
Abstract
Objectives
To investigate the long-term outcomes, including risk factors, for exacerbation between monotherapy and combination therapy in patients with interstitial pneumonia with autoimmune features (IPAF).
Methods
We assessed 672 patients between April 2009 and March 2019 who were evaluated using high-resolution computed tomography (HRCT) of the chest. We applied the IPAF criteria. Fifty-two patients who visited our department for at least 6 months were diagnosed with IPAF. Clinical, laboratory, and imaging data were collected from medical records and statistically analyzed.
Results
Among the 52 cases of IPAF, we compared the characteristics at diagnosis between treated (n = 28) and untreated patients (n = 24). The exacerbation rates were 42.9% (n = 12) and 8.3% (n = 2) (P = 0.0051), respectively. Among the treated patients, smoking history, high titer of KL-6, and the duration from diagnosis to the start of treatment were significant risk factors for exacerbation (P = 0.0062, 0.011, and 0.019, respectively). The number of risk factors was significantly and positively associated with exacerbation rate (P = 0.0053). Among the treated patients, glucocorticoid (GC) monotherapy was used in 13 cases, and GC and immunosuppressant (IS) combination therapy was used in 14 patients. There was no significant difference in the treatment methods between patients with and without risk factors (P = 0.47). When comparing the long-term outcomes between the monotherapy and combination therapy groups, the 3-year non-exacerbation rates were 72.9 and 45.9% (P = 0.020), respectively.
Conclusions
IPAF patients with risk factors had a high exacerbation rate, regardless of the type of treatment. New interventions aimed at preventing exacerbations in these patients are required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
The prognosis of IPAF varies among studies. Nonetheless, no study has compared the long-term outcome, and the efficacy and safety profiles of different treatment methods for IPAF have not been reported. |
We investigated the long-term outcomes, including risk factors, for exacerbation between monotherapy and combination therapy in patients with IPAF |
What was learned from the study? |
The exacerbation rate of IPAF patients with risk factors was high, regardless of treatment. |
It is important to start treatment early. |
New interventions aimed at preventing exacerbations in these patients are required. |
Introduction
Some patients with idiopathic interstitial pneumonia (IIP) may have features of connective tissue disease (CTD) [1]. In the past, this disorder was referred to as undifferentiated connective tissue disease-associated interstitial lung disease (UCTD-ILD), lung-dominant CTD, or autoimmune-featured ILD [2,3,4]. There were discrepancies in the inclusion criteria among published reports. Therefore, the term interstitial pneumonia with autoimmune features (IPAF) has been proposed to define patients with ILD with autoimmune features but who do not meet the established CTD criteria by the European Respiratory Society (ERS)/American Thoracic Society (ATS) in 2015 to unify the inclusion criteria. These advocated criteria are composed of three domains: clinical, serological, and morphological [5] (Table 1).
The prognosis of IPAF varies among studies. According to one study [1], IPAF is associated with worse survival than CTD-ILD while showing slightly better survival than idiopathic pulmonary fibrosis (IPF). Other studies showed no significant difference between IPAF and IPF [6], and IPAF survival was better than IIP survival and similar to CTD-ILD survival [7]. To date, no study has compared the long-term outcome or the efficacy and safety profiles of different treatment methods for IPAF.
This study investigated the long-term outcomes, including risk factors, for exacerbation between monotherapy and combination therapy in patients with IPAF.
Methods
Patients
In this observational cohort study, the data of 672 patients who visited the Division of Pulmonary Medicine, Allergy, and Rheumatology, Iwate Medical University Hospital, between April 2009 and March 2019 and were evaluated by chest high-resolution computed tomography (HRCT) scans were submitted to the IPAF criteria. All procedures were approved by the medical ethics committee of Iwate Medical University Hospital (registration no. MH2018-505) and followed the tenets of the Declaration of Helsinki. This is a retrospective study and written consent is not required. The method of disclosure to patients is as follows. The method of disclosure to patients will be explained in writing at the outpatient clinic of the Department of Respiratory, Allergy, and Collagen Diseases, and subjects will be given the opportunity to refuse the use of their data.
