
Abstract
Introduction
To evaluate long-term efficacy of once-daily baricitinib 2 mg in patients with active rheumatoid arthritis who had an inadequate response (IR) to conventional synthetic disease-modifying antirheumatic drugs (csDMARD) or biologic DMARDs (bDMARD).
Methods
Data from patients treated with baricitinib 2 mg daily in two 24-week, phase III studies, RA-BUILD (csDMARD-IR; NCT01721057) and RA-BEACON (bDMARD-IR; NCT01721044), and one long-term extension study (RA-BEYOND; NCT01885078), were analyzed (120 weeks). The main outcomes were achievement of low-disease activity (LDA; Simple Disease Activity Index [SDAI] ≤ 11), clinical remission (SDAI ≤ 3.3), Health Assessment Questionnaire Disability Index (HAQ-DI) ≤ 0.5 and improvement from baseline of ≥ 0.22, and safety. Analysis populations included (1) all patients and (2) never-rescued patients. Completer and non-responder imputation (NRI) analyses were conducted on each population.
Results
In RA-BUILD, 684 were randomized (229 to baricitinib 2 mg, 180 of whom completed RA-BUILD and entered RA-BEYOND). In RA-BEACON, 527 were randomized (174 to baricitinib 2 mg, 117 of whom completed RA-BEACON and entered RA-BEYOND). In RA-BUILD-BEYOND, 85.1% (63/74, completer) and 27.5% (63/229, NRI) of csDMARD-IR patients treated with baricitinib 2 mg achieved SDAI LDA; 40.5% (30/74, completer) and 13.1% (30/229, NRI) were in SDAI remission; 62.2% (46/74, completer) and 20.1% (46/229, NRI) had HAQ-DI ≤ 0.5 and 81.1% (60/74, completer); and 26.2% (60/229, NRI) achieved ≥ 0.22 change from baseline at week 120. In RA-BEACON-BEYOND, 86.5% (32/37, completer) and 18.4% (32/174, NRI) of bDMARD-IR patients treated with baricitinib 2 mg achieved SDAI LDA; 24.3% (9/37, completer) and 5.2% (9/174, NRI) were in SDAI remission; 50.0% (19/38, completer) and 10.9% (19/174, NRI) had HAQ-DI ≤ 0.5; and 73.7% (28/38, completer) and 16.1% (28/174, NRI) achieved ≥ 0.22 change from baseline at week 120. Rates of adverse events of special interest were consistent with previous reports.
Conclusions
Long-term treatment with baricitinib 2 mg demonstrated efficacy for up to 120 weeks and was well tolerated.
Trial registration
ClinicalTrials.gov identifier, NCT01721057, NCT01721044, and NCT01885078.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Baricitinib is a selective Janus kinase 1 and 2 inhibitor approved for the treatment of adults with moderately-to-severely active rheumatoid arthritis. |
Data reported in this manuscript are especially important to healthcare providers and patients making treatment decisions in Canada and the United States, where baricitinib 2 mg is the approved dose for the treatment of rheumatoid arthritis, and China, where baricitinib 2 mg and (recently) 4 mg are the approved doses. |
What did the study ask? |
This study evaluated the achievement and maintenance of low disease activity, remission, and a normative state of physical functioning in patients with active rheumatoid arthritis and an inadequate response to conventional synthetic disease-modifying antirheumatic drugs or biologic disease-modifying antirheumatic drugs treated with baricitinib 2 mg for up to 120 weeks. |
What has been learned from the study? |
Long-term treatment with baricitinib 2 mg daily demonstrated efficacy for up to 120 weeks and was well tolerated. |
A more comprehensive look at the safety and efficacy profile for baricitinib gives healthcare providers and patients crucial data to inform on the long-term use of this therapy for rheumatoid arthritis. |
Digital Features
This article is published with digital features, including a summary slide to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14555526.
Introduction
Rheumatoid arthritis (RA) is a systemic inflammatory autoimmune disease characterized by joint damage, loss of physical function, and progressive disability [1, 2]. RA is a chronic and progressive disease that can greatly impact a patient’s quality of life; therefore, there is a need for treatments that are both safe and efficacious in the long term.
Baricitinib is a selective Janus kinase 1 and 2 inhibitor [3] approved for the treatment of adult patients with moderately-to-severely active RA at the 2-mg dose in Canada and the United States, and China at the 2-mg and (recently) 4-mg doses. It is administered orally and provides a convenient and practical once-a-day treatment option for patients.
