
Abstract
Introduction
This phase III trial (NCT04178850) evaluated the efficacy, safety, and immunogenicity of GB242, an infliximab biosimilar, vs. infliximab (Remicade®) reference product in patients with moderate-to-severe active rheumatoid arthritis (RA) combination with methotrexate (MTX) therapy.
Methods
Patients were randomized in a 1:1 ratio to receive either GB242 or INF (3 mg/kg). Therapeutic equivalence of clinical response according to the American College of Rheumatology 20% (ACR20) response rate at week 30 was declared if the two-sided 95% CI for the treatment difference was within ± 14%. The comparison of GB242 with INF also included the proportion of patients achieving a week 30 ACR 50 response, ACR70 response, change in Disease Activity Score 28 (DAS28), as well as safety and immunogenicity.
Results
A total of 570 subjects were randomized into GB242 (N = 285) or INF (N = 285) and 283 subjects in each group were analyzed. At week 30, the ACR20 was 62.54% for the GB242 group (95% CI 56.62–68.20%) and 56.89% for the INF group (95% CI 50.90–62.74%). The difference between the two groups was 5.65% with a 95% CI of – 2.48 to 13.74. ACR50 response was 37.12% for GB242 and 32.86% for INF at week 30. ACR70 response was 19.79% for GB242 and 16.96% for INF at week 30, respectively. The incidence of treatment-emergent adverse events was comparable (77.4% in GB242 vs. 80.2% in INF) and detection of antidrug antibodies (ADA) to infliximab up to week 30 (60.8% in GB242 vs. 59.4% in INF) was comparable.
Conclusions
GB242 demonstrated equivalent efficacy to INF at week 30. Moreover, GB242 was well tolerated, with a similar immunogenicity and safety profile comparable to INF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Globally, several clinical studies are ongoing to find a more economical therapy of rheumatoid arthritis than infliximab. |
Preclinical data and phase I study of GB242 support that GB242 could be an effective therapy in patients with moderate to severe active rheumatoid arthritis. |
What was learned from the study? |
GB242 phase III clinical study evaluated the efficacy and safety of GB242, an infliximab biosimilar, vs infliximab (Remicade®) reference product in patients with moderate to severe active rheumatoid arthritis (RA). The data show that GB242 demonstrate equivalent efficacy to INF at week 30, with similar immunogenicity. Moreover, GB242 is well tolerated, with a safety profile comparable to infliximab. |
Our findings suggest GB242 is clinically active and well-tolerated in patients with moderate to severe active RA. GB242 is a low-cost biosimilar drug with the potential to have a large impact on RA outcomes globally. |
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease characterized by high morbidity and disabilities leading to a significant cost to both individual and society [1, 2]. Methotrexate (MTX) remains the anchor drug in RA according to ACR-EULAR 2019. However, only half of the patients respond adequately to MTX [3, 4]. While tumor necrosis factor(TNF)-alpha inhibitors such as infliximab have shown clinical benefit in patients with RA, the high economic burden due to hospitalization and long-term therapy is considerable [5, 6].
A biosimilar is a biologic drug that contains a high similarity of the structure and function to a reference or original biological medicinal product [7,8,9]. The definition of a biosimilar include three aspects: Firstly, the similarity of molecular level in terms of a comprehensive physicochemical and biological characterization was demonstrated [10]. Secondly, bioequivalence of pharmacokinetic (PK) need to be proved [11], and finally, the safety and efficacy of clinical equivalence are done to compare with the reference product [12]. Several studies have already shown that infliximab biosimilars could be a good choice in terms of reduction of financial burden on patients and maintaining excellent efficacy [11,12,13,14,15].
GB242 is an immunoglobulin (Ig) G1 chimeric human–murine monoclonal antibody biosimilar to infliximab reference product (INF). GB242 is produced in the same type of cell line and has an identical amino acid sequence to INF. The phase I study had shown that GB242 and the INF were similar on the molecular level and bioequivalent in the healthy adult volunteers in China [16].
Based on the data described, this study now reports a phase III study to assess efficacy equivalence and overall safety of GB242 versus INF in active moderate-to-severe RA patients.
