
Abstract
Introduction
Upadacitinib is a Janus kinase inhibitor under investigation in patients with psoriatic arthritis (PsA). This study assessed the 56-week efficacy and safety of upadacitinib in patients with PsA and an inadequate response or intolerance to biologic therapy.
Methods
In the phase 3 SELECT-PsA 2 study, patients were randomized to 56 weeks of blinded treatment with oral upadacitinib 15 or 30 mg once daily, or placebo switched to upadacitinib 15 or 30 mg once daily at week 24. Efficacy endpoints included the proportion of patients achieving 20/50/70% improvement in American College of Rheumatology criteria (ACR20/50/70), 75/90/100% improvement in Psoriasis Area and Severity Index (PASI75/90/100), and minimal disease activity. Safety was assessed throughout the study.
Results
Of 641 patients who received ≥ 1 dose of study drug, 479 (74.7%) completed 56 weeks of treatment. Improvements in the proportion of patients achieving ACR20/50/70, PASI75/90/100, and minimal disease activity were maintained with both doses of upadacitinib through 56 weeks. Week 56 results for patients who switched from placebo to upadacitinib at week 24 were similar to those for patients originally randomized to the upadacitinib groups. The exposure-adjusted event rate for serious infections was 2.6 and 6.1 events/100 patient-years in the upadacitinib 15 and 30 mg groups, respectively. Herpes zoster occurred more frequently with upadacitinib 30 versus 15 mg; most cases were non-serious.
Conclusion
In patients with PsA who had an inadequate response or intolerance to biologic therapy, the efficacy of upadacitinib was maintained over 56 weeks with no new significant safety signals observed.
Trial registration
NCT03104374.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Many patients with psoriatic arthritis (PsA) do not adequately respond and maintain response to currently recommended therapies, highlighting an unmet need for additional therapeutic agents that can effectively control disease activity. |
Upadacitinib, an oral Janus kinase inhibitor, is currently under investigation for the treatment of PsA; during the 24-week, placebo-controlled period of the SELECT-PsA 2 study, upadacitinib demonstrated efficacy and acceptable safety in patients who had an inadequate response or intolerance to ≥1 biologic disease-modifying anti-rheumatic drugs. |
The purpose of this 56-week analysis of SELECT-PsA 2 data was to explore the longer-term safety and maintenance of efficacy of upadacitinib in patients with PsA and to describe the safety and efficacy in those who switched from placebo to upadacitinib at week 24. |
What was learned from the study? |
In patients with PsA and prior inadequate response or intolerance to biologic therapy, the efficacy of upadacitinib across manifestations of PsA, including musculoskeletal symptoms, psoriasis, and patient-reported outcomes, remained consistent or improved through 56 weeks, with no new significant safety signals observed compared with the known safety profile of upadacitinib. |
By week 56, the efficacy of upadacitinib 15 mg approached or was similar to that of upadacitinib 30 mg, and responses for patients who switched from placebo to upadacitinib at week 24 had a similar trajectory to those for patients originally randomized to upadacitinib. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14216975.
Introduction
Psoriatic arthritis (PsA) is a chronic inflammatory joint disease that is characterized by clinical manifestations such as psoriasis, nail dystrophy, enthesitis, dactylitis, and involvement of the spine or sacroiliac joint (axial PsA) [1]. Treatment guidelines for PsA advocate an approach whereby the goal of treatment is to achieve remission, inactive PsA, or low disease activity [2,3,4]. Available treatment options include non-steroidal anti-inflammatory drugs (NSAIDs); local corticosteroid injections; conventional synthetic disease-modifying antirheumatic drugs (DMARDs), such as methotrexate (MTX); biologic DMARDs (bDMARDs), such as tumor necrosis factor inhibitors, interleukin-12/23 inhibitors, and interleukin-17 inhibitors; and targeted synthetic DMARDs, such as phosphodiesterase 4 inhibitors or Janus kinase (JAK) inhibitors [2,3,4]. Despite the emergence of these treatments with distinct mechanisms of action, many patients (> 70%) do not achieve 70% improvement in American College of Rheumatology (ACR) criteria (ACR70) [5,6,7,8,9], highlighting an unmet need for additional therapeutic agents that can effectively control disease activity. This is especially important for patients with PsA who have experienced inefficacy (primary or secondary) or intolerance to biologic therapy.
