacy and Safety of Risankizumab in Patients with Active Psoriatic Arthritis: Results from a 76-Week Phase 2 Randomized Trial. Rheumatol Ther 9, 1361–1375 (2022). https://doi.org/10.1007/s40744-022-00474-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-022-00474-5