Assessment of IPAF
Chest HRCT scans were assessed by a trained radiologist and pulmonologist looking for adherence to the guidelines for IPF [8] and the ATS/ERS statement of IIP 2013 [9]. Then, a trained rheumatologist classified patients as IPAF if they met the IPAF criteria. We defined ‘exacerbation’ as an acute, clinically significant respiratory deterioration characterized by evidence of new ground-glass opacification, consolidation, or reticulation on HRCT occurring less than 1 month before starting or increasing of glucocorticoid (GC) and/or immunosuppressant (IS) [10]. We defined ‘severity of interstitial pneumonia’ by a three-point scale (stage I–III) according to a partially modified from the guidelines for systemic sclerosis of the Japanese Dermatological Association [11], as follows: “stage I, disease extent on HRCT ≤ 20%; stage II, disease extent on HRCT > 20% without oxygen therapy; stage III, disease extent on HRCT > 20% with oxygen therapy”. We also defined ‘treated patients’ as patients treated at our department for the first time, although they had never been treated.
Statistical Analysis
The association of prognostic factors suggesting exacerbation to clinical, laboratory, and radiographic information at baseline characteristics was analyzed using Fisher’s exact test, the Wilcoxon rank-sum test, or the Kruskal–Wallis test. We compared these variables between groups using the Chi-square test. Results were considered significant at P < 0.05. Baseline variables (P < 0.05) in univariate analysis were included in the multivariable models. Kaplan–Meier curves and log-rank tests were used to compare non-exacerbation rates between the groups. Results were considered significant at P < 0.05. All statistical analyses were performed using JMP software v. 13.2.1 (SAS Institute Inc., Cary, NC, USA).
Results
Characteristics of IPAF
Sixty-eight cases (17.1%, among all patients excluding without ILD) were clinically, serologically, and morphologically diagnosed as IPAF. Among them, 52 patients who visited our department for at least 6 months were enrolled (Supplementary Material). Of the 52 cases of IPAF, the exacerbation rate was 27% (n = 14), including 3.8% (n = 2) mortality. The clinical characteristics of the 52 patients are shown in Table 2. The mean age at diagnosis was 63.6 ± 13.9 years, and 55.8% were women. The mean observation period was 45.0 ± 34.6 months. The proportion of current or past smoking was 46.2% (n = 24). The proportion of severity of interstitial pneumonia was 55.8% (n = 29) with stage I, 32.7% (n = 17) with stage II and 11.5% (n = 6) with stage III, respectively. In the baseline values of pulmonary function test, which was conducted on a small number of patients, mean percentage of predicted forced vital capacity (%FVC) was within the normal range (80.2 ± 19.5) and mean percentage of predicted diffusing capacity of the lung for carbon monoxide (%DLCO) was a mild reduction (73.0 ± 17.5). There were relatively few severe interstitial pneumonia cases. On HRCT, the proportion of morphological pattern was 11.5% (n = 6) with UIP, 78.8% (n = 41) with NSIP, 7.7% (n = 4) with OP and 1.9% (n = 1) with NSIP with OP overlap, respectively. Almost all of the patients had NSIP patterns. The proportions of diagnostic domains in IPAF were 48.1, 36.5, 88.5, and 36.5% for the clinical and serological domain, clinical and morphological domain, serological and morphological domain, and all domains, respectively. The features within each IPAF domain were follows. The most frequently identified clinical findings were arthritis or morning stiffness (28.8%), followed by Raynaud's phenomenon (19.2%) and unexplained digital edema (15.4%). The most frequently identified serological findings were ANA positive ≥ 320 (36.5%) and anti-Ro (SS-A) positive (36.5%). The most frequently identified morphological finding was NSIP pattern by HRCT (78.8%) while only one patient showed pericardial effusion or pericardial thickening within multicompartment involvement.