Upadacitinib is an oral reversible JAK inhibitor engineered for increased selectivity for JAK1 over JAK2, JAK3, and tyrosine kinase 2 that is currently under investigation for the treatment of PsA [10]. In the 24-week placebo-controlled period of the SELECT-PsA 2 study, the primary endpoint was met: a significantly greater proportion of patients with PsA who had an inadequate response or intolerance to bDMARDs achieved a 20% improvement in ACR criteria (ACR20) at week 12 with upadacitinib 15 mg and 30 mg once daily (QD) compared with placebo [11]; ACR20 and 50/70% improvement in ACR criteria (ACR50/70) response rates were maintained for both upadacitinib doses at week 24 [12]. Key ranked secondary outcomes also demonstrated significant improvements with both doses of upadacitinib compared with placebo, including: changes from baseline in Health Assessment Questionnaire-Disability Index (HAQ-DI) at 12 weeks; Static Investigator’s Global Assessment of Psoriasis score of 0 or 1 and ≥ 2-point improvement (sIGA 0/1) at 16 weeks; Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) score at 12 weeks; Self-Assessment of Psoriasis Symptoms (SAPS) score at 16 weeks and 36-item Short Form Health Survey (SF-36) score at 12 weeks; and proportions of patients achieving 75% improvement in the Psoriasis Area and Severity Index (PASI75) at 16 weeks and minimal disease activity (MDA) at 24 weeks [12]. In a post hoc analysis of SELECT-PsA 2, patients also achieved a higher rate of tight disease control (based on Disease Activity in Psoriatic Arthritis and PsA Disease Activity Score criteria) with upadacitinib compared with placebo at weeks 12 and 24 [13]. Here, we report the safety and efficacy of upadacitinib over 56 weeks in SELECT-PsA 2.
Methods
Patients and Study Design
The methodology of SELECT-PsA 2 (ClinicalTrials.gov Identifier: NCT03104374) has been published previously [11]. In brief, eligible patients were adults (≥ 18 years old), had active PsA with symptom duration ≥ 6 months, fulfilled the Classification Criteria for Psoriatic Arthritis [14], had historical or current plaque psoriasis, had ≥ 3 swollen joints (swollen joint count [SJC] of 66) and ≥ 3 tender joints (tender joint count [TJC] of 68), and had an inadequate response or intolerance to ≥ 1 bDMARD. Patients meeting these criteria were randomized in a 2:2:1:1 ratio to receive upadacitinib 15 mg QD, upadacitinib 30 mg QD, or placebo switched to either upadacitinib 15 mg (placebo to upadacitinib 15 mg) or 30 mg (placebo to upadacitinib 30 mg) QD at week 24. An unblinded analysis was conducted after all patients had completed week 24 or prematurely discontinued the study. Investigator and patient blinding were maintained until week 56 at the study sites, at which point patients could enter an open-label extension period through to week 152.
During the study, although background therapy was not required, patients were permitted to receive stable background treatment with NSAIDs, corticosteroids (equivalent to ≤ 10 mg/day prednisone), and/or ≤ 2 non-bDMARDs (conventional synthetic DMARDs or apremilast). Concomitant treatments specifically for psoriasis (e.g., topical therapy, light therapy, or retinoids) were not permitted prior to week 16. At week 16, background medications were initiated or adjusted in patients who did not achieve ≥ 20% improvement in TJCs and SJCs versus baseline at weeks 12 and 16. From week 36 onwards, patients who did not achieve ≥ 20% improvement in TJCs and SJCs versus baseline at two consecutive visits discontinued the study drug. Initiation or adjustment of background PsA medication was permitted for all patients after week 36.
The study was conducted according to the International Conference on Harmonisation guidelines, applicable regulations and guidelines governing clinical trial conduct, and the Declaration of Helsinki 1964 and its later amendments. The trial protocol was approved by independent ethics committees and institutional review boards. All patients provided written informed consent.
Outcome Measures
Clinical efficacy endpoints assessed through week 56 included the proportion of patients achieving ACR20/50/70; MDA (defined as per [15]); PASI75 and PASI 90/100% improvement (PASI90/100; among patients with ≥ 3% body surface area of psoriasis at baseline) [16]; sIGA 0/1 for patients with baseline sIGA ≥ 2 [17]; Psoriatic Arthritis Response Criteria (improvement in 2 of the following 4 criteria, 1 of which must be TJC68 or SJC66, without worsening of any measure: ≥ 30% improvement in TJC68 or SJC66, and improvement in Patient’s Global Assessment of Disease Activity [PtGA] [18] and Physician’s Global Assessment of Disease Activity on a numeric rating scale [range 0–10, with higher scores indicating greater disease activity]); resolution of enthesitis [defined by Leeds Enthesitis Index (LEI) = 0] for patients with baseline LEI > 0 [19]; and resolution of dactylitis [defined by Leeds Dactylitis Index (LDI) = 0] for patients with baseline LDI > 0 [20]. Change from baseline in individual components of the ACR criteria was also assessed.