Methods
Patients
Chinese patients who were 18–75 years old with RA according to the 2010 American College of Rheumatology (ACR) classification criteria were recruited. The patients had to fulfil the following criteria: they had to have ≥ 4 swollen and ≥ 6 tender joints, and with erythrocyte sedimentation rate (ESR) > 28 mm/h or serum C reactive protein (CRP) concentration > 1.0 mg/dl. The patients had to have been receiving methotrexate (MTX) therapy for ≥ 3 months (stable dose of 10–15 mg/week for ≥ 4 weeks prior to screening). The patients had to have discontinued other disease-modifying antirheumatic drugs (DMARDs) for at least 4 weeks except for MTX prior to the first dose of study drug. The use of leflumide also required a washout period prior to the first dose of study drug. Patients were permitted to receive oral glucocorticoids of stable dose (equivalent to ≤ 10 mg daily prednisolone) for ≥ 4 weeks prior to screening. Patients who experienced bDMARD treatment within 3 months should be excluded. More detailed eligibility criteria are available in the supplementary material.
Study Design
This study is a phase III, randomized, double-blind, multicenter parallel group study. The study (ClinicalTrials.gov NCT04178850) was conducted according to the Declaration of Helsinki and the International Committee on Harmonisation good clinical practice and all applicable regulatory requirements. The study protocol was reviewed and approved by independent ethics committees of each study site. All patients provided written informed consent.
The study was conducted at 29 centers in China. Patients were randomized 1:1 to receive 2-h intravenous infusion of either 3 mg/kg of GB242 (Yuxi Genor Biotechnology Co., Ltd.) or INF(Cliag AG) at weeks 0, 2, 6, 14, 22, in total five times. MTX was given as an oral weekly dose of 10–25 mg/week with folic acid of 5–10 mg/week, 30 times totally. Non-steroidal anti-inflammatory drugs and corticosteroids (≤ 10 mg prednisolone) were allowed if taken for a stable dose for 4 weeks before randomization. Other disease-modifying anti-rheumatic drugs (except for MTX) were prohibited during the study. Other therapeutic biologic agents should be banned 3 months before and throughout the trial.
In this study, the dynamic random variance minimization random method was used, and the electronic central random system was used to calculate and assign random numbers and drugs.
Blind design The test drug and the reference drug use the same packaging, and the outer packaging is the same. Each research center needs to be equipped with non-blind nurses for the preparation of medicines (the drug preparation nurses shall not contact the patients), and record the screening number of the subject and the date of medication on the used outer packaging box. After use, the packaging of the medicine needs to be sealed with a disposable adhesive strip to prevent others from viewing the medicine in the box. The used drug packaging will be recycled to the sponsor on a regular basis, and the non-participating clinical research staff will check and sort, submit the screening number and the number of bottles used to the non-blind monitor, and check with the medication record.
Endpoints
The primary endpoint was the percentage of patients achieving ACR criteria for ≥ 20% clinical improvement (ACR20) at week 30 in the per-protocol set (PPS) and the full analysis set (FAS), as determined by ACR20 response criteria. Equivalence of efficacy was concluded if the 95% CIs for treatment difference were within ± 14% at week 30.
Second endpoints at weeks 2, 4, 6, 12, 14, and 22 included ACR20 (other than week 30), ACR50 (≥ 50% clinical improvement), and ACR70 (≥ 70% clinical improvement) response rates; the changes from baseline in DAS28 (CRP) at weeks 14 and 30. Disease Activity was measured by DAS28 in RA. Patients were considered to be in DAS remission when DAS28-CRP was < 2.6, LDA is defined as DAS28 2.6 ≤ to < 3.2, moderate is defined as DAS28 3.2 ≤ to ≤ 5.1, and high is defined as DAS28 > 5.1 [17]. ACR remission was defined based on scores for the tender joint count, swollen joint count, CRP, ESR, and HAQ-DI. Additional secondary endpoints included immunogenicity and safety [18].
Immunogenicity tests were evaluated by serum antidrug antibodies (ADAs) and neutralizing antibodies (NAbs) to infliximab and GB242 at weeks 2, 6, 14, and 22 before infusion and weeks 30, respectively. ADAs were measured by a bridging electrochemiluminescent (Bridging-ECL) immunoassay utilizing the Meso Scale Discovery platform (MSD, Shanghai, China). Those who were ADA-positive were additionally assessed for neutralizing antibodies.
We used the L-929 cell proliferation endpoint method to detect the neutralizing antibodies of the test drug GB242 in human serum. The principle of the method is as follows: using mouse fibroblasts (L-929 cells) as target cells, under the action of actinomycin D, L-929 cells are highly sensitive to the killing and inhibition of rh TNFα. First, add the drug GB242 and the pretreated system suitability samples/validation samples/samples to be tested. Incubate at room temperature for about 2 h. Then plate the L-929 cells and add the incubated system suitability samples/validation samples/samples to be tested, and add rh TNF α containing actinomycin D at the same time. If there is no anti-GB242 neutralizing antibody in the sample, GB242 in the system can neutralize the killing effect of rh TNF α on L-929 cells, so that the cells grow and proliferate normally. Add CellTiter-Glo reagent to quantitatively detect the ATP of living cells after 20 h of culture. The instrument response value (RLU) read on the chemiluminescence detection instrument is high; if the sample contains anti-GB242 neutralizing antibody, then the RLU value is low.
Safety endpoints included incidence and type of adverse events (AEs) and serious AEs, clinical laboratory abnormalities, including incidence, severity, type, and infusion-related reactions. Other safety assessments included vital signs and abnormalities of other physical examinations. AEs were coded by the Medical Dictionary for Regulatory Activities, version 22.0 and severity was classified as 1-5 according to Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. Treatment-emergent AEs (TEAEs) were assessed throughout the study and defined as any AE that occurred, or any pre-existing AE that worsened, after the beginning of study treatment. Latent or active tuberculosis (TB) was screened by an interferon γ-release assay using QuantiFERON-TB Gold in tube (QTF-TB Gold-IT, Genor, China) and chest X-ray at screening, weeks 14 and 30.
Statistical Analysis
All primary efficacy analyses were performed on both the PPS and the FAS. Sample size was calculated according to the following criterion: the randomized equivalence trial to INF is based on the expected response rate of 60% [19]. Sample size was calculated by specifying a two-sided α level of 0.05, power of 80% and a two-sided equivalence margin of 15%. A sample size required 516 patients in the per-protocol (PP) population final analysis. With a 10% dropout rate, a sample size of at least 568 randomized patients were required for the PPS. The equivalence margin was determined using data from several INF studies [20, 21] and regulatory guidelines [22,23,24].
All efficacy outcome was analyzed using the FAS [25]. FAS included all randomized patients who received at least one dose of GB242 or INF. The primary efficacy was to measure equivalence of ACR20 of GB242 and INF at week 30. Analysis of ACR50 and ACR70 was also done in the FAS; DAS28 was performed in the FAS and DAS28 was analyzed by using MMRM to calculate the mean value and the corresponding 95% confidence interval. The PPS statistical analysis method was described in Supplemental files.
Safety analysis was done in the number of patients who had a particular AE in the safety analysis set (SS; those who received at least one dose of GB242 or INF). ADA analysis was also done in those patients having incident ADA up to week 30 from the SS.
Results
Patient Characteristics at Baseline
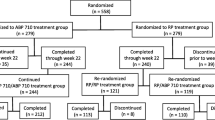
The first patient was screened in October 2017; the last week 30 evaluation was performed in March 2020. Baseline demographics and disease status were comparable between GB242 and INF (Table 1). Of the 570 randomized patients, 491 completed the 30-week study period and, of these, 65 patients were excluded from the PP population due to major protocol violations. Discontinuation in randomized patients was primarily due to AEs (8.9%) and patient withdrawal of consent (4.1%) (Fig. 1).