We compared the baseline characteristics between patients with IPAF, CTD-ILD, and IIP (Supplementary Material). The mean age and titer of Krebs von den Lungen-6 (KL-6) at diagnosis in the IPAF group were significantly higher than those in the CTD group (P = 0.018 and 0.019, respectively). The proportion of treated patients with IPAF, including those treated with IS, was significantly lower than that with CTD-ILD (P = 0.0001). Comparing the long-term outcomes among patients with IPAF, CTD-ILD, and IIP, the 3-year non-exacerbation rates were 71.8, 67.5, and 47.2%, respectively (Supplementary Material). There was no significant difference in long-term outcomes between patients with IPAF and CTD-ILD (P = 0.61).
Prognostic Factors for Exacerbation of IPAF
We divided IPAF patients into two groups: those with exacerbation and those without exacerbation. We evaluated risk factors for exacerbation. On univariate analysis, smoking history, high titer of KL-6, high titer of pulmonary surfactant protein-D (SP-D), duration from diagnosis to the start of treatment, and treatment with oral GC and/or IS use were significantly elevated in the exacerbation group (Table 2). On multivariate analysis, smoking history, high titer of KL-6 and SP-D, duration from diagnosis to the start of treatment, and treatment with oral GC and/or IS were significantly elevated in the exacerbation group. In the multivariate logistic regression model, exacerbation of IPAF was significantly associated with smoking history, high titers of KL-6 and SP-D, and the duration from diagnosis to the start of treatment (Table 3).
Characteristics of Treated and Untreated IPAF
Among the 52 patients with IPAF, 28 were treated with GC and/or IS therapy. The proportions of each therapy were 51.9% (n = 27) for GC and 28.8% (n = 15), respectively. One patient was treated with IS only (Table 2). The remaining 24 patients were not treated. We compared the characteristics at diagnosis between the treated (n = 28) and untreated (n = 24) groups (Table 4). The treated group was significantly positively associated with titer of KL-6 and severity of interstitial pneumonia (P = 0.042 and 0.0015, respectively) (Table 4). The treated group was also significantly associated with arthritis, anti-SS-A antibody positivity, and anti-ARS antibody positivity (P = 0.016, 0.03, and 0.016, respectively) characterized by polymyositis/dermatomyositis (Table 4). In contrast, anti-Scl-70 antibody positivity was significantly higher (P = 0.025), characterized by systemic scleroderma (Table 4).
Prognostic Factors for Exacerbation of Treated IPAF
Among the 28 patients treated with IPAF, the exacerbation rate was 42.9% (n = 12) (Table 4). We evaluated the risk factors for exacerbation in the treated groups. Smoking history, high KL-6 titer, and the duration from diagnosis to the start of treatment (> 1 month) were significant risk factors for exacerbation (P = 0.0062, 0.011, and 0.019, respectively) (Table 4). The number of risk factors was positively and significantly associated with the exacerbation rate (P = 0.0053) (Fig. 1).

Cumulative rate of exacerbation in 28 treated patients with IPAF. The positivity of three risk factors, namely smoking history, high titer of KL-6, and the duration from diagnosis to the start of treatment, were counted. P values were calculated using the Kruskal–Wallis test
Treatment Method and the Long-Term Outcome in IPAF
We divided the treated group into two subgroups: those treated with monotherapy (GC) and those treated with combination therapy (GC and IS). GC monotherapy was administered in 46.4% (n = 13), and GC + IS combination therapy was administered in 50% (n = 14) of the treatments. The combination therapy group was significantly associated with ‘mechanic’s hands’ (P = 0.037) (Table 4). In contrast, anti-SS-A antibody positivity was significantly higher (P = 0.012) (Table 4). Combination therapy was significantly higher in patients with more than two risk factors than monotherapy (P = 0.030) (Table 4). GC dose at the start of treatment in the monotherapy and combination therapy groups was 30.4 ± 13.0 mg/day and 32.1 ± 17.2 mg/day, compared with 9.2 ± 7.2 mg/day and 8.1 ± 3.6 mg/day at the last visit, showing no significant difference between groups (P = 0.96 and 0.98, respectively) (Table 4). The immunosuppressants included calcineurin inhibitors tacrolimus and cyclosporine (29%, n = 4; 64%, n = 9). Azathioprine was administered to one patient (Fig. 2). There was no significant difference in the rate of exacerbations among patients with IS (P = 0.16). Comparing the long-term outcomes between the monotherapy and combination therapy groups, it turned out that the 3-year non-exacerbation rates were 72.9 and 45.9%, respectively (P = 0.020) (Fig. 3). The combination therapy group had a significantly worse prognosis than the monotherapy group, and the rate of severe adverse events (SAE) requiring hospitalization due to infections such as pneumonia was 23.1 and 0%, respectively (P = 0.057) (Table 4). The combination therapy group tended to have fewer SAEs.

IPAF treatment type. The GC monotherapy subgroup included 13 participants (46.4%). Immunosuppressant monotherapy (tacrolimus) was administered to one patient (3.6%). Immunosuppressants included tacrolimus, cyclosporine, and azathioprine (29%, n = 4; 64%, n = 9; and 7%, n = 1) in the GC + IS combination therapy group

Kaplan–Meier survival curves in each treatment for IPAF. Interstitial pneumonia with autoimmune features (IPAF) with monotherapy (n = 13), combined therapy (n = 14), and without treatment (n = 24). IPAF-Combo vs. IPAF-Mono; P = 0.020, IPAF-Combo vs. IPAF-untreated; P = 0.00070, IPAF-Mono vs. IPAF-untreated; P = 0.0088
Discussion
This study found that the risk factors for exacerbation of IPAF were smoking history, high titer of KL-6, and high titer of SP-D. Particularly in the treated group, smoking history, high titer of KL-6, and the duration from diagnosis to the start of treatment were significantly associated with exacerbations. Regarding long-term outcomes, IPAF was better than IIP and similar to CTD-ILD. This is comparable to the results of a previous study [7]. Patients with IPAF with risk factors had a high exacerbation rate regardless of treatment; however, there was no significant difference between the monotherapy and combination therapy groups.
Older age, male sex, ever-smokers, radiological and/or pathological UIP pattern, %FVC, and %DLCO have been reported as risk factors for exacerbation [1, 6, 7, 12, 13]. In our study, ever-smoking was a risk factor for exacerbation, similar to previous reports [1, 6, 7, 12, 13]. In contrast, older age, male sex, and radiological UIP pattern were not risk factors for exacerbation in our study. Regarding %FVC and %DLCO, the rates of these examinations were only 57 and 26%, respectively, so their association with exacerbation could not be assessed. Our study revealed that KL-6 and SP-D were risk factors for exacerbation. KL-6 and SP-D are active markers of interstitial pneumonia in Japan. Although one study reported KL-6 as a poor prognostic factor in IPAF [13], SP-D has not been reported previously. A high SP-D titer at diagnosis was a poor prognostic factor in patients with IIP [14]. SP-D has never been reported as a poor prognostic factor in patients with IPAF, but we speculated that SP-D might predict a poor prognosis. When treated with IPAF, a high SP-D titer was not a risk factor for exacerbation. Our study also revealed that the duration from diagnosis to the start of treatment was a risk factor for exacerbation. These findings suggest that early diagnosis and early start of treatment are necessary. We speculated that the start of treatment as early as possible may improve the prognosis.