The short-term efficacy and safety of baricitinib have been demonstrated in multiple patient populations [4,5,6,7]. The efficacy and safety of baricitinib 2 mg and 4 mg were assessed in two 24-week clinical trials, RA-BUILD, which studied patients with an IR to conventional synthetic DMARDs (csDMARD) [6] and RA-BEACON, which studied patients with an IR to biologic DMARDs (bDMARD) [7].
These studies provided an initial safety and efficacy profile of baricitinib. In an integrated analysis of safety, incidence rates of death, adverse events leading to discontinuation, malignancies, major adverse cardiovascular events (MACE), serious infections, and deep vein thrombosis (DVT)/pulmonary embolism (PE) were consistent over time, with no increase observed with prolonged exposure [8, 9].
A more comprehensive look at the safety and efficacy profile for baricitinib will give healthcare providers and patients crucial information on the long-term use of this therapy for RA. This study evaluated the achievement and maintenance of low disease activity (LDA), remission, and normalization of physical functioning in csDMARD-IR and bDMARD-IR patients treated with baricitinib 2 mg for up to 120 weeks.
Methods
Study Design and Patients
RA-BUILD (NCT01721057) and RA-BEACON (NCT01721044) are completed phase III clinical studies that evaluated the efficacy and safety of baricitinib over 24 weeks in adults (≥ 18 years) with moderate-to-severely active RA [6, 7]. Patients enrolled in RA-BUILD had an IR or intolerance to ≥ 1 csDMARD (including methotrexate) and had not previously been treated with a bDMARD. Patients enrolled in RA-BEACON had an IR to prior treatment with ≥ 1 tumor necrosis factor (TNF) inhibitor and were on stable doses of concomitant conventional DMARD therapy.
RA-BEYOND (NCT01885078) is an ongoing, phase III long-term extension (LTE) study to assess the efficacy and safety of baricitinib in patients who completed RA-BUILD and RA-BEACON. Patients were not eligible for participation in RA-BEYOND if they demonstrated laboratory abnormalities or significant uncontrolled medical conditions that, in the opinion of the investigator, created additional risk with the administration of baricitinib.
Patients enrolled in the originating studies were initially randomized 1:1:1 to receive once-daily doses of placebo or baricitinib 2 mg or 4 mg added to any stable background therapies (Fig. 1). Rescue treatment (to baricitinib 4 mg) was assigned at week 16 for non-responders (patients whose tender joint counts [TJC] and swollen joint counts [SJC] had less than a 20% improvement from baseline at weeks 14 and 16). After week 16, rescue was at the discretion of the investigator based on TJCs and SJCs.

The treatment course is described for both originating studies. aPatients enrolled in RA-BUILD had an inadequate response or intolerance to ≥ 1 conventional synthetic disease-modifying antirheumatic drugs (DMARD) and had not previously been treated with a biologic DMARD; bPatients enrolled in RA-BEACON had an inadequate response to prior treatment with ≥ 1 tumor necrosis factor inhibitor and were on stable doses of concomitant conventional DMARD therapy; cRescue treatment (baricitinib 4 mg) was assigned at week 16 for non-responders (patients whose tender and swollen joint counts had improved by less than 20% from baseline at both week 14 and week 16). After week 16, rescue was at the discretion of the investigator based on tender and swollen joint counts. Patients who completed RA-BUILD or RA-BEACON and who were receiving baricitinib 2 mg and 4 mg continued blinded treatment in the long-term extension (LTE) study. Treatment was switched to open-label baricitinib 4 mg once patients were rescued or switched from placebo upon entry to the LTE study. Please note, the focus of this manuscript was the treatment course of baricitinib 2 mg, shown in light blue; dRescue therapy was available for patients with a Clinical Disease Activity Index score > 10 at 3 months or later following the study entry; eTotal duration of the ongoing LTE study, RA-BEYOND, is 7 years
Patients who completed RA-BUILD or RA-BEACON and who were receiving baricitinib 2 mg or 4 mg continued blinded treatment in RA-BEYOND. Treatment was switched to open-label baricitinib 4 mg once patients were rescued or switched from placebo upon entry to the LTE study. Rescue therapy (baricitinib 4 mg) was available in RA-BEYOND for patients with a Clinical Disease Activity Index (CDAI) score > 10 at 3 months or later following the LTE study entry.
Studies included in this analysis were conducted (RA-BUILD, RA-BEACON) or are being conducted (RA-BEYOND) in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice Guidelines and were approved by each center’s institutional review board (IRB) of ethics committee (Quorum Review IRB, #27258, #27259, and #28020). Written informed consent was provided by all patients.