Disposition flow chart of the study population. A total of 905 patients were screened for the study, and 566 eligible patients were randomized into a GB242 group (n = 283) or an infliximab reference product (INF) group (n = 283) to receive 3 mg/kg of GB242 or INF, respectively, coadministered with methotrexate (MTX) and folic acid. The full analysis set (FAS) for GB242 is n = 283 and infliximab reference product (INF) n = 283. The per-protocol set (PPS) for GB242 is n = 237 and INF n = 233
Efficacy
The ACR20 response over time is shown in Fig. 2. The ACR20 response at each visit was similar between GB242 and INF at all time points through week 30. The curve in Fig. 2 is fitted by generalized linear mixed models linked with the binomial mean with logit function. The upper limit of the 95% CI for the two curves was 11.03%, which was below the prespecified equivalence margin of 15%. The p value for two-treatment comparison is 0.5259. Therefore, the two time–response curves were determined to be equivalent. Moreover, the primary endpoint of ACR20 response at week 30 was equivalent between GB242 and INF group. The ACR20 response for the FAS was 62.54% for GB242 and 56.89% for INF, respectively. The 95% CI for the rate difference was − 2.48% to 13.74%, which was within the prespecified equivalence margin of ± 14%. The ACR20 response was also similarly shown in the PPS; ACR20 was 71.73% for GB242 and 66.52% for INF, respectively. Thus, the equivalence of GB242 compared with INF was concluded. Other efficacy outcomes such as ACR50 or ACR70 were also similar in the FAS and PPS (Fig. 3). ACR50 response for the FAS was 37.10% for GB242 and 32.86% for INF, ACR50 response for the PPS was 42.62% for GB242 and 38.63% for INF. ACR70 response for the FAS was 19.79% for GB242 and 16.96% for INF, ACR70 response for the PPS was 22.78% for GB242 and 20.60% for INF, respectively.

ACR20 response pattern over time. INF infliximab reference product

American College of Rheumatology (ACR) response rates at week 30. A ACR20, 50 and 70 responses for GB242 and INF in the full analysis set (FAS). B ACR20, 50 and 70 responses for GB242 and infliximab reference product (INF) in the per-protocol set (PPS)
The data show that from baseline to week 30, the changes of each efficacy component used for calculating ACR responses or DAS28 activity were similar between GB242 and INF (Supplementary Material S1). The overall ACR20 response rate was lower in the ADA-positive subgroup compared with the ADA-negative subgroup, but was also similar between GB242 and INF within each ADA subgroup (58.67 vs. 54.26%, ADA-positive subgroup; 74.68 vs. 65.88%, ADA-negative subgroup(supplementary material S2), and there was not significant difference (P = 0.7408) between the ADA- positive subgroup and ADA-negative subgroup).
The response for DAS28 over time, the proportion of remission, low disease activity (LDA), and moderate and high disease activity by DAS28 are shown in Fig. 4. The change of DAS28 (CRP) and DAS28 (ESR) over time was similar between GB242 and INF (Fig. 4A), and the proportion of remission, LDA, moderate and high disease activity by DAS28 (CRP) at weeks 30 was 43% for GB242 and 42.5% for INF, 19.8% for GB242 and 18% for INF, 34.6% for GB242 and 36.9% for INF, 2.5% for GB242 and 2.6% for INF, respectively (Fig. 4B). Overall, the efficacy of GB242 appeared to be slightly better than that of INF, but the difference was not statistically significant.

DAS28 responses in the per-protocol population. A Mean DAS28 score based on C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) at baseline, weeks 14 and 30 for GB242 and infliximab reference product (INF). B Disease activity classification by DAS28 (CRP). Remission is defined as DAS28 < 2.6, LDA is defined as DAS28 2.6 ≤ to < 3.2, moderate is defined as DAS28 3.2 ≤ to ≤ 5.1, and high is defined as DAS28 > 5.1
Immunogenicity
The incidence of ADA was similar between GB242 and INF at all measured time points (supplementary material S3). At baseline, 2.1% (6/283) patients in GB242 and 2.8% (8/283) patients in INF group tested positive for ADA. Patients who developed ADA up to week 6 were 7.4% (21/283) in the GB242 treatment group and 11.0% (31/283) in the INF treatment group and 60.8% (172/283) in the GB242 treatment group and 59.4% (168/283) in the INF treatment group, respectively, at week 30. Patients were also tested for NAb-positivity. The incidence of NAb was similar between GB242 and INF treatment group at all measured time points (Supplementary Material S3). There was a similar trend between two treatment groups. NAb-positive rate at baseline was 0.7% in the GB242 treatment group and 0 in the INF treatment group, at week 30, 25.4% (72/283) in the GB242 treatment group and 23% (65/283) in the INF treatment group, respectively.
Safety
In the safety analysis set, overall treatment-emergent AEs (TEAEs) occurred in 166 (58.7%) patients in the GB242 treatment group and 174 (61.5%) patients in the INF treatment group (Table 2). The most common TEAEs that occurred were upper respiratory tract infection, increase of white blood cell count, and urinary tract infection. The majority of TEAEs were mild to moderate in severity.
Overall treatment-related serious AEs (SAEs) occurred in ten (3.5%) patients in the GB242 treatment group and 12 (4.2%) patients in the INF treatment group (Table 3). The most common treatment-related SAEs that occurred were lung infection, shingles, cryptococcal pneumonia, drug-induced hypersensitivity, immediate anaphylactic shock, decreased oral sensation, gastritis, lung inflammation, and liver damage.
All patients with latent TB had undergone prophylactic TB medication, and only one patient with latent TB developed active TB, who was in the INF group.
Infusion-related reactions occurred in 27 (9.5%) and 23 (8.1%) patients for GB242 and INF, respectively. There was one death reported in GB242 (dead of traumatic brain injury) and one case of suicide from INF. Overall, the safety profile was comparable between GB242 and INF.
Results of Immune Response
Patients who had DAS28 (CRP) < 2.6 in weeks 30 were associated with a rapid increase and then stabilization of CD19 + peripheral B cells in GB242 group as well as in the INF group (Fig. 5A). Differently, CD19 + peripheral B cell percentage rose slowly and then remained stable in the GB242 group and increased firstly and then fell in the INF group in patients with DAS28(CRP) ≥ 2.6 in week 30 (Fig. 5B). In both the two groups, CD19 + B cell in the DAS28 (CRP) < 2.6 group showed a greater increase at week 30 than that at baseline (p < 0.05). Meanwhile, mean immunoglobulin (IgG and IgA) levels showed significant decrease between week 30 and baseline in DAS28 < 2.6 group (p < 0.05).

CD19 response pattern overtime. A Mean CD19 percentage based on DAS28 < 2.6 at weeks 30 for GB242 and infliximab reference product (INF). B Mean CD19 percentage based on DAS28 ≥ 2.6 at weeks 30 for GB242 and INF
Discussion
In this randomized, double-blind study, the equivalence of efficacy, safety, and immunogenicity profiles between GB242 and INF was demonstrated. The primary endpoint was ACR20 response at week 30, where GB242 was shown to be slightly higher than that of INF in the FAS and PPS, the 95% CIs for treatment difference were within the predefined margins for equivalence of ± 14%. Moreover, the efficacy endpoints at all visits were compared, and this study has demonstrated the equivalence of ACR20 response over time. Comparable to the results of TNF inhibitor biosimilars, we performed a meta-analysis of 290 patients with RA treated with INF biosimilars in four studies (Yoo DH, 2013, Choe J-Y, 2015, Cohen, 2018, Genovese, 2020) [12, 13, 26, 27]. ACR responses of GB242 were also higher than the results at week 30 in the meta-analysis (ACR20, 50 and 70: 62.54, 37.12, and 19.79% for GB242 and 59.2, 32.9, and 16.0% for biosimilars, respectively).
To ensure a credible comparison between GB242 and INF data, we also observe other efficacy outcomes besides the ACR 20 response such as ACR50, ACR70 responses. Moreover, DAS28 was similar between GB242 and INF, further supporting the biosimilarity of GB242 to INF. Efficacy criteria was also measured according to ADA-positive rate and NAb-positive rate at week 30, and no statistically significant differences in responses between GB242 and INF groups were found.
The objective with regard to safety was to demonstrate a comparable safety profile of GB242 and INF. Overall, the incidence of AEs associated with treatments such as upper respiratory tract infection, an increase in white blood cell counts, and latent TB, were comparable between GB242 and INF. It is notable that the incidence of active TB was very low (only one patient in INF group), which might be attributed to universal TB prophylaxis for patients with latent TB at screening. The rate of infusion reactions in both treatment groups (4.6 vs. 6.4%) was comparable. The safety results were similar to those previous studies.
In terms of immunogenicity, GB242 has demonstrated a comparable profile at all measured time points to that of INF. The incidence of ADA (∼60%) is similar to recent INF studies [12,13,14,15].
In terms of results of immune response, an increase of CD19 + B cells was regarded as a reliable clinical response biomarker of RA patients [24]. In patients who achieved disease remission at week 30, the increase in CD19 from baseline at week 30 was statistically significant (p < 0.05), while in patients who did not achieve disease remission at week 30, the increase in CD19 from baseline at week 30 was not statistically significant (p = 0.273). This suggests that the increase of CD19 expression was predictive of a faster control of pathological activity in patients treated with GB242 and INF, especially in patients who achieved DAS28 < 2.6 at week 30.
Conclusions
In conclusion, GB242 and INF were shown to be equivalent in terms of ACR20, ACR50, and ACR70 at week 30 in active moderate-to-severe RA patients. Other efficacy end points also show consistently similar results when compared to the originator product. Moreover, GB242 was well tolerated, and the safety and immunogenicity profiles were comparable with those of INF.
References
Birnbaum H, Pike C, Kaufman R, Marynchenko M, Kidolezi Y, Cifaldi M. Societal cost of rheumatoid arthritis patients in the US. Curr Med Res Opin. 2010;26(1):77–90.
Cross M, Smith E, Hoy D, et al. The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73(7):1316–22.
Smolen JS, Landewé RBM, Bijlsma JWJ, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2020;79(6):685–99.
Maini R, St Clair EW, Breedveld F, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet. 1999;354(9194):1932–9.
Dörner T, Strand V, Cornes P, et al. The changing landscape of biosimilars in rheumatology. Ann Rheum Dis. 2016;75(6):974–82.
Modena V, Bianchi G, Roccatello D. Cost-effectiveness of biologic treatment for rheumatoid arthritis in clinical practice: an achievable target? Autoimmun Rev. 2013;12(8):835–8.
Chadwick L, Zhao S, Mysler E, et al. Review of biosimilar trials and data on etanercept in rheumatoid arthritis. Curr Rheumatol Rep. 2018;20(12):84.
US Food and Drug Administration (FDA). Scientific considerations in demonstrating biosimilarity to a reference product guidance for industry. Silver Spring: FDA; 2015.
Gulácsi L, Brodszky V, Baji P, et al. Biosimilars for the management of rheumatoid arthritis: economic considerations. Expert Rev Clin Immunol. 2015;11(Suppl 1):S43-52.
Jung SK, Lee KH, Jeon JW, et al. Physicochemical characterization of Remsima. MAbs. 2014;6(5):1163–77.
Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72(10):1605–12.
Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72(10):1613–20.
Cohen SB, Alten R, Kameda H, et al. A randomized controlled trial comparing PF-06438179/GP1111 (an infliximab biosimilar) and infliximab reference product for treatment of moderate to severe active rheumatoid arthritis despite methotrexate therapy. Arthritis Res Ther. 2018;20(1):155.
Bendtzen K, Geborek P, Svenson M, et al. Individualized monitoring of drug bioavailability and immunogenicity in rheumatoid arthritis patients treated with the tumor necrosis factor alpha inhibitor infliximab. Arthritis Rheum. 2006;54:3782–9.
Arstikyte I, Kapleryte G, Butrimiene I, Venalis A. Influence of immunogenicity on the efficacy of long-term treatment with TNF-α blockers in rheumatoid arthritis and spondyloarthritis patients. Biomed Res Int. 2015; 2015:604872.
Zhang T, Chen G, Liu C, et al. A phase I study comparing the pharmacokinetics, safety, and immunogenicity of proposed biosimilar GB242 and reference infliximab in healthy subjects. BioDrugs. 2019;33(1):93–100.
Felson DT, Smolen JS, Wells G, et al. American College of Rheumatology/European League Against Rheumatism Provisional Definition of Remission in Rheumatoid Arthritis for Clinical Trials. Ann Rheum Dis. 2011;70(3):404–13.
Anderson J, Caplan L, Yazdany J, Robbins ML, Neogi T, Michaud K, et al. Rheumatoid arthritis disease activity measures: American College of Rheumatology recommendations for use in clinical practice. Arthritis Care Res (Hoboken). 2012;64(5):640–7.
Maini R, St Clair EW, Breedveld F, et al. In fliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study. Group Lancet. 1999;354:1932–9.
Abe T, Takeuchi T, Miyasaka N, et al. A multicenter, double-blind, randomized, placebo controlled trial of infliximab combined with low dose methotrexate in Japanese patients with rheumatoid arthritis. J Rheumatol. 2006;33(1):37–44 (PMID: 16395748).
Westhovens R, Yocum D, Han J, et al. The safety of infliximab, combined with background treatments, among patients with rheumatoid arthritis and various comorbidities: a large, randomized, placebo-controlled trial. Arthritis Rheum. 2006;54:1075–86.
EMA/CHMP. Guideline on the choice of the non-inferiority margin. 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003636.pdf. Accessed 6 Mar 2015.
US FDA. Guidance for Industry: non-inferiority clinical trials. 2010. http://www.fda.gov/downloads/Drugs/Guidances/UCM202140.pdf. Accessed 6 Mar 2015.
ICH Harmonised Tripartite Guideline. Statistical principles for clinical trials. International Conference on Harmonisation E9 Expert Working Group. Stat Med. 1999;18(15):1905–42.
Zecevic L, Mekic M, Subasic D, et al. Expression of NK (CD16+ 56+) and B cells (CD19) receptor molecules as a reliable clinical response biomarkers of SLE and RA patients under the rituximab treatment. Med Arch. 2019;73(6):374–7.
Choe JY, Prodanovic N, Niebrzydowski J, Staykov I, Dokoupilova E, Baranauskaite A, et al. A randomised, double-blind, phase III study comparing SB2, an infliximab biosimilar, to the infliximab reference product Remicade in patients with moderate to severe rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2017;76(1):58–64.
Genovese MC, Sanchez-Burson J, Oh M, Balazs E, Neal J, Everding A, Hala T, et al. Comparative clinical efficacy and safety of the proposed biosimilar ABP 710 with infliximab reference product in patients with rheumatoid arthritis. Arthritis Res Ther. 2020;22(1):60.
Acknowledgements
The authors thank the patients and their families who were involved in this study, and the sponsor Genor Biopharma Co., Ltd., China.
Funding
This study and the journal’s Rapid Service Fee were funded by Genor Biopharma Co., Ltd., China.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
Zhanguo Li and Yanying Liu contributed to study conception and design. Yanying Liu and Zhanguo Li contributed to drafting the manuscript and final approval of the version to be published. All authors contributed significantly to data collection and approved the manuscript.
Disclosures
This trial was sponsored by Genor Biopharma Co. QianGuo, Yingxue Cathy Liu, are employees at Genor Biopharma Co. The following authors (Yanying Liu, Shengyun Liu, Lin Liu, Xiaowei Gong, Ju Liu, Lingyun Sun, Xiumei Liu, Lijun Wu, Linjie Chen, Ling Wang, Li Luo, Jinying Lin, Ning Tie, Zhenyu Jiang, Jian Wu, Fuai Lu, Hongsheng Sun, Xiaomei Li, Niansheng Yang, Kexia Chai, Hua Wei, Zhanyun Da, Cheng Zhao, Lie Dai, Youlian Wang, Guixiu Shi, Zhenchun Zhang, Hui Song, Zhanguo Li) have nothing to disclose.
Compliance with Ethics Guidelines
This study complied with the provisions of the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice Guidelines, and it was reviewed and approved by an institutional review board or independent ethics committees at each of the participating investigational sites (see supplementary material S4). Informed consent was provided to all patients prior to undergoing any screening procedures.
Data Availability
All data generated or analyzed during this study are included in this published article as supplementary information files.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Liu, Y., Liu, S., Liu, L. et al. Fine Comparison of the Efficacy and Safety Between GB242 and Infliximab in Patients with Rheumatoid Arthritis: A Phase III Study. Rheumatol Ther 9, 175–189 (2022). https://doi.org/10.1007/s40744-021-00396-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-021-00396-8