To date, there have been no promising treatments. GC with or without IS, such as mycophenolate mofetil, azathioprine, calcineurin inhibitors, or rituximab, has been used for CTD treatment [15,16,17,18,19,20,21,22,23,24,25]. However, no studies have comprehensively described the long-term outcomes of patients with IPAF by comparing different treatment methods. Our study compared the efficacy and safety profiles of GC monotherapy and GC + IS combination therapy. The reduction in GC dose was not significantly different between monotherapy and combination therapy. In the combination therapy group, ‘mechanic’s hands’ was significantly higher. This suggests ‘anti-ARS antibody syndrome’ [26,27,28] and has a favorable response to GC [29]. Therefore, we speculated that this was why there was no significant difference in the reduction of GC dose between monotherapy and combination therapy groups, regardless of the addition of IS. Although there was no significant difference in the duration from diagnosis to the start of treatment, the GC dose at the start of treatment, and the last visit between the two groups, the combination therapy group had significantly worse long-term outcomes than the monotherapy group. We speculated that this was because the combination therapy group had more risk factors than the monotherapy group. On the other hand, age at diagnosis, GC dose at the start of treatment, and the last visit were not significantly different between the two groups. Despite the use of IS in combination, SAE tended to be lower in the combination therapy group. This may be because IS was administered to patients who were judged to be tolerant of IS.
This study has some limitations. First, this is a single-center retrospective observational cohort study, which makes selection bias and limits generalizability. However, many previous studies were also single-center retrospective studies [1, 6, 7, 13, 30, 31]. Second, we evaluated the small number of IPAF patients. Therefore, we may not be able to evaluate the effect of some variables accurately. However, the number of IPAF patients in this study is not different from previous studies [6, 30, 31]. Third, this study did not examine the risk of exacerbations related to lung function (%FVC and %DLCO) because we could not collect the data on pulmonary function tests for all patients from their medical records, as the data were collected from actual clinical practice.
In conclusion, our study revealed that patients with IPAF with more than two risk factors had a high exacerbation rate regardless of treatment. Moreover, the later the start of treatment, the worse the prognosis. Therefore, it is important to start treatment early. To the best of our knowledge, this is the first report comparing the significance of the effectiveness of IPAF treatment between monotherapy and combination therapy. Further studies should clarify new interventions aimed at preventing exacerbations in these patients.
References
Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J. 2016;47:1767–75.
Kinder BW, Collard HR, Koth L, Daikh DI, Wolters PJ, Elicker B, et al. Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease? Am J Respir Crit Care Med. 2007;176:691–7.
Fischer A, West SG, Swigris JJ, Brown KK, du Bois RM. Connective tissue disease-associated interstitial lung disease: a call for clarification. Chest. 2010;138:251–6.
Vij R, Noth I, Strek ME. Autoimmune-featured interstitial lung disease: a distinct entity. Chest. 2011;140:1292–9.
Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46:976–87.
Ahmad K, Barba T, Gamondes D, Ginoux M, Khouatra C, Spagnolo P, et al. Interstitial pneumonia with autoimmune features: clinical, radiologic, and histological characteristics and outcome in a series of 57 patients. Respir Med. 2017;123:56–62.
Kim HC, Lee JH, Chae EJ, Song JS, Song JW. Long-term clinical course and outcome of interstitial pneumonia with autoimmune features. Respirology. 2020;25:636–43.
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824.
Travis WD, Costabel U, Hansell DM, King TE, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med. 2016;194(3):265–75.
Asano Y, Jinnin M, Kawaguchi Y, Kuwana M, Goto D, Sato S, et al. Japanese dermatological association guidelines: systemic scleroderma diagnostic criteria, severity classification, and clinical practice guidelines. Jpn J Dermatol. 2016;126(10):1831–96.
Dai J, Wang L, Yan X, Li H, Zhou K, He J, et al. Clinical features, risk factors, and outcomes of patients with interstitial pneumonia with autoimmune features: a population-based study. Clin Rheumatol. 2018;37:2125–32.