Efficacy and Physical Function
Efficacy was assessed by the proportion of patients who achieved and maintained Simple Disease Activity Index (SDAI) LDA (SDAI ≤ 11) and remission (SDAI ≤ 3.3). Efficacy was assessed by SDAI because it reflects a stringent measure of disease activity and includes patient, physician, and inflammation (C-reactive protein) assessments. Normalization of physical function was assessed by the proportion of patients who reported scores that met or exceeded the population normative value of ≤ 0.5 based on the Health Assessment Questionnaire-Disability Index (HAQ-DI) as well as a minimum clinically important difference (MCID) of HAQ-DI, which is defined as an improvement from baseline ≥ 0.22. Rates of rescue and discontinuation (including reasons) were summarized.
Statistical Analyses
Efficacy and physical function analyses were based on two analysis populations: (1) the modified intention-to-treat (mITT) population (hereafter, referred to as “All Patients”) who were randomized to baricitinib 2 mg or placebo in the RA-BUILD and RA-BEACON studies and had received ≥ 1 dose of the study drug after randomization; data after rescue were set as missing and (2) all patients who were never rescued from baricitinib 2–4 mg at any time during the 120 weeks (hereafter, referred to as “Never Rescued” to reflect the population with clinical response to the dose of interest).
For each analysis population, two sets of post hoc analyses were conducted for the categorical measures (SDAI ≤ 11, SDAI ≤ 3.3, HAQ-DI ≤ 0.5, and HAQ-DI MCID improvement ≥ 0.22): (1) a non-responder imputation (NRI) analysis, which considered missing data (including data set as missing after rescue) as non-responders, and (2) a completer analysis based on patients who had non-missing data available on baricitinib 2 mg at the time of analysis. All analysis results are only descriptive with no statistical comparison. This approach is consistent with the European League Against Rheumatism recommendations for reporting extension studies [10].
The data cut-off for these analyses was February 13, 2018.
Results
Patient Disposition
In RA-BUILD, 684 patients were randomized; 229 patients were randomized to baricitinib 2 mg, 180 of whom completed the study and entered RA-BEYOND. Of the 90 patients never rescued, 74 (82.2%) patients completed 120 weeks of treatment. Figure 2 displays comprehensive disposition of patients randomized in RA-BUILD and RA-BEYOND.

The patient disposition for RA-BUILD describes the frequency and reasons for discontinuation in the originating study and in the long-term extension study. The percentages are calculated based on the overall modified intention-to-treat patients included in the originating study
In RA-BEACON, 527 patients were randomized; 174 patients were randomized to baricitinib 2 mg, 117 of whom completed the study and entered RA-BEYOND. Of the 49 patients never rescued, 36 (73.5%) patients completed 120 weeks of treatment. Figure 3 displays comprehensive disposition of patients randomized in RA-BEACON and RA-BEYOND.

The patient disposition for RA-BEACON describes the frequency and reasons for discontinuation in the originating study and in the long-term extension study. The percentages are calculated based on the overall modified intention-to-treat patients included in the originating study
Efficacy
SDAI LDA
In both studies, response trends were similar between the NRI and completer analyses; however, response rates based on the completer analysis were consistently higher. This was anticipated given that patients who discontinued from the study were defined as non-responders in the NRI analysis but excluded from the completer analysis.
In RA-BUILD, a greater proportion of patients in the baricitinib 2 mg treatment group achieved SDAI LDA compared to the group that received placebo (Fig. 4a, b). At week 24, completer analyses showed that 58.6% of patients in the baricitinib 2 mg (All Patients) group, 78.4% of patients in the baricitinib 2 mg (Never Rescued) cohort, and 45.5% of patients in the placebo group were in SDAI LDA. Based on NRI analysis, 47.6, 64.4, and 28.5% of patients treated with baricitinib 2 mg (All Patients), baricitinib 2 mg (Never Rescued), and placebo (All Patients), respectively, were in SDAI LDA. At week 120, completer analyses showed that 85.1% of patients in the baricitinib 2 mg (All Patients) group and 86.1% of the baricitinib 2 mg (Never Rescued) cohort were in SDAI LDA. The NRI analysis at week 120 showed that 27.5% of patients in the baricitinib 2 mg (All Patients) group and 52.5% of the baricitinib 2 mg (Never Rescued) cohort were in SDAI LDA.

Patients who achieved Simple Disease Activity Index ≤ 11 in RA-BUILD and RA-BEACON trials. a, b The efficacy over time based on the non-responder imputation (NRI) and completer analysis in RA-BUILD. c, d The efficacy over time based on the NRI and completer analysis in RA-BEACON
In RA-BEACON, a greater proportion of patients in the baricitinib 2 mg treatment group achieved SDAI LDA vs. the placebo group (Fig. 4c, d). At week 24, completer analyses showed that 35.1% of patients in the baricitinib 2 mg (All Patients) group, 54.0% of patients in the baricitinib 2 mg (Never Rescued) cohort, and 29.2% of patients in the placebo group were in SDAI LDA. Based on NRI analysis, 23.0, 39.7, and 14.8% of patients in the baricitinib 2 mg (All Patients), baricitinib 2 mg (Never Rescued), and placebo (All Patients) groups, respectively, were in SDAI LDA. At week 120, completer analyses showed that 86.5% of patients in the baricitinib 2 mg (All Patients) group and 88.6% of the baricitinib 2 mg (Never Rescued) cohort were in SDAI LDA. The NRI analysis at week 120 showed that 18.4% of baricitinib 2 mg All Patients and 45.6% of Never Rescued patients achieved SDAI LDA.
SDAI Remission
In both studies, response trends were similar between the NRI and completer analyses; however, response rates based on the completer analysis were consistently higher. This was anticipated given that patients who discontinued from the study were defined as non-responders in the NRI analysis but excluded from the completer analysis.
A greater proportion of patients in the baricitinib 2 mg treatment group from RA-BUILD achieved SDAI clinical remission vs. the placebo group (Fig. 5a, b). At week 24, completer analyses showed that 19.9% of patients in the baricitinib 2 mg (All Patients) group, 29.9% of patients in the baricitinib 2 mg (Never Rescued) cohort, and 7.0% of patients in the placebo group were in SDAI remission. The percentages of patients in SDAI remission based on NRI analysis were 16.2% (baricitinib 2 mg, All Patients), 24.6% (baricitinib 2 mg, Never Rescued), and 4.4% placebo (All Patients). At week 120, completer analyses showed that 40.5% of patients in the baricitinib 2 mg (All Patients) group and 41.7% of the baricitinib 2 mg (Never Rescued) cohort were in SDAI remission. Based on the NRI analysis, 13.1% and 25.4% of patients in the baricitinib 2 mg (All Patients) and baricitinib 2 mg (Never Rescued) groups, respectively, were in SDAI remission. The responses at week 24 in patients initially treated with baricitinib 2 mg were generally maintained (NRI analysis) or further improved (completer analysis) at week 120.

Patients who achieved Simple Disease Activity Index ≤ 3.3 in RA-BUILD and RA-BEACON trials. a, b The efficacy over time based on the non-responder imputation (NRI) and completer analysis in RA-BUILD. c, d The efficacy over time based on the NRI and completer analysis in RA-BEACON
In RA-BEACON, a greater proportion of patients in the baricitinib 2 mg treatment group achieved SDAI clinical remission, compared to those who received placebo (Fig. 5c, d). At week 24, completer analyses showed that 7.9% of patients in the baricitinib 2 mg (All Patients) group, 12.0% of patients in the baricitinib 2 mg (Never Rescued) cohort, and 4.5% of patients in the placebo group were in SDAI remission. Based on NRI analysis, 5.2, 8.8, and 2.3% of patients treated with baricitinib 2 mg (All Patients), baricitinib 2 mg (Never Rescued), and placebo (All Patients), respectively, were in SDAI remission. At week 120, completer analyses showed that 24.3% of patients in the baricitinib 2 mg (All Patients) group and 25.7% of the baricitinib 2 mg (Never Rescued) cohort were in SDAI remission. Based on the NRI analysis, 5.2% of patients in the baricitinib 2 mg (All Patients) group and 13.2% of those in the baricitinib 2 mg (Never Rescued) cohort were in SDAI remission. The responses at week 24 in patients initially treated with baricitinib 2 mg were, in general, maintained NRI analysis) or further improved (completer analysis) at week 120.
Physical Function (HAQ-DI)
In both trials, response rates were consistently higher based on the completer analysis relative to NRI. Patients who were discontinued or rescued were included in the NRI analysis as non-responders but excluded from the completer analysis. Maintenance of response was similar between the NRI and completer analysis groups.
In RA-BUILD, a greater proportion of patients in the baricitinib 2 mg treatment group achieved HAQ-DI ≤ 0.5 vs. the placebo group (Fig. 6a, b). At week 24, completer analyses showed that 39.2% of patients in the baricitinib 2 mg (All Patients) group, 51.5% of the baricitinib 2 mg (Never Rescued) cohort, and 30.3% of patients in the placebo group had HAQ-DI ≤ 0.5. Based on the NRI analysis, 32.3, 43.2, and 19.3% of patients treated with baricitinib 2 mg (All Patients), baricitinib 2 mg (Never Rescued), and placebo (All Patients), respectively, had HAQ-DI ≤ 0.5. At week 120, completer analyses showed that 62.2% of patients in the baricitinib 2 mg (All Patients) group and 62.5% of the baricitinib 2 mg (Never Rescued) cohort had HAQ-DI ≤ 0.5. Based on the NRI analysis, 20.1% of baricitinib 2 mg All Patients and 38.1% of patients in the baricitinib 2 mg (Never Rescued) cohort had HAQ-DI ≤ 0.5.

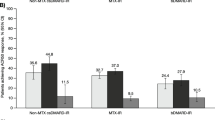
Patients who reached Health Assessment Questionnaire-Disability Index (HAQ-DI) ≤ 0.5 in RA-BUILD and RA-BEACON trials. a, b The percent of patients who met or exceeded HAQ-DI ≤ 0.5 at week 120 based on the non-responder imputation (NRI) and completer analysis in RA-BUILD. c, d The percent of patients who met or exceeded HAQ-DI ≤ 0.5 at week 120 based on the NRI and completer analysis in RA-BEACON
A greater proportion of patients in the baricitinib 2 mg treatment group in RA-BEACON achieved HAQ-DI ≤ 0.5, compared to the placebo treatment group (Fig. 6c, d). At week 24, completer analyses showed that 26.7% of patients in the baricitinib 2 mg (All Patients) group, 30.8% of the baricitinib 2 mg (Never Rescued) cohort, and 12.1% of patients in the placebo group had HAQ-DI ≤ 0.5. Based on the NRI analysis, 18.4% (baricitinib 2 mg, All Patients), 23.5% (baricitinib 2 mg, Never Rescued), and 6.3% (placebo, All Patients had HAQ-DI ≤ 0.5. At week 120, completer analyses showed that 50.0% of patients in the baricitinib 2 mg (All Patients) group and 50.0% of the baricitinib 2 mg (Never Rescued) cohort had HAQ-DI ≤ 0.5. Based on the NRI analysis, 10.9% of patients in the baricitinib 2 mg (All Patients) group and 26.5% of patients in the baricitinib 2 mg (Never Rescued) group had HAQ-DI ≤ 0.5.
In RA-BUILD, a greater proportion of patients in the baricitinib 2 mg treatment group achieved HAQ-DI MCID improvement ≥ 0.22 vs. the placebo group (Fig. 7a, b). At week 24, completer analyses showed that 78.8% of patients in the baricitinib 2 mg (All Patients) group, 83.8% of the baricitinib 2 mg (Never Rescued) cohort, and 67.1% of patients in the placebo group had HAQ-DI MCID improvement ≥ 0.22. Based on the NRI analysis, 65.1, 70.3, and 42.1% of patients treated with baricitinib 2 mg (All Patients), baricitinib 2 mg (Never Rescued), and placebo (All Patients), respectively, had HAQ-DI MCID improvement 0.22. At week 120, completer analyses showed that 81.1% of patients in the baricitinib 2 mg (All Patients) group and 81.9% of the baricitinib 2 mg (Never Rescued) cohort had HAQ-DI MCID improvement ≥ 0.22. Based on the NRI analysis, 26.2% of baricitinib 2 mg All Patients and 50.0% of patients in the baricitinib 2 mg (Never Rescued) cohort had HAQ-DI MCID improvement ≥ 0.22.

Patients who achieved Health Assessment Questionnaire-Disability Index (HAQ-DI) improvement ≥ 0.22 in RA-BUILD and RA-BEACON trials. a, b The efficacy over time based on the non-responder imputation (NRI) and completer analysis in RA-BUILD. c, d The efficacy over time based on the NRI and completer analysis in RA-BEACON
A greater proportion of patients in the baricitinib 2 mg treatment group in RA-BEACON achieved HAQ-DI MCID improvement ≥ 0.22, compared to the placebo treatment group (Fig. 7c, d). At week 24, completer analyses showed that 74.2% of patients in the baricitinib 2 mg (All Patients) group, 76.9% of the baricitinib 2 mg (Never Rescued) cohort, and 58.2% of patients in the placebo group had HAQ-DI MCID improvement ≥ 0.22. Based on the NRI analysis, 51.2% (baricitinib 2 mg, All Patients), 58.8% (baricitinib 2 mg, Never Rescued), and 30.1% (placebo, All Patients had HAQ-DI MCID improvement ≥ 0.22. At week 120, completer analyses showed that 73.7% of patients in the baricitinib 2 mg (All Patients) group and 77.8% of the baricitinib 2 mg (Never Rescued) cohort had HAQ-DI MCID improvement ≥ 0.22. Based on the NRI analysis, 16.1% of patients in the baricitinib 2 mg (All Patients) group and 41.2% of patients in the baricitinib 2 mg (Never Rescued) group had HAQ-DI MCID improvement ≥ 0.22.