Patient-reported endpoints assessed through week 56 included change from baseline in HAQ-DI score (range 0–3, with higher scores indicating greater disability) [21]; FACIT-F score (range 0–52, with higher scores indicating less fatigue) [22]; SF-36 Physical Component Summary and Mental Component Summary scores (norm-based scores were used, with higher scores indicating better health-related quality of life) [23]; SAPS score (range 0–110, with higher scores indicating more severe psoriasis symptoms) [24]; score on the overall work impairment domain of the Work Productivity and Activity Impairment (WPAI) questionnaire (range 0–100%, with higher scores indicating greater impairment) for patients employed at baseline; morning stiffness (mean score of Bath Ankylosing Spondylitis Disease Activity Index [BASDAI] questions 5 and 6, with higher scores indicating more severe and longer duration of morning stiffness) [25]; PtGA score on a numeric rating scale (range 0–10, with higher scores indicating greater disease activity) [26]; and patient assessment of pain based on a numeric rating scale (range 0–10, with higher scores indicating greater pain). The proportion of patients achieving a clinically meaningful improvement from baseline in HAQ-DI of ≥ 0.35 was also assessed [27].
In patients with axial PsA at baseline (as assessed by the investigator), change from baseline in the Ankylosing Spondylitis Disease Activity Score (ASDAS) and BASDAI, and the proportion of patients achieving 50% improvement in BASDAI were also reported through week 56.
Adverse events (AEs) and clinical laboratory measures were reported through week 56. AEs were coded per the Medical Dictionary for Regulatory Activities version 22.0 (https://www.meddra.org/how-to-use/support-documentation/english); AEs and laboratory changes were graded using the National Cancer Institute Common Toxicity Criteria versions 5.0 and 4.03 (https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm), respectively. Major adverse cardiovascular events (MACE) and venous thromboembolic (VTE) events were blindly adjudicated by an independent cardiovascular adjudication committee per pre-defined event definitions. An internal gastrointestinal perforation adjudication committee blindly adjudicated reported gastrointestinal perforation events as stated in the gastrointestinal perforation charter.
Statistical Analysis
Efficacy analyses were conducted on the full analysis set, including all randomized patients who had received ≥ 1 dose of study drug. There were no formal statistical comparisons between groups for long-term efficacy. For binary endpoints, frequencies and percentages are summarized. Non-responder imputation (NRI) was used for missing data for binary endpoints. In addition, as-observed data without any imputation are shown for binary endpoints at week 56.
For continuous endpoints, mixed model for repeated measures analysis was performed on the as-observed data, with least-squares means and 95% confidence intervals (CIs) provided for each randomized treatment group sequence. Missing data were handled using a mixed model for repeated measures assuming the data were missing at random.
For safety analyses, the upadacitinib 15 mg and 30 mg groups included patients who were originally randomized to placebo and switched to upadacitinib at week 24. Treatment-emergent AEs are summarized for events occurring while exposed to upadacitinib (i.e., onset after the first dose of upadacitinib and no more than 30 days after the last dose of study drug). Exposure-adjusted event rates (EAERs) per 100 patient-years (PY; E/100 PY) are summarized as events based on the treatment received at the time of each AE. Multiple AEs occurring in the same patient are included in the numerators, and 95% CIs were calculated based on normal approximation to the Poisson distribution. Exposure-adjusted incidence rates per 100 PY are summarized as the number of patients with ≥ 1 event/100 PY (n/100 PY), with exposure calculated up to onset of the first event; multiple events occurring in the same patient are not included in the numerator, and 95% CIs were based on normal approximation to the Poisson distribution.
Results
Patients
Of 642 randomized patients, 641 received ≥ 1 dose of the study drug; of these, 560 (87.2%) and 479 (74.7%) completed 24 and 56 weeks of treatment, respectively (Fig. 1). The most common reasons for discontinuation at weeks 24 and 56 were patient withdrawal (5.0 and 8.6%, respectively) and AEs (3.9 and 6.2%, respectively). As reported previously [11], baseline characteristics were well balanced across the treatment groups (Electronic Supplementary Material [ESM] Table S1). Of the 641 patients, 61.0% had failed one prior bDMARD, 18.1% had failed two prior bDMARDs, and 12.9% had failed ≥ 3 prior bDMARDs. At baseline, 53.8% of patients were not using concomitant non-bDMARDs. Approximately one-third of patients were receiving MTX alone, 8.7% were receiving a non-bDMARD other than MTX, and a small proportion (2.8%) were receiving MTX plus another non-bDMARD (ESM Table S1).

Patient disposition at week 56. aOne patient did not receive study drug. AE adverse event, PBO placebo, QD once daily, UPA upadacitinib
Efficacy
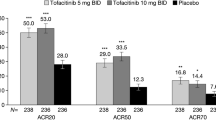
Musculoskeletal Outcomes and Composite Outcomes
At week 56, the proportion of patients achieving ACR20/50/70 using NRI was 59.7/40.8/24.2% with upadacitinib 15 mg and 59.2/38.5/26.6% with upadacitinib 30 mg (Fig. 2). In both placebo to upadacitinib groups, responses at week 56 approached or were similar to those for patients who received upadacitinib from baseline. ACR response rates and response rates in other binary efficacy endpoints tended to be higher in the as-observed analysis (ESM Table S2) than in the NRI analysis at week 56. Individual patient responses for ACR20/50/70 over 56 weeks for all treatment groups, including the time course of achievement and sustainability of these responses, are presented in ESM Figs. S1–S3. Numerically greater proportions of patients who received upadacitinib at baseline compared with the placebo to upadacitinib groups achieved MDA through week 56 (Fig. 3). Individual patient responses for MDA over time for all treatment groups, including the time course of achievement and sustainability of these responses, are presented in ESM Fig. S4. More patients receiving upadacitinib 15 mg and 30 mg from baseline achieved MDA at week 24 than those receiving placebo, and most patients who achieved MDA maintained their response through week 56. Numerically greater proportions of patients who received upadacitinib from baseline than those in the placebo to upadacitinib groups for each of the doses met Psoriatic Arthritis Response Criteria through week 56 using NRI (ESM Fig. S5).

Patients achieving a ACR20, b ACR50, and c ACR70 over 56 weeks (non-responder imputation). The gray dotted line represents PBO prior to patients switching to UPA. ACR20/50/70 American College of Rheumatology criteria 20/50/70% improvement, CI confidence interval

Patients achieving minimal disease activity over 56 weeks (non-responder imputation). The gray dotted line represents PBO prior to patients switching to UPA. MDA minimal disease activity
In patients with dactylitis at baseline (LDI > 0), complete resolution (LDI = 0) of previous dactylitis was observed in 50.9 and 58.0% of patients treated with upadacitinib 15 mg and 30 mg, respectively, by week 56 using NRI; of those with enthesitis at baseline (LEI > 0), complete resolution (LEI = 0) of previous enthesitis was observed in 42.9 and 42.8% of patients, respectively (ESM Fig. S6). Rates of resolution of dactylitis and enthesitis in both placebo to upadacitinib groups increased after the switch to upadacitinib and approached those for patients who received upadacitinib from baseline. In addition, patients assessed by the investigator to have axial PsA at baseline showed improvements in the ASDAS and BASDAI from baseline to week 56 using as-observed data (Table 1).
Skin Outcomes
At week 56, the proportions of patients achieving PASI75/90/100 were 52.3/40.8/26.9% and 58.8/47.3/35.1% using NRI with upadacitinib 15 mg and 30 mg, respectively. In both placebo to upadacitinib groups, PASI75/90/100 responses at week 56 approached or were similar to those for patients who received upadacitinib from baseline, with the exception of PASI100 in the placebo to upadacitinib 15 mg group (Fig. 4). Individual patient responses for PASI75 and PASI90 over 56 weeks for all treatment groups, including the time course of achievement and sustainability of these responses, are presented in ESM Figs. S7 and S8. Numerically greater proportions of patients who received upadacitinib from baseline than those in the placebo to upadacitinib groups for each of the doses achieved sIGA 0 or 1 and ≥ 2 point improvement from baseline (Fig. 4).

Patients achieving a PASI75, b PASI90, c PASI100, and d sIGA 0 or 1 and ≥ 2-point improvement from baseline over 56 weeks (non-responder imputation). The gray dotted line represents PBO prior to patients switching to UPA. PASI75/90/100 75/90/100% improvement in Psoriasis Area and Severity Index, sIGA Static Investigator’s Global Assessment
Patient-Reported Outcomes
Improvements in several patient-reported outcomes were observed through week 56 using as-observed data (Table 1). Least square mean improvements from baseline to week 56 in physical function, as measured by HAQ-DI, were − 0.35 and − 0.49 for patients originally randomized to upadacitinib 15 mg and 30 mg, respectively (Table 1; ESM Fig. S9). Patients in the placebo to upadacitinib groups achieved similar results at week 56. Clinically meaningful improvement in HAQ-DI (≥ 0.35) [27] based on NRI data was achieved by 47.3 and 56.0% of patients treated with upadacitinib 15 mg and 30 mg, respectively, during the first 24 weeks of the study, and this decreased to 42.9 and 46.6%, respectively, by 56 weeks (ESM Fig. S9). The proportions of patients who achieved a clinically meaningful improvement in HAQ-DI in the placebo to upadacitinib groups at week 56 approached those of the upadacitinib from baseline groups. Mean improvements from baseline to week 56 were observed for patients randomized to upadacitinib from baseline for the FACIT-F, PtGA, pain, morning stiffness, SF-36 Physical and Mental Component Summary, SAPS, and WPAI overall work impairment (Table 1). Results were generally comparable at week 56 in the placebo to upadacitinib groups.
Safety
The rate of treatment-emergent AEs was lower with upadacitinib 15 mg than with upadacitinib 30 mg (260.6 vs. 334.4 E/100 PY, respectively), although the rates of serious AEs and AEs leading to discontinuation were similar between the groups (for upadacitinib 15 mg and 30 mg, respectively: serious AEs: 14.3 and 15.8 E/100 PY; AEs leading to discontinuation: 10.0 and 11.1 E/100 PY). During the 56-week period of this trial, two deaths were reported: one related to a motor vehicle accident in a patient receiving placebo; and one in a patient receiving upadacitinib 30 mg, with the cause of death reported as acute respiratory distress syndrome and right pneumothorax per the death certificate, and as pancytopenia per investigator. The patient was hospitalized due to the right pneumothorax, with a subsequent complicated hospital course that included pancytopenia, cytomegalovirus infection, disseminated intravascular coagulation, bleeding duodenal ulcers, Pneumocystis jirovecii pneumonia, and interstitial pneumonia (all considered by the investigator to have a reasonable possibility of relationship to the study drug; after consideration of risk factors, only Pneumocystis jirovecii pneumonia was considered by the sponsor to have a reasonable possibility of relationship to the study drug), in addition to mechanical ventilation and thoracoscopic pneumonectomy, bilateral pneumothoraces, and cerebral infarction.
The most commonly reported AEs in both dose groups were nasopharyngitis and upper respiratory tract infection (ESM Table S3). The EAER for serious infections was 2.6 and 6.1 E/100 PY in the upadacitinib 15 mg and 30 mg groups, respectively (Fig. 5). The most common serious infections were cellulitis (0.5 E/100 PY in each group) and pneumonia (0.7 and 1.2 E/100 PY in the upadacitinib 15 mg and 30 mg groups, respectively). Opportunistic infections (excluding tuberculosis and herpes zoster) were infrequent and consisted mostly of mucosal candidiasis in both groups, along with coccidioidomycosis in the upadacitinib 15 mg group, and cytomegalovirus infection and Pneumocystis jirovecii pneumonia (both in the same patient who subsequently died, as discussed above) in the upadacitinib 30 mg group. No cases of active tuberculosis were reported.

a EAERs and b EAIRs of treatment-emergent adverse events through week 56. a Excluding tuberculosis and herpes zoster. b Four cases of basal cell carcinoma and 1 case of squamous cell carcinoma of the skin in the UPA 15 mg group. c Three cases of basal cell carcinoma and 3 cases of squamous cell carcinoma of the skin in the UPA 30 mg group. d Two cases of prostate cancer and single cases of malignant melanoma, ovarian cancer, and rectal cancer in the UPA 15 mg group. e Single cases of basosquamous carcinoma (considered NMSC after medical review), malignant melanoma, oropharyngeal squamous cell carcinoma, and rectal adenocarcinoma, as well as endometrial cancer and ovarian cancer (occurred in the same patient), in the UPA 30 mg group. f Two events of treatment-emergent abnormal lymphocyte morphology were identified in the UPA 15 mg group; abnormal lymphocytes were not reported in subsequent laboratory testing. CPK Creatine phosphokinase, E/100 PY events per 100 patient-years, EAIR exposure-adjusted incidence rate, EAER exposure-adjusted event rate, GI gastrointestinal, MACE major adverse cardiovascular event (defined as non-fatal myocardial infarction, non-fatal stroke, or cardiovascular death), n/100 PY number per 100 patient-years, NMSC non-melanoma skin cancer, PY patient-year, VTE venous thromboembolic event (defined as deep vein thrombosis or pulmonary embolism)
Herpes zoster was less frequent in the upadacitinib 15 mg group than in the upadacitinib 30 mg group (3.8 vs. 8.5 E/100 PY). Most events in either dose group were non-disseminated and did not lead to discontinuation of the study drug. None of the events in the upadacitinib 15 mg group were serious. In the upadacitinib 30 mg group, one case of herpes zoster was serious, and two non-serious cases led to discontinuation of the study drug. Eight events (3 in the upadacitinib 15 mg group and 5 in the upadacitinib 30 mg group) were reported to involve ≥ 3 dermatomes, one of which in the upadacitinib 30 mg group led to discontinuation of the study drug.
Ten patients developed malignancies in the upadacitinib 15 mg group (2.4 n/100 PY), including five patients with non-melanoma skin cancer (NMSC), and eight patients developed malignancies in the upadacitinib 30 mg group (1.9 n/100 PY), including four patients with NMSC.
Adjudicated MACEs were reported in one patient in the upadacitinib 15 mg group (non-fatal myocardial infarction) and one patient in the upadacitinib 30 mg group (non-fatal stroke). Two patients developed adjudicated VTE, one each in the upadacitinib 15 mg and 30 mg groups (both 0.2 n/100 PY); both were non-fatal pulmonary embolisms and both patients had notable risk factors for VTE, including obesity in both patients and hypertension in one patient. There were no cases of adjudicated gastrointestinal perforation.
Hepatic disorders were less common in the upadacitinib 15 mg group (4.8 E/100 PY) than in the upadacitinib 30 mg group (17.7 E/100 PY) and the majority were non-serious, transient transaminase elevations that did not lead to study drug discontinuation. Grade 3 increases in alanine aminotransferase and aspartate aminotransferase (AST) were seen in three (1.0%) and three (1.0%) patients, respectively, in the upadacitinib 15 mg group and in three (1.0%) and five (1.6%) patients, respectively, in the upadacitinib 30 mg group, most of which were isolated and not confirmed upon repeat testing (ESM Table S4). No Grade 4 increases in alanine aminotransferase or AST were observed in either group. One patient from the upadacitinib 30 mg group met the laboratory criteria for Hy’s law based on AST and total bilirubin values at a single visit, but an alternative etiology of fatty liver disease (hepatic steatosis) was identified and thus it was not confirmed as a true case of Hy’s law. Similarly, rates of anemia and creatine phosphokinase (CPK) elevation were slightly lower with upadacitinib 15 mg than with upadacitinib 30 mg (ESM Table S3). Grade 3 increases in CPK were seen in five patients each in the upadacitinib 15 mg (1.7%) and 30 mg (1.6%) groups, with three (1.0%) patients in each group experiencing Grade 4 increases. Two patients in the upadacitinib 30 mg group discontinued the study drug due to CPK elevation. There were no cases of rhabdomyolysis.
The EAERs of neutropenia, lymphopenia, and renal dysfunction were relatively low in both groups. Grade 3 decreases in hemoglobin, lymphocytes, and neutrophils were relatively infrequent, occurring in ≤ 2% of patients in either group (ESM Table S4), and most were isolated and not confirmed upon repeat testing. No Grade 4 decreases in lymphocytes or neutrophils were observed.
Discussion
In the 24-week, placebo-controlled phase of SELECT-PsA 2, upadacitinib at doses of 15 mg and 30 mg QD improved the signs and symptoms of PsA versus placebo with no new significant safety signals identified [11]. In this report, we found that the efficacy of upadacitinib across manifestations of PsA remained consistent or improved through 56 weeks in SELECT-PsA 2; furthermore, patients who switched from placebo to upadacitinib at week 24 showed improvements at week 56 that approached or were similar to those in patients originally randomized to upadacitinib. Improvements were observed across endpoints assessing musculoskeletal symptoms (peripheral arthritis, enthesitis, dactylitis, and spondylitis), psoriasis, and patient-reported outcomes (physical function, pain, fatigue, overall work impairment, and quality of life). Furthermore, achievement of comprehensive disease control as measured by MDA was maintained over 56 weeks, with most patients remaining in MDA once achieved. In general, across the efficacy endpoints evaluated, improvements observed for the upadacitinib 15 mg dose were similar to or approached those of the 30 mg dose over 56 weeks.
The safety profile of upadacitinib was comparable with that observed in the 24-week double-blind period [11] and in the rheumatoid arthritis clinical development program [28,29,30,31,32,33]. Rates of serious infections and herpes zoster events appeared to be dose dependent, whereas no dose-dependent risks were observed for adjudicated MACE, VTE, or malignancies.
VTE has recently emerged as a safety concern in patients receiving JAK inhibitors. In this study there were two cases of adjudicated pulmonary embolism (1 each in the upadacitinib 15 mg and 30 mg arms), both of which were non-fatal and occurred in patients who had risk factors for VTE. The incidence of adjudicated VTEs through week 56 in this study (1 case each in the upadacitinib 15 mg and 30 mg groups) was similar to that reported at week 24 in the SELECT-PsA 1 study (NCT03104400) of patients with PsA who had an inadequate response or were intolerant to non-bDMARDs (1 in the upadacitinib 30 mg arm) [34].
The risk of malignancy is also a safety concern for patients receiving JAK inhibitors [35]. In this study, the incidence of malignancies was low. Most of the NMSCs in either dose group were basal cell carcinomas, which is the most common malignancy worldwide [36]. A study that used data from the Consortium of Rheumatology Researchers of North America registry found the types of malignancies with the highest incidence rates in patients with PsA were NMSC and breast cancer, followed by prostate cancer, hematologic malignancies, colorectal cancer, and melanoma [37]. The types of malignancy reported here are consistent with those most commonly seen in the PsA population, except no cases of breast cancer or hematologic malignancies were reported. There were no notable patterns to the types of malignancies observed, and the rates of malignancies with upadacitinib were comparable with those reported with other JAK inhibitors [38,39,40]. Moreover, a systematic review and meta-analysis of clinical trials in which adults with immune-mediated disease (including ankylosing spondylitis and psoriasis) received a JAK inhibitor suggested that there is no increased risk of malignancy with JAK inhibitors [41].
A limitation of this study is that there was no placebo control from week 24 onwards. There was also no assessment of radiographic progression outcomes, although the effect of upadacitinib on radiographic progression in patients with PsA has been evaluated in the SELECT-PsA 1 trial. Another limitation is that this study was not powered or designed to include a pre-specified statistical comparison of efficacy between the upadacitinib treatment arms through week 56. Lastly, due to the relatively short study duration, limited safety conclusions can be drawn around MACE, VTE, and malignancy, which may have longer latency, as well as rare events.
Conclusions
In summary, this study shows that the efficacy of upadacitinib 15 mg and 30 mg QD was maintained over 56 weeks and was comparable between upadacitinib doses in patients with PsA and an inadequate response to biologic treatment. The safety profile of upadacitinib over 56 weeks was comparable with that in the double-blind period and with that reported in the upadacitinib rheumatoid arthritis trials, with no new significant safety signals identified. As PsA is a chronic condition, longer observation of the benefits of upadacitinib is warranted. However, these results suggest that upadacitinib could be a favorable long-term treatment option in patients with PsA who are refractory to biologic therapy.
References
Ritchlin CT, Colbert RA, Gladman DD. Psoriatic arthritis. N Engl J Med. 2017;376:2095–6.
Gossec L, Baraliakos X, Kerschbaumer A, de Wit M, McInnes I, Dougados M, et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann Rheum Dis. 2020;79:700–12.
Singh JA, Guyatt G, Ogdie A, Gladman DD, Deal C, Deodhar A, et al. Special article: 2018 American college of rheumatology/national psoriasis foundation guideline for the treatment of psoriatic arthritis. Arthritis Care Res (Hoboken). 2019;71:2–29.
Coates LC, Kavanaugh A, Mease PJ, Soriano ER, Laura Acosta-Felquer M, Armstrong AW, et al. Group for research and assessment of psoