
Abstract
Rheumatoid arthritis (RA) is an articular disease with extra-articular manifestations. Pulmonary manifestations are not uncommon and can involve all compartments of the lungs with airway disease in the form of bronchiectasis or bronchiolitis, interstitial lung disease (ILD), pleural effusions and parenchymal lung nodules. The pulmonary features may present synchronously or after the articular disease, but, importantly, it may be the first presentation in 10% of patients in the absence of articular symptoms. Here we discuss the pathogenesis of RA lung involvement, particularly interstitial lung disease and bronchiectasis, focusing on the role anti-CCP antibodies (ACPAs). We highlight the complex interplay among genetic, environmental and immune factors. Furthermore, we explore the relationship of citrullination and smoking as well as the concept of interstitial pneumonia with autoimmune features (IPAF), where patients do not have evidence of another known cause of interstitial pneumonia and have incomplete features of connective tissue disease (CTD). We surmise that the frequency and titers of rheumatoid factor (RF) and ACPAs are increased in bronchiectasis and RA-bronchiectasis compared to RA patients without lung disease. ACPA is associated with more severe disease in both RA-ILD and RA-bronchiectasis even in the absence of articular symptoms. There is no clear prediction of development of articular RA with high ACPA levels in the context of positive ACPA and ILD; however, in RA-bronchiectasis, patients with positive antibodies can develop RA within a year after diagnosis of bronchiectasis. Though the primary focus of this narrative is to highlight the role of ACPA in pathogenesis and clinical practice, we also discuss the current treatment options and trials in RA-ILD and RA-bronchiectasis. Currently, there are no clear treatment guidelines. The treatments are now focusing on using a combination of immunosuppression and antifibrotic agents. Combination treatment targets both the fibrotic and inflammatory components of the disease process. Further studies are needed to identify the use of ACPA as a biomarker to tailor the treatment in RA-ILD and RA-bronchiectasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
There is significant disease burden in RA-ILD and RA-bronchiectasis. A closer look at the pathogenesis is needed to guide clinical management |
What did the study ask? |
We aimed to investigate the pathogenesis of RA-ILD and RA-bronchiectasis concentrating on the role of autoimmunity and ACPA |
What was learned from the study? |
(1) The pathogenesis of RA-ILD and RA-bronchiectasis is complex with MUC5B gene polymorphism playing a potential role in both conditions |
(2) Current studies suggest ACPA levels can predict the severity of lung disease in both RA-ILD and bronchiectasis, and often this predates the articular manifestations |
Study outcome |
More research is needed to identify ACPA as a biomarker/phenotype to personalize treatment in bronchiectasis and ILD associated with RA. Currently there are no clear treatment pathways, but an MDT approach is advised |
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune inflammatory disease with a global prevalence of approximately 0.46% that presents with mostly articular but also extraarticular manifestations [1]. Pulmonary manifestations of rheumatoid arthritis include pulmonary nodules, pleural effusions, bronchiolitis, bronchiectasis and interstitial lung disease (ILD) [2]. The latter two are the most common pulmonary manifestations and in 10% of cases can be the first presentation of RA [3]. In some cases, patients may present with respiratory symptoms and positive antibodies but without articular features of rheumatoid arthritis [4], while in a minority of cases, the ILD may precede the development of anti-citrullinated peptide antibodies (ACPA) by several years [4, 5]. In this review article, we provide an update on the pathogenesis of rheumatoid arthritis-associated bronchiectasis (RA-bronchiectasis) and ILD, including the role of ACPA [6].
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Pathogenesis of RA-ILD and RA-Bronchiectasis
Pathogenesis of RA-ILD
Though the exact etiology of RA -associated ILD (RA-ILD) is not known, several risk factors have been identified. These include cigarette smoking, ACPA, genetic variants, including HLA alleles, male gender, > 60 years of age, duration of RA 0–5 years and high LDH. No correlation has been observed between standard disease-modifying antirheumatoid drugs (DMARDS) and ILD [7, 8].
All the known histological interstitial pneumonia patterns are seen in RA, on high-resolution CT (HRCT) scan or histology, often with evidence of multicompartment involvement including airways, pleura or vasculature. Lung biopsies are rarely performed in RA-ILD, as CT patterns provide prognostic separation. The pattern most frequently observed is usual interstitial pneumonia (UIP) (on CT characterized by peripheral distribution of reticulation, with traction bronchiectasis with/without honeycombing, and on histology patchy fibrosis, microscopic honeycombing and fibroblastic foci), where fibrosis dominates over inflammation. Non-specific interstitial pneumonitis (NSIP) is the second most common pattern, characterized by a homogeneous distribution of fibrosis admixed with inflammation. More inflammatory patterns, including organizing pneumonia and lymphocytic interstitial pneumonia, are observed less frequently. A CT UIP pattern has the worst prognosis, with an idiopathic pulmonary fibrosis (IPF)-like survival in patients with extensive disease on CT. Features inconsistent with UIP are associated with a more favorable prognosis [9]. Features of small airway involvement are manifested on CT by the presence of a mosaic attenuation pattern and are often seen in RA-ILD [9]. Finally, concomitant emphysema is seen in a subset of RA-ILD patients and is associated with a UIP pattern and a worse outcome. Jacob et al. report a prevalence of emphysema in 27% of never smokers with RA-ILD, suggesting shared pathways, possibly autoimmune, between ILD and emphysema in RA [9].
Recently, there has been renewed interest in the role of ACPAs in disease pathogenesis. The presence of citrullinated peptides in the serum and synovial fluid in RA with high specificity and the facts that ACPAs can develop 3–5 years before the onset of arthritis [10] and that they are often related to more severe disease have suggested a key role in the pathogenesis of RA [11].
The lung is considered a potential site of initiation in ACPA-positive RA [12], possibly triggered by processes such as smoking and microbial dysbiosis [13,14,15]. Furthermore, IgA and IgG RA-associated antibodies are found in the sputum of individuals at high risk of RA (e.g., with a first-degree relative with RA) even in the absence of serum ACPA or RF. Through loss of integrity of the mucosal barrier, these antibodies may subsequently leak into the systemic circulation [16, 17].
The citrullination process converts arginine to citrulline, which results in an immune response. The process relies on the extracellular release of peptidyl-arginine deiminase 4 (PAD4) by neutrophils or macrophages. The full range of recognized citrullinated proteins is not known. ACPA can be present in other diseases, including systemic lupus erythematosus (SLE) (16.6%), ankylosing spondylitis (4%), psoriatic spondyloarthropathy (PSA) (10.6%), unclassified rheumatism (33%) and tuberculosis [18, 19]. Higher ACPA titers are associated with an increased prevalence of ILD, even when corrected for confounders such as RA and smoking [20]. Approximately half of individuals with RA-ILD have evidence of citrullinated proteins on examination of their lung tissue [21]. Interestingly, the proportion with evidence of citrullination with idiopathic interstitial pneumonia is similar to that with RA-ILD and double that seen in control populations [21].
Some patients with RA have a genetic predisposition to developing ILD. The presence of HLA-B54, HLADQ1B*0601, HLA-B40 and HLA-DR4 is associated with increased risk of ILD [22]. The MUC5B gene encodes mucin 5B, a macromolecule glycoprotein secreted by airway cells in the airway mucus barrier. A promoter polymorphism of this gene, rs37505950, associated with increased MUC5B expression, confers the greatest risk of developing IPF [23]. MUC5B is overexpressed in the distal airways and honeycomb cysts of IPF lungs [24]. Interestingly, the MUC5B promoter variant is associated with the development of RA-ILD, and specifically with a UIP pattern, but is not a risk factor for the development of RA per se [25]. The mechanisms underlying the contribution of the MUC5B polymorphism to the pathogenesis of IPF and RA-UIP are not known, although aberrant mucociliary clearance in the small airways may be associated with a more detrimental impact of inhalational exposures such as smoking, known to be strongly associated with both IPF and RA-ILD [26]. Research is ongoing so that this genotype may guide better understanding of the pathogenesis and management of this condition.
Cigarette smoking is an independent risk factor in the development of RA and ILD. It may play a role in inducing antibody formation and has been linked to higher titers of rheumatoid factor. Smoking may also play a specific role in RA-ILD by promoting citrullination of lung proteins by increasing PAD 4 levels, thus leading to the development of ACPA [27,28,29]. This seems to be especially the case for individuals who have the shared epitope HLA-DRB1. Male sex has also been identified as a risk factor for RA-ILD, although the exact reason is not understood [22]. A study has found that the proportion of smokers and non-smokers with ACPA antibodies was not statistically different but smokers did have higher levels of ACPA and more severe diseases with poorer response to treatments [30].
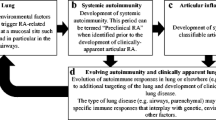
Overall, there is a complex interplay among immune, genetic and environmental factors contributing to the pathogenesis of RA ILD (Fig. 1) [22].

Pathogenesis of RA-ILD
Pathogenesis of RA-Bronchiectasis
Bronchiectasis is defined as the irreversible dilatation and damage of the bronchi [31]. It is associated with rheumatoid arthritis and can be either isolated or secondary to traction, as an expression of surrounding fibrosis, often seen in ILD. Although traction bronchiectasis is not considered to be associated with a productive cough and suppurative lung disease, in clinical practice this is seen, particularly when individuals are immunosuppressed and have bacterial colonization [32]. The prevalence of symptomatic free-standing bronchiectasis with RA is between 2 and 12% [33] but asymptomatic disease prevalent on a HRCT is much higher, between 30 and 50% [34]. Interestingly, bronchiectasis may precede articular manifestations of RA [35] but is most often seen as a delayed complication of RA, possibly related to immunosuppression predisposing to recurrent infection [36]. However, the fact that patients with other CTDs such as systemic sclerosis or anti-synthetase syndrome-associated lung involvement only very rarely develop free standing bronchiectasis makes this less likely as the main mechanism.
Of note, small airway involvement (bronchiolitis), even in the absence of free-standing bronchiectasis, is common in RA and is observed on chest high-resolution computed tomography (HRCT) in almost two-thirds of patients, with or without associated ILD [37].
The exact pathogenesis of RA-bronchiectasis remains poorly understood. In addition to the vicious cycle hypothesis of chronic infection leading to recurrent inflammation and airway damage (Fig. 2), risk factors such as positive autoantibodies (e.g., RF and ACPA), CFTR mutations (F508Del) and HLA associations (particularly HLADQB1) have been identified [38, 39].

Cole’s vicious cycle hypothesis
The role of chronic bacterial infections in inducing autoimmunity in bronchiectasis alone and RA-associated bronchiectasis has also been explored. The frequency and titers of RF and ACPA are increased in bronchiectasis and RA-bronchiectasis compared to RA patients without lung disease, after adjusting for smoking history [40, 41]. Furthermore, citrulline specificity of ACPA was increased approximately two-fold in RA-bronchiectasis compared to bronchiectasis alone [40]. Overall, this suggests that presence of these antibodies in bronchiectasis may potentiate the exacerbation of the autoimmune response in RA even before the articular manifestations develop.
The Bronchiectasis, Asthma, Control, Rheumatoid Arthritis (BRAC) study prospectively looked at the relationship between bronchiectasis and RA antibodies. Comparison was made among three groups of patients with bronchiectasis, asthma and a group of controls [40]. RF positivity was 25%, 16% and 10.3%, respectively, in each group. In the RF-positive bronchiectasis group, 13% (4 patients) were also ACPA positive compared to no ACPA identified in the asthma/control groups. The ACPA response in patients with bronchiectasis was not citrulline specific, suggesting epitope spreading to citrullinated peptides in patients who subsequently develop RA. Importantly, half of the individuals with strongly positive ACPA subsequently developed RA within 12 months. This supports the role of autoimmunity in some individuals with bronchiectasis and subsequent development of RA-bronchiectasis. A multicenter randomized control trial [42] has identified RA-bronchiectasis overlap to be an independent risk factor for mortality (28% in RA-bronchiectasis overlap vs. 18% in RA alone and 9% in bronchiectasis alone over a period of 48 months). These findings suggest that antibody positivity in RA-bronchiectasis may be associated with a poorer prognosis, but further randomized control trials are needed to compare mortality and prognosis with non-antibody-positive RA-bronchiectasis.
Genetic predisposition to RA-bronchiectasis has been investigated, and the role of cystic fibrosis transmembrane regulator (CFTR) mutations has been identified. In a small French study, a heterozygous CFTR mutation in F508Del was identified in 15.4% of patients with RA-bronchiectasis versus none in the RA or bronchiectasis groups individually [43].
In a family-based study by the same group [44], the frequency of general CFTR mutations was higher in family members with RA-bronchiectasis or bronchiectasis only compared to unaffected relatives and to unrelated healthy controls but not to family members with RA only, suggesting an association between CFTR mutations with bronchiectasis and RA. CFTR mutations were five-fold more frequent in family members with RA-associated bronchiectasis than in those with RA only [44]. Similarly to RA-ILD, certain HLA alleles have been linked with RA-associated bronchiectasis. These include DQB1*0601, DQB1*0301, DQB1*0201 and DQA1*0501 [45]. Their clinical significance is not yet clear and requires further research.
Role of ACPA in Clinical Practice
ACPAs have been shown to develop up to 14 years before the onset of clinical RA [10]. However, there are no clear predictors as to who will develop clinical manifestations of disease or when these features will be detected. In a group of patients with positive ACPA and connective tissue disease, 6.1% developed RA over 8.9 years and titers were higher in patients with a diagnosis of RA compared to the non-RA/CTD group [46]. The presence of ACPA in the context of RA is important as it can be used as a marker of prognosis for erosions and disease severity [47]. In clinical practice, when ACPAs are detected in individuals with only ILD or bronchiectasis, it is unclear whether antibody levels correlate with pulmonary disease severity or if the treatment strategy needs to be altered. A research group first explored this in 2012 in a group of subjects with lung disease and autoimmune antibodies but no extrapulmonary manifestations of autoimmune disease. These included individuals with isolated airway disease, isolated ILD, mixed airways disease and ILD, and combined ILD and emphysema. There was no correlation between ACPA presence and HRCT changes in this study. In the cohort of patients with high titer ACPA, 9% of subjects developed articular rheumatoid arthritis over 1.2 years of median follow-up, while no patients in the medium or low ACPA titer groups developed RA [4]. Interestingly most patients in the total cohort had a smoking history, which as previously described is linked to ACPA. The presence of high ACPA titers is also associated with shared epitope alleles, higher inflammatory markers (IL-6 and c-reactive protein) and presence of rheumatoid nodules [48].
A recent systematic review surmised that individuals with RA-ILD have higher ACPA titers than those with RA alone. Higher ACPA titers are associated with more extensive lung involvement. The quality of evidence however was rated as low or very low [48,49,50]. Reticulation, honeycombing or traction bronchiectasis on CT scan seems to be two-fold more prominent in APCA-positive patients compared to those who were negative, suggesting that usual interstitial pneumonitis (UIP) is more common in those with higher ACPA levels. Furthermore, higher ACPA levels are also associated with worse lung function [48].
Interstitial pneumonia with autoimmune features (IPAF) has been suggested as a research entity to include those patients with ILD without evidence of another known cause of interstitial pneumonia and incomplete features of CTD [51]. To meet criteria for IPAF at least one feature of at least two out of three domains must be reached (serological, clinical, morphological). All the idiopathic interstitial pneumonia patterns except for UIP count as a feature in the morphologic domain, whereas in the presence of a UIP pattern, at least one feature in the clinical and serologic domains needs to be present, unless there is an additional morphologic feature [51]. IPAF criteria are met in up to 25% of ILD patients and will include patients presenting with ILD and ACPA but no articular manifestations of RA, provided there is an additional feature from the clinical or morphological domain if the pattern is UIP. Individuals with CTD-ILD are more likely female and with higher incidence of joint pain, sicca symptoms and Raynaud’s phenomenon; patients with IPAF are more likely have high Ro-52 titers and more honeycombing and pleural thickening on HRCT and to present primarily with respiratory symptoms [52]. Interestingly, a MUC5B minor allele is associated with a worse survival in the context of IPAF, whereas it has been associated with a better survival in IPF in some, but not all, studies [53].
The presence of ACPA in the context of lung disease may lead to intensification of the search for underlying RA and raises questions as to treatment choices, whether patients with ILD and ACPA should be treated as RA ILD rather than idiopathic pulmonary fibrosis/ILD. The treatment challenge lies in the fact that ILD can occur at any point in a patient's journey with RA. Treatment for the joints may not be effective for treating ILD and vice versa [6]. ACPA titers in the context of RA can be used to monitor disease activity. Up to 25% reduction in levels has been noted in half of the patients on treatment. Both rituximab and abatacept have been shown to reduce ACPA levels in responders in RA patients [54, 55]. Some reports have noted this with anti-TNF therapy but not consistently. The usefulness of assessing ACPA levels in the context of RA-ILD is less well understood, and further research is warranted.
Treatment of RA-ILD and RA-Bronchiectasis
Pharmacological Interventions in RA-ILD
There are no treatment guidelines for the management of RA-ILD. Current evidence is based on retrospective data and small studies. Corticosteroids have been shown to improve lung function, particularly in those with NSIP, but their use increases the risk of infection at doses > 10 mg [53, 55, 56]. In the context of a UIP pattern, it is unclear whether immunosuppression may have similar adverse outcomes as seen in the PANTHER trial for IPF [53]. On the other hand, Song et al. [55] describe a small retrospective series where half of patients with RA-UIP treated with immunosuppression either improved or had stable lung function. Recent large case control studies have suggested that treatment with methotrexate may delay the onset of ILD in patients with RA [57].
Cyclophosphamide and mycophenolate have been investigated with RCTs in systemic sclerosis-associated ILD, with MMF having a better side effect profile compared to cyclophosphamide [55, 56, 58,59,60]. Azathioprine is also an option [56, 61]. However, none of these immunosuppressants have been evaluated in controlled clinical trials in RA-ILD. There are conflicting data regarding the safety of anti-TNF therapy, with concerns about its association with acute exacerbations of the ILD. Retrospective studies have suggested abatacept as a safe treatment for RA-ILD, although controlled prospective trials are needed; one is currently ongoing (APRIL Trial) [6, 62, 63] (Table 1). Rituximab has been used for the treatment of RA-ILD, although rituximab-induced pulmonary toxicity is a concern and needs to be evaluated prospectively [64]. Additionally, rituximab results in depletion of B cells, secondary immunodeficiency, recurrent infections and de novo bronchiectasis [65,66,67]. Tofacitinib has been noted to be associated with a low incidence of RA-ILD (0.18 per 100 patient-years) and is used to treat articular manifestations. It may therefore be a good treatment option in the future. Trials are ongoing comparing tofacitinib versus methotrexate in the treatment of ILD [55, 68] (Table 1).
Antifibrotic agents (e.g., nintedanib and pirfenidone) remain a treatment possibility in this cohort. As described, there is considerable overlap between IPF/IPAF and RA-ILD with the MUC5B gene being a potential link. Both are more frequent in older male patients with a smoking history. Treating both the fibrotic and inflammatory components may provide additional benefit; therefore, several trials are now underway looking at the safety and efficacy of these agents in RA-ILD. Efficacy data are available for nintedanib from the INBUILD trial. A significant reduction in the rate of FVC decline was seen across ILD entities, including RA-ILD, which had progressed despite conventional treatment [6, 69]. The RCT TRAIL 1 using pirfenidone in treatment of RA-ILD is still underway [53, 70] (Table 1).
RA-ILD has a 10-year mortality of 60% [71] promoting the need to “treat to target” as we do with patients with florid synovitis. It is unclear what the future holds, but it is likely that treatment regimens will include antifibrotic agents alongside immunosuppressive medication in selected subgroups. Treatment will require an MDT approach and will need to be individualized based on the patient demographics and pattern of disease involvement as well as comorbidities. The challenge of treating both articular and pulmonary symptoms remains with few agents having an impact on both.
Pharmacological Interventions in RA-Bronchiectasis
Research is needed in this area to improve management of RA-bronchiectasis. The identification of ACPA is useful to phenotype individuals with the hope of future personalization of bronchiectasis treatments. It is plausible that systemic inflammation can drive bronchiectasis progression and severity and that suppressing RA-related systemic inflammation could attenuate this. In contrast, airway inflammation is thought to be a trigger for citrullination; optimal control of bronchiectasis-related airway inflammation may attenuate antibody formation. However, targeting both airway and/or systemic inflammation may be associated with bacterial colonization, overgrowth and infections that further drive bronchiectasis pathogens. Bronchiectasis management in the context of associated RA involves interventions aimed at breaking the cycle in Cole’s vicious cycle hypothesis. Infective exacerbations are treated with 10–14-day courses of antibiotics. Azithromycin is used for its immunomodulatory properties in those with frequent exacerbations. In the presence of bacterial colonization and frequent exacerbations, prophylactic oral or nebulized antibiotics are often used. Individuals with RA-bronchiectasis are often receiving DMARDs and biological therapies that suppress immune function, which results in frequent infections. The impact of biological therapies on respiratory infection risk has been reviewed elsewhere [50]. Prophylactic antibiotics are useful but in some cases modification of immunosuppression therapy is needed. It is also important to evaluate individuals for the presence of secondary antibody deficiency as immunoglobulin replacement may be necessary.
Non-Pharmacological Interventions in RA-ILD and RA-Bronchiectasis
Regular airway clearance with physiotherapy techniques, airway adjuncts and mucolytics is the mainstay of treatment in bronchiectasis.
Annual influenza vaccination is recommended, and consideration should be given for pneumococcal vaccination with PCV13 followed by PPV23 2 months later at least 2 weeks prior initiation of immunosuppressive medication in both RA-ILD and RA-bronchiectasis [72]. As smoking is an important factor in the pathogenesis of both conditions, advising smoking cessation is recommended to all patients with RA-ILD and RA-bronchiectasis.
Conclusions
The current literature suggests that ACPA levels are higher in patients with RA-ILD and RA-bronchiectasis and associated with more severe lung disease. Patients with RA-bronchiectasis and positive ACPA may go on to develop articular manifestations of RA within a year of diagnosis. Higher ACPA titers do not predict the development of articular RA, but the presence of ACPA alone is a predictor of articular RA over several years. Although ACPA levels drop with some treatments, it is still unclear whether they can be used as biomarkers of lung disease activity. There are no clear treatment pathways for patients with RA-ILD, and on every occasion an MDT approach is advised.
Change history
27 September 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40744-021-00376-y
References
Almutairi K, Nossent J, Preen D, Keen H, Inderjeeth C. The global prevalence of rheumatoid arthritis: a meta-analysis based on a systematic review. Rheumatol Int. 2021;41(5):863–77.
Mackintosh JA, Stainer A, De Sadeleer LJ, Stock C, Wuyts WA, Renzoni EA. Pulmonary involvement in rheumatoid arthritis. In: Pulmonary manifestations of systemic diseases, vol. 86. Sheffield: European Respiratory Society; 2019. p 44–67.
Chan E, Chapman K, Kelly C. Interstitial lung disease in rheumatoid arthritis: a review. Arthritis Res Top Rev Ser. 2013;7:1–4.
Fischer A, Solomon JJ, du Bois RM, Deane KD, Olson AL, Fernandez-Perez ER, et al. Lung disease with anti-CCP antibodies but not rheumatoid arthritis or connective tissue disease. Respir Med. 2012;106(7):1040–7.
Demoruelle MK, Solomon JJ, Fischer A, Deane KD. The lung may play a role in the pathogenesis of rheumatoid arthritis. Int J Clin Rheumtol. 2014;9(3):295–309.
Cassone G, Manfredi A, Vacchi C, Luppi F, Coppi F, Salvarani C, et al. Treatment of rheumatoid arthritis-associated interstitial lung disease: lights and shadows. J Clin Med. 2020;9(4):1082.
Li L, Liu R, Zhang Y, Zhou J, Li Y, Xu Y, et al. A retrospective study on the predictive implications of clinical characteristics and therapeutic management in patients with rheumatoid arthritis-associated interstitial lung disease. Clin Rheumatol. 2020;39(5):1457–70.
Choi W-I, Dauti S, Kim HJ, Park SH, Park JS, Lee CW. Risk factors for interstitial lung disease: a 9-year Nationwide population-based study. BMC Pulm Med [Internet]. 2018;18(1):1–7.
Jacob J, Hirani N, van Moorsel CHM, Rajagopalan S, Murchison JT, van Es HW, et al. Predicting outcomes in rheumatoid arthritis related interstitial lung disease. Eur Respir J. 2019;53(1):1800869.
Nielen MMJ, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MHMT, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50(2):380–6.
Kurowska W, Kuca-Warnawin EH, Radzikowska A, Maśliński W. The role of anti-citrullinated protein antibodies (ACPA) in the pathogenesis of rheumatoid arthritis. Cent J Immunol. 2017;42(4):390–8.
Perry E, Kelly C, Eggleton P, De Soyza A, Hutchinson D. The lung in ACPA-positive rheumatoid arthritis: an initiating site of injury? Rheumatology (Oxford). 2014;53(11):1940–50.
Hensvold AH, Magnusson PKE, Joshua V, Hansson M, Israelsson L, Ferreira R, et al. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: an epidemiological investigation in twins. Ann Rheum Dis. 2015;74(2):375–80.
Catrina AI, Deane KD, Scher JU. Gene, environment, microbiome and mucosal immune tolerance in rheumatoid arthritis. Rheumatology (Oxford). 2016;55(3):391–402.
Scher JU, Joshua V, Artacho A, Abdollahi-Roodsaz S, Öckinger J, Kullberg S, et al. The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome. 2016;4(1):60.
Holers VM, Demoruelle MK, Kuhn KA, Buckner JH, Robinson WH, Okamoto Y, et al. Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction. Nat Rev Rheumatol. 2018;14(9):542–57.
Jorgensen C, Moynier M, Bologna C, Youinou P, Sany J. Rheumatoid factor associated with a secretory component in rheumatoid arthritis. Br J Rheumatol. 1995;34(3):236–40.
Payet J, Goulvestre C, Bialé L, Avouac J, Wipff J, Job-Deslandre C, et al. Anticyclic citrullinated peptide antibodies in rheumatoid and nonrheumatoid rheumatic disorders: experience with 1162 patients. J Rheumatol. 2014;41(12):2395–402.
Ariza-Prota M, Pando-Sandoval A, García-Clemente M, Casan P. Poncet’s disease mimicking rheumatoid arthritis in a patient with suspected Crohn’s disease. Clin Case Rep. 2016;4:72–5.
Correia CS, Briones MR, Guo R, Ostrowski RA. Elevated anti-cyclic citrullinated peptide antibody titer is associated with increased risk for interstitial lung disease. Clin Rheumatol. 2019;38(4):1201–6.
Bongartz T, Cantaert T, Atkins SR, Harle P, Myers JL, Turesson C, et al. Citrullination in extra-articular manifestations of rheumatoid arthritis. Rheumatology (Oxford). 2007;46(1):70–5.
Shaw M, Collins BF, Ho LA, Raghu G. Rheumatoid arthritis-associated lung disease. Eur Respir Rev. 2015;24(135):1–16.
Helling BA, Gerber AN, Kadiyala V, Sasse SK, Pedersen BS, Sparks L, et al. Regulation of MUC5B expression in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2017;57(1):91–9.
Conti C, Montero-Fernandez A, Borg E, Osadolor T, Viola P, De Lauretis A, et al. Mucins MUC5B and MUC5AC in distal airways and honeycomb spaces: comparison among idiopathic pulmonary fibrosis/usual interstitial pneumonia, fibrotic nonspecific interstitial pneumonitis, and control lungs. Am J Respir Crit Care Med. 2016;193:462–4.
Juge P-A, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N Engl J Med. 2018;379(23):2209–19.
Michalski JE, Schwartz DA. Genetic risk factors for idiopathic pulmonary fibrosis: insights into immunopathogenesis. J Inflamm Res. 2020;13:1305–18.
Tuomi T, Heliövaara M, Palosuo T, Aho K. Smoking, lung function, and rheumatoid factors. Ann Rheum Dis [Internet]. 1990;49(10):753–6.
Källberg H, Ding B, Padyukov L, Bengtsson C, Rönnelid J, Klareskog L, et al. Smoking is a major preventable risk factor for rheumatoid arthritis: estimations of risks after various exposures to cigarette smoke. Ann Rheum Dis. 2011;70(3):508–11.
Makrygiannakis D, Hermansson M, Ulfgren A-K, Nicholas AP, Zendman AJW, Eklund A, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis. 2008;67(10):1488–92.
Chirea G, Sarbu I, Mangaloiu D. Smoking habits and anti-CCP antibodies in patients with Rheumatoid Arthritis. Gabriela Chirea. Eur J Publ Heal. 2017;27(suppl_3):ckx186.046.
José RJ, Brown JS. Bronchiectasis. Br J Hosp Med (Lond). 2014;75(Suppl 1):C146–51.
José RJ, Manuel A, Gibson-Bailey K, Lee L. Post COVID-19 bronchiectasis: a potential epidemic within a pandemic. Expert Rev Respir Med. 2020;14:1183–4.
Allain J, Saraux A, Guedes C, Valls I, Devauchelle V, Le Goff P. Prevalence of symptomatic bronchiectasis in patients with rheumatoid arthritis. Rev Rhum Engl Ed. 1997;64(10):531–7.
Despaux J, Manzoni P, Toussirot E, Augé B, Cedoz JP, Wendling D. Prospective study of the prevalence of bronchiectasis in rheumatoid arthritis using high-resolution computed tomography. Rev Rhum Engl Ed [Internet]. 1998;65(7–9):453–61.
Despaux J, Polio JC, Toussirot E, Dalphin JC, Wendling D. Rheumatoid arthritis and bronchiectasis. A retrospective study of fourteen cases. Rev Rhum Engl Ed. 1996;63(11):801–8.
Shadick NA, Fanta CH, Weinblatt ME, O’Donnell W, Coblyn JS. Bronchiectasis. A late feature of severe rheumatoid arthritis. Medicine (Baltimore). 1994;73(3):161–70.
Lieberman-Maran L, Orzano IM, Passero MA, Lally EV. Bronchiectasis in rheumatoid arthritis: report of four cases and a review of the literature—implications for management with biologic response modifiers. Semin Arthritis Rheum. 2006;35(6):379–87.
Duarte AC, Porter J, Leandro MJ. Bronchiectasis in rheumatoid arthritis. A clinical appraisial. Jt Bone Spine. 2020;87(5):419–24.
Quirke A-M, Perry E, Cartwright A, Kelly C, De Soyza A, Eggleton P, et al. Bronchiectasis is a model for chronic bacterial infection inducing autoimmunity in rheumatoid arthritis. Arthritis Rheumatol (Hoboken, NJ). 2015;67(9):2335–42.
Perry E, Eggleton P, De Soyza A, Hutchinson D, Kelly C. Increased disease activity, severity and autoantibody positivity in rheumatoid arthritis patients with co-existent bronchiectasis. Int J Rheum Dis. 2017;20(12):2003–11.
De Soyza A, McDonnell MJ, Goeminne PC, Aliberti S, Lonni S, Davison J, et al. Bronchiectasis Rheumatoid Overlap Syndrome is an independent risk factor for mortality in patients with bronchiectasis: A Multicenter Cohort Study. Chest. 2017;151(6):1247–54.
Puéchal X, Fajac I, Bienvenu T, Desmazes-Dufeu N, Hubert D, Kaplan JC, et al. Increased frequency of cystic fibrosis deltaF508 mutation in bronchiectasis associated with rheumatoid arthritis. Eur Respir J. 1999;13(6):1281–7.
Puéchal X, Bienvenu T, Génin E, Berthelot J-M, Sibilia J, Gaudin P, et al. Mutations of the cystic fibrosis gene in patients with bronchiectasis associated with rheumatoid arthritis. Ann Rheum Dis. 2011;70(4):653–9.
Hillarby MC, McMahon MJ, Grennan DM, Cooper RG, et al. HLA associations in subjects with rheumatoid arthritis and Bronchiectasis but not with other pulmonary complications of rheumatoid disease. Rheumatology. 1993;32(9):794–7.
Burska AN, Hunt L, Boissinot M, Strollo R, Ryan BJ, Vital E, et al. Autoantibodies to posttranslational modifications in rheumatoid arthritis. Mediat Inflamm. 2014;2014:492873.
Meyer O, Labarre C, Dougados M, Goupille P, Cantagrel A, Dubois A, et al. Anticitrullinated protein/peptide antibody assays in early rheumatoid arthritis for predicting five year radiographic damage. Ann Rheum Dis. 2003;62(2):120–6.
Giles JT, Danoff SK, Sokolove J, Wagner CA, Winchester R, Pappas DA, et al. Association of fine specificity and repertoire expansion of anticitrullinated peptide antibodies with rheumatoid arthritis associated interstitial lung disease. Ann Rheum Dis. 2014;73(8):1487–94.
Rocha-Muñoz AD, Ponce-Guarneros M, Gamez-Nava JI, Olivas-Flores EM, Mejía M, Juárez-Contreras P, et al. Anti-cyclic citrullinated peptide antibodies and severity of interstitial lung disease in women with rheumatoid arthritis. J Immunol Res. 2015;2015:151626.
Kamiya H, Panlaqui OM. Systematic review and meta-analysis of the risk of rheumatoid arthritis-associated interstitial lung disease related to anti-cyclic citrullinated peptide (CCP) antibody. BMJ Open. 2021;11(3): e040465.
Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. 2015;46(4):976–87.
Tian M, Huang W, Ren F, Luo L, Zhou J, Huang D, et al. Comparative analysis of connective tissue disease-associated interstitial lung disease and interstitial pneumonia with autoimmune features. Clin Rheumatol. 2020;39(2):575–83.
Newton CA, Oldham JM, Ley B, Anand V, Adegunsoye A, Liu G, et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur Respir J. 2019;53(4):1801641.
Raghu G, Anstrom KJ, King TEJ, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–77.
Taylor P, Gartemann J, Hsieh J, Creeden J. A systematic review of serum biomarkers anti-cyclic citrullinated peptide and rheumatoid factor as tests for rheumatoid arthritis. Cutolo M, editor. Autoimmune Dis [Internet]. 2011;2011:815038.
Song JW, Lee H-K, Lee CK, Chae EJ, Jang SJ, Colby TV, et al. Clinical course and outcome of rheumatoid arthritis-related usual interstitial pneumonia. Sarcoidosis Vasc Diffus Lung Dis Off J WASOG. 2013;30(2):103–12.
Zamora-Legoff JA, Krause ML, Crowson CS, Ryu JH, Matteson EL. Risk of serious infection in patients with rheumatoid arthritis-associated interstitial lung disease. Clin Rheumatol. 2016;35(10):2585–9.
Juge P-A, Lee JS, Lau J, Kawano-Dourado L, Rojas Serrano J, Sebastiani M, et al. Methotrexate and rheumatoid arthritis associated interstitial lung disease. Eur Respir J. 2021;57(2):2000337.
Zhang G, Xu T, Zhang H, Ye S, Wang Q, Zhang L, et al. Randomized control multi-center clinical study of mycophenolate mofetil and cyclophosphamide in the treatment of connective tissue disease related interstitial lung disease. Zhonghua Yi Xue Za Zhi. 2015;95(45):3641–5.
Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354(25):2655–66.
Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4(9):708–19.
Kelly C, Young A, Ahmad Y, Dawson J, Carty S, Nisar M, et al. 092 The effect of steroids, azathioprine and mycophenolate on the risk of death in rheumatoid lung disease. Rheumatology [Internet]. 2016;55(suppl_1):99–100.
Fernández-Díaz C, Loricera J, Castañeda S, López-Mejías R, Ojeda-García C, Olivé A, et al. Abatacept in patients with rheumatoid arthritis and interstitial lung disease: a national multicenter study of 63 patients. Semin Arthritis Rheum. 2018;48(1):22–7.
Cassone G, Manfredi A, Atzeni F, Venerito V, Vacchi C, Picerno V, et al. Safety of abatacept in Italian patients with rheumatoid arthritis and interstitial lung disease: a multicenter retrospective study. J Clin Med. 2020;9(1):277.
Franzen D, Ciurea A, Bratton DJ, Clarenbach CF, Latshang TD, Russi EW, et al. Effect of rituximab on pulmonary function in patients with rheumatoid arthritis. Pulm Pharmacol Ther. 2016;37:24–9.
José RJ, Hall J, Brown JS. De novo bronchiectasis in haematological malignancies: patient characteristics, risk factors and survival. ERJ Open Res. 2019;5(4):00166–2019.
José R, Mouyis M. Biological therapies in the treatment of inflammatory disease and cancer: impact on pulmonary infection. Ann Res Hosp. 2017;1:1.
Santos VA, Tobón GJ, Cañas CA. Development of bronchiectasis during long-term rituximab treatment for rheumatoid arthritis. Adv Respir Med. 2018. https://doi.org/10.5603/ARM.a2018.0050.
Citera G, Mysler E, Madariaga H, Cardiel MH, Castañeda O, Fischer A, et al. Incidence Rates of Interstitial Lung Disease Events in Tofacitinib-Treated Rheumatoid Arthritis Patients: Post Hoc Analysis From 21 Clinical Trials. J Clin Rheumatol. 2020. https://doi.org/10.1097/rhu.0000000000001552. PMID: 32826657.
Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718–27.
Solomon JJ, Danoff SK, Goldberg HJ, Woodhead F, Kolb M, Chambers DC, et al. The design and rationale of the trail1 trial: a randomized double-blind phase 2 clinical trial of pirfenidone in rheumatoid arthritis-associated interstitial lung disease. Adv Ther. 2019;36(11):3279–87.
Hyldgaard C, Hilberg O, Pedersen AB, Ulrichsen SP, Løkke A, Bendstrup E, et al. A population-based cohort study of rheumatoid arthritis-associated interstitial lung disease: comorbidity and mortality. Ann Rheum Dis. 2017;76(10):1700–6.
Froneman C, Kelleher P, José RJ. Pneumococcal vaccination in immunocompromised hosts: an update. MDPI AG. 2021;9(6):536.
Acknowledgements
Thanks to William Henderson of Bedfordshire Hospitals Library and Knowledge Service with his help in the literature search.
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
MM and RJ contributed to the conception and design of the article as well as text and editing. TK helped to draft and edit the manuscript. ER overviewed the drafts, reviewed the concept and provided great insight and expertise.
Disclosures
Tanjila Khan, Ricardo J. Jose and Maria Mouyis have nothing to disclose. Elisabetta Renzoni has received lecture fees paid to the Institution by Roche and Boehringer and Roche.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Khan, T., Jose, R.J., Renzoni, E.A. et al. A Closer Look at the Role of Anti-CCP Antibodies in the Pathogenesis of Rheumatoid Arthritis-Associated Interstitial Lung Disease and Bronchiectasis. Rheumatol Ther 8, 1463–1475 (2021). https://doi.org/10.1007/s40744-021-00362-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-021-00362-4