Safety
Recently, an updated assessment of baricitinib safety in patients with RA through a median of 3.1 years of treatment (maximum 7 years) was published using pooled data from 3770 patients, totaling 10,127 years of patient exposure [8]. In the “all-bari-RA” dataset, which included all patients who were given ≥ 1 dose of baricitinib, incidence rates (IRs) of serious infections (2.8/100 patient years [PY]), herpes zoster (3.3/100 PY), MACE (0.5/100 PY), DVT (0.3/100 PY), PE (0.2/100 PY), venous thromboembolism (i.e., DVT and PE combined) (0.5/100 PY), and malignancy (excluding non-melanoma skin cancer, 0.8/100 PY) were similar to those previously reported for baricitinib [11] and consistent with those observed in other RA therapeutic programs. Among the patients in this dataset, there were a total of 479 patients originally randomized and treated with baricitinib 2 mg totaling 675.6 years of patient exposure. In patients treated with baricitinib 2 mg, IRs of serious infections (3.1/100 patient years [PY]), herpes zoster (2.7/100 PY), MACE (0.3/100 PY), DVT (0.6/100 PY), PE (0.2/100 PY), venous thromboembolism (i.e., DVT and PE combined) (0.6/100 PY), and malignancy (excluding non-melanoma skin cancer, 0.4/100 PY) were also similar to those previously reported for baricitinib [11].
Discussion
RA is a chronic and progressive disease. The main treatment goal of RA is clinical remission, with LDA as the best possible alternative in patients with established disease [12]. Despite a variety of approved treatments for RA, complete and sustained disease remission is rare. Disease activity in RA can fluctuate and flares are common—up to 30% of patients experience them on a regular basis [13].
Additional treatment goals of RA include prevention of joint damage accumulation, maximization of physical function, and improvement in quality of life [12]. Validated clinical outcome parameters are critical to assess the overall efficacy of RA therapies in clinical trials. Assessments of disease activity include the SDAI [14], clinical disease activity index (CDAI [15]), and the Disease Activity Score (DAS-28 [16]). Assessments of physical function include the HAQ-DI [17] and 36-Item Short-Form Health Survey (SF-36 [18]). Remission and normalization of physical function are frequently elusive goals in patients with established and long-standing or refractory RA or prior biologics use and LDA is acceptable goal. Despite this, the results from this study demonstrated the long-term maintenance of clinically relevant treatment goals achieved with baricitinib 2 mg including LDA (SDAI ≤ 11), remission (SDAI ≤ 3.3), and normative physical function (HAQ-DI ≤ 0.5). These data are especially important to healthcare providers and patients making treatment decisions in Canada and the United States, where baricitinib 2 mg is the approved dose for the treatment of rheumatoid arthritis, and China, where baricitinib 2 mg and (recently) 4 mg are the approved doses. Data presented here specific to the baricitinib 2-mg dose adds to the body of published literature focusing on Janus kinase inhibitors used for the treatment of RA [19,20,21,22,23].
LDA and remission were achieved in a greater proportion of patients treated with baricitinib 2 mg vs. placebo after 24 weeks. Responses at week 120 in patients initially treated with baricitinib 2 mg were generally maintained from week 24. The response rates based on the completer analysis in both studies remained high during the LTE. However, responses at week 120 decreased based on the NRI analysis, mainly in csDMARD-IR patients enrolled in RA-BUILD. One potential explanation for this observation may be the stringent rescue criteria for a non-responder employed in the LTE, which potentially contributed to a high rate of rescue (the rescue rate in RA-BEYOND [50.0%] was much higher than in RA-BUILD [9.2%] and all rescued patients were analyzed as non-responders in the NRI analysis).
Higher SDAI LDA and remission rates were also observed in csDMARD-IR patients in RA-BUILD compared to bDMARD-IR patients in RA-BEACON (at week 120, 27.5% of csDMARD-IR and 18.4% of bDMARD-IR patients treated with baricitinib 2 mg were in SDAI LDA; 13.1% of csDMARD-IR and 5.2% bDMARD-IR patients were in SDAI remission). This was anticipated given the composition of the analysis population. RA-BUILD included patients who had failed csDMARDs (25% with ≥ 3 prior csDMARD experience) and RA-BEACON enrolled patients who had failed at least one biologic TNF inhibitor (27% with ≥ 3 prior biologics experience) [6, 7]. In addition, the time from RA symptom onset was approximately 14 years for patients enrolled in RA-BEACON [5] versus 7.5 years for patients enrolled in RA-BUILD [6], suggesting patients in RA-BEACON had more advanced disease. Patients with RA who have had an inadequate response to TNF inhibitors, such as those in RA-BEACON, usually achieve lower response rates to other biologics than patients never exposed to bDMARDs [24,25,26,27,28,29].
There are limitations to this analysis that should be noted. Although the originating studies were placebo-controlled and all patients received background csDMARDs, no active comparators were included in the study designs. Additionally, in this post hoc analysis, not all patients had data available from their originally randomized treatment during the analysis period. This was due to discontinuation or rescue, and, in these instances, data imputation was required in the analysis. Another limitation is the small number of never rescued patients included in the analysis attributed to the prompted rescue criterion (i.e., CDAI > 10) employed in RA-BEYOND. Despite these limitations, these efficacy data, which assessed parameters that are the current goals of treating RA in the context of safety, are of clinical relevance to patients and providers to inform on the benefit-to-risk of long-term use of baricitinib 2 mg.
Conclusions
In conclusion, the long-term efficacy of baricitinib 2 mg was demonstrated and maintained for up to 120 weeks in patients with inadequate response to csDMARDs or TNFi. Baricitinib 2 mg treatment continues to be well tolerated as evidenced by low discontinuation rates 15% for csDMARDs-IR; 17% for TNFi-IR) through 120 weeks of treatment.
References
Smolen J, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov. 2003;2:473–88.
Colmegna I, Ohata BR, Menard HA. Current understanding of rheumatoid arthritis therapy. Clin Pharmacol Ther. 2012;91:607–20.
Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184:5298–307.
Fleischmann R, Schiff M, van der Heijde D, Ramos-Remus C, Spindler A, Stanislav M, et al. Baricitinib, methotrexate, or combination in patients with rheumatoid arthritis and no or limited prior disease-modifying antirheumatic drug treatment. Arthritis Rheumatol. 2017;69:506–17.
Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, del Carmen ML, Gonzaga JR, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. 2017;376:652–62.
Dougados M, van der Heijde D, Chen YC, Greenwald M, Drescher E, Liu J, et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA-BUILD study. Ann Rheum Dis. 2017;76:88–95.
Genovese MC, Kremer J, Zamani O, Ludivico C, Krogulec M, Xie L, et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med. 2016;374:1243–52.
Genovese MC, Smolen JS, Takeuchi T, Burmester G, Brinker D, Rooney TP, et al. Safety profile of baricitinib for the treatment of rheumatoid arthritis over a median of 3 years treatment: an updated integrated safety analysis. Lancet Rheumatol. 2020;2(6):E347–57.
Taylor PC, Weinblatt ME, Burmester GR, Rooney TP, Witt S, Walls CD, et al. Cardiovascular safety during treatment with baricitinib in rheumatoid arthritis. Arthritis Rheumatol. 2019;71:1042–55.
Buch MH, Silva-Fernandez L, Carmona L, Aletaha D, Christensen R, Combe B, et al. Development of EULAR recommendations for the reporting of clinical trial extension studies in rheumatology. Ann Rheum Dis. 2015;74:963–9.
Smolen JS, Genovese MC, Takeuchi T, Hyslop DL, Macias WL, Rooney T, et al. Safety profile of baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J Rheumatol. 2019;46:7–18.
Smolen JS, Landewé RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2019;2020:22.
Bykerk VP, Shadick N, Frits M, Bingham CO III, Jeffery I, Iannaccone C, et al. Flares in rheumatoid arthritis: frequency and management. A report from the BRASS registry. J Rheumatol. 2014;41:227–34.
Smolen JS, Breedveld FC, Schiff MH, Kalden JR, Emery P, Eberl G, et al. A simplified disease activity index for rheumatoid arthritis for use in clinical practice. Rheumatology (Oxford). 2003;42:244–57.
Aletaha D, Nell VP, Stamm T, Uffmann M, Pflugbeil S, Machold K, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther. 2005;7:R796-806.
Prevoo ML, van ’tHof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38:44–8.
Wolfe F, Michaud K, Pincus T. Development and validation of the health assessment questionnaire II: a revised version of the health assessment questionnaire. Arthritis Rheum. 2004;50:3296–305.
Ware JE, Sherbourne CD. The MOS 36-Item Short-Form Health Survey (SF-36). I. Conceptual framework and item selection. Med Care. 1992;30:473–83.
van der Heijde D, Landewé RBM, Wollenhaupt J, et al. Assessment of radiographic progression in patients with rheumatoid arthritis treated with tofacitinib in long-term studies. Rheumatology (Oxford). 2020. https://doi.org/10.1093/rheumatology/keaa476.
Cohen SB, Tanaka Y, Mariette X, et al. Long-term safety of tofacitinib up to 9.5 years: a comprehensive integrated analysis of the rheumatoid arthritis clinical development programme. RMD Open. 2020;6(3):e001395.
Álvaro-Gracia JM, García-Llorente JF, Valderrama M, et al. Update on the safety profile of tofacitinib in rheumatoid arthritis from clinical trials to real-world studies: a narrative review. Rheumatol Ther. 2021;8(1):17–40.
Nash P, Kerschbaumer A, Dörner T, et al. Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: a consensus statement. Ann Rheum Dis. 2021;80(1):71–87.
El Jammal T, Sève P, Gerfaud-Valentin M, et al. State of the art: approved and emerging JAK inhibitors for rheumatoid arthritis. Expert Opin Pharmacother. 2021;22(2):205–18.
Burmester GR, Blanco R, Charles-Schoeman C, et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet. 2013;381:451–60.
Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at 24 weeks. Arthritis Rheum. 2006;54:2793–806.
Emery P, Keystone E, Tony HP, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67:1516–23.
Genovese MC, Schiff M, Luggen M, et al. Efficacy and safety of the selective co-stimulation modulator abatacept following 2 years of treatment in patients with rheumatoid arthritis and an inadequate response to anti-tumour necrosis factor therapy. Ann Rheum Dis. 2008;67:547–54.
Smolen JS, Aletaha D. Rheumatoid arthritis therapy reappraisal: strategies, opportunities and challenges. Nat Rev Rheumatol. 2015;11:276–89.
Smolen JS, Kay J, Doyle MK, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet. 2009;374:210–21.
Acknowledgements
The authors thank the patients and investigators who participated in the baricitinib studies. The authors also thank Yun-Fei Chen, Ph.D., for her input on the analysis strategy for the manuscript.
Funding
This study and the journal’s Rapid Service Fee were funded by Eli Lilly and Company and Incyte Corporation. Medical writing and editorial support were provided by Shannon E. Gardell, Ph.D., of Evidera/PPD and Colleen Dumont, of Evidera/PPD, and was funded by Eli Lilly and Company.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
All authors and Eli Lilly and Company were involved in data interpretation and reviewed and approved the manuscript. The authors maintained control over the final content.
Disclosures
AF Wells has served as a consultant for Takeda Global Research & Development Center, Inc., Abbott Laboratories, Amgen, Inc., Bristol-Myers Squibb Company, Centocor, Inc., Eli Lilly and Company, GlaxoSmithKline, Pfizer, Inc., Wyeth Pharmaceuticals, and Genentech, Inc. B Jia, L Xie, AK Quebe, K Griffing, and S Otawa are employees and stockholders of Eli Lilly and Company. GJ Valenzuela. EC Keystone reports funding for research from AbbVie, Amgen, Bristol-Myers Squibb, F. Hoffmann-La Roche Inc, Gilead, Janssen, Lilly Pharmaceuticals, Pfizer Pharmaceuticals, and Sanofi-Aventis; consulting agreements with AbbVie, Amgen, AstraZeneca Pharma, Biotest, Bristol-Myers Squibb Company, Celltrion, Crescendo Bioscience, F. Hoffmann-La Roche Inc, Genentech Inc, Gilead, Janssen Inc, Lilly Pharmaceuticals, Merck, Pfizer Pharmaceuticals, Sandoz and UCB; and has received speaker honoraria from Amgen, AbbVie, Bristol-Myers Squibb Canada, F. Hoffmann-La Roche Inc., Janssen, Merck, Pfizer Pharmaceuticals, Sanofi Genzyme and UCB. Z Li. B Haraoui has received honoraria for participation in advisory boards and/or research grants from AbbVie, Amgen, Bristol-Myers Squibb, Janssen, Pfizer, Roche and UCB.
Compliance with Ethics Guidelines
Studies included in this analysis were conducted (RA-BUILD, RA-BEACON) or are being conducted (RA-BEYOND) in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice Guidelines and were approved by each center’s institutional review board (IRB) of ethics committee (Quorum Review IRB, #27258, #27259, and #28020). Written informed consent was provided by all patients.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wells, A.F., Jia, B., Xie, L. et al. Efficacy of Long-Term Treatment with Once-Daily Baricitinib 2 mg in Patients with Active Rheumatoid Arthritis: Post Hoc Analysis of Two 24-Week, Phase III, Randomized, Controlled Studies and One Long-Term Extension Study. Rheumatol Ther 8, 987–1001 (2021). https://doi.org/10.1007/s40744-021-00317-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-021-00317-9