Ito Y, Arita M, Kumagai S, Takei R, Noyama M, Tokioka F, et al. Serological and morphological prognostic factors in patients with interstitial pneumonia with autoimmune features. BMC Pulm Med. 2017;17:111.
Takahashi H, Kuroki Y, Tanaka H, Saito T, Kurokawa K, Chiba H, et al. Serum levels of surfactant proteins A and D are useful biomarkers for interstitial lung disease in patients with progressive systemic sclerosis. Am J Respir Crit Care Med. 2000;162:258–63.
Oddis CV, Aggarwal R. Treatment in myositis. Nat Rev Rheumatol. 2018;14:279–89.
Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4:708–19.
Morganroth PA, Kreider ME, Werth VP. Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis Care Res (Hoboken). 2010;62:1496–501.
Kurita T, Yasuda S, Oba K, Odani T, Kono M, Otomo K, et al. The efficacy of tacrolimus in patients with interstitial lung diseases complicated with polymyositis or dermatomyositis. Rheumatol Oxf Engl. 2015;54:39–44.
Jordan S, Distler JH, Maurer B, Huscher D, van Laar JM, Allanore Y, et al. Effects and safety of rituximab in systemic sclerosis: an analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann Rheum Dis. 2015;74:1188–94.
Keir GJ, Maher TM, Ming D, Abdullah R, de Lauretis A, Wickremasinghe M, et al. Rituximab in severe, treatment-refractory interstitial lung disease. Respirology. 2014;19:353–9.
Hervier B, Devilliers H, Stanciu R, Meyer A, Uzunhan Y, Masseau A, et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev. 2012;12:210–7.
Kawasumi H, Gono T, Kawaguchi Y, Yamanaka H. Recent treatment of interstitial lung disease with idiopathic inflammatory myopathies. Clin Med Insights Circ Respir Pulm Med. 2015;9(Suppl 1):9–17.
Labirua-Iturburu A, Selva-O’Callaghan A, Vincze M, Dankó K, Vencovsky J, Fisher B, et al. Anti-PL-7 (anti-threonyl-tRNA synthetase) antisynthetase syndrome: clinical manifestations in a series of patients from a European multicenter study (EUMYONET) and review of the literature. Med (Baltim) 2012;91:206–11.
Marie I, Josse S, Decaux O, Diot E, Landron C, Roblot P, et al. Clinical manifestations and outcome of anti-PL7 positive patients with antisynthetase syndrome. Eur J Intern Med. 2013;24:474–9.
Abe Y, Matsushita M, Tada K, Yamaji K, Takasaki Y, Tamura N. Clinical characteristics and change in the antibody titres of patients with anti-MDA5 antibody-positive inflammatory myositis. Rheumatol Oxf Engl. 2017;56:1492–7.
Tanizawa K, Handa T, Nakashima R, Kubo T, Hosono Y, Watanabe K, et al. The long-term outcome of interstitial lung disease with anti-aminoacyl-tRNA synthetase antibodies. Respir Med. 2017;127:57–64.
Katzap E, Barilla-LaBarca ML, Marder G. Antisynthetase syndrome. Curr Rheumatol Rep. 2011;13:175–81.
Lega JC, Fabien N, Reynaud Q, Durieu I, Durupt S, Dutertre M, et al. The clinical phenotype associated with myositis-specific and associated autoantibodies: a meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun Rev. 2014;13:883–91.
Takato H, Waseda Y, Watanabe S, Inuzuka K, Katayama N, Ichikawa Y, et al. Pulmonary manifestations of anti-ARS antibody positive interstitial pneumonia–with or without PM/DM. Respir Med. 2013;107:128–33.
Lim JU, Gil BM, Kang HS, Oh J, Kim YH, Kwon SS. Interstitial pneumoniae with autoimmune features show better survival and less exacerbations compared to idiopathic pulmonary fibrosis. BMC Pulm Med. 2019;19(1):120.
Chartrand S, Swigris JJ, Stanchev L, Lee JS, Brown KK, Fischer A. Clinical features and natural history of interstitial pneumonia with autoimmune features: a single center experience. Respir Med. 2016;119:150–4.
Acknowledgements
We express our gratitude to all the participants in this study.
Funding
This work was supported by the Division of Pulmonary Medicine, Allergy, and Rheumatology, Department of Internal Medicine, Iwate Medical University School of Medicine. The journal's Rapid Service Fee was funded by a special research grant from the Division of Pulmonary Medicine, Allergy, and Rheumatology, Department of Internal Medicine, Iwate Medical University School of Medicine.
Editorial Assistance
We thank Editage Corp. (www.editage.com) for editing the manuscript drafts.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
O.M. designed the study, had full access to all the data used for analyses in this study, contributed to the acquisition, analysis, and interpretation of data, and wrote the initial draft of the manuscript. K.S. designed the study, contributed to the analysis and interpretation of data, and assisted in drafting the manuscript. T.T. and M.M. contributed to the analysis and interpretation of data.
Disclosures
Katsuya Suzuki and Tsutomu Takeuchi have nothing to disclosure. Okinori Murata has changed affiliation: Division of Allergy and Rheumatology, Department of Internal Medicine, Iwate Medical University School of Medicine, Morioka, Japan. Makoto Maemondo has changed affiliation: Division of Pulmonary Medicine, Department of Internal Medicine, Iwate Medical University School of Medicine, Morioka, Japan.
Compliance with Ethics Guidelines
All procedures were approved by the Medical Ethics Committee of Iwate Medical University Hospital (registration no. MH2018-505) and adhered to the tenets of the Declaration of Helsinki. This is a retrospective study and written consent is not required. The method of disclosure to patients is as follows. The method of disclosure to patients will be explained in writing at the outpatient clinic of the Department of Respiratory, Allergy, and Collagen Diseases, and subjects will be given the opportunity to refuse the use of their data.
Data Availability
All data generated or analyzed during this study are included in this published article or as supplementary information files.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
40744_2021_371_MOESM1_ESM.pdf
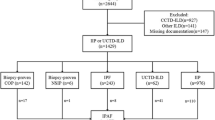
Supplementary Table S1. Baseline characteristics of patients with IPAF, CTD-ILD, and IIP.aAs prednisolone, *: P value < 0.05, **: P value < 0.01, ***: P value < 0.001, DLCO % pred: percent predicted diffusion capacity for carbon monoxide, FVC % pred: percent predicted forced vital capacity, KL-6: Krebs von den Lungen 6, SP-D: surfactant protein-D, Data represented the median (IQR), the mean ± SD or % (number); P value was calculated using the Fisher’s exact test, the Wilcoxon rank-sum test, or Kruskal–Wallis test. The observed duration was calculated from the diagnosis to the point of the last follow-up between April 2009 and March 2019. Supplementary Figure S1. Overview of enrolled patients. IIP, idiopathic interstitial pneumonia; ILD, interstitial lung disease; CTD, connective tissue disease; UCTD, undifferentiated connective tissue disease; IPAF, interstitial pneumonia with autoimmune features. Supplementary Figure S2. Kaplan–Meier survival curves of IPAF, CTD-ILD, and IIP. Interstitial pneumonia with autoimmune features (IPAF) (n = 52), CTD-ILD (n = 92), and idiopathic pulmonary fibrosis (IIP) (n = 174). IPAF vs. CTD; P = 0.61, IPAF vs. IIP; P = 0.0005, IIP vs. CTD; P = 0.0001 (PDF 121 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Murata, O., Suzuki, K., Takeuchi, T. et al. The Risk Factors of Exacerbation in Interstitial Pneumonia With Autoimmune Features: A Single-Center Observational Cohort Study. Rheumatol Ther 8, 1693–1710 (2021). https://doi.org/10.1007/s40744-021-00371-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-021-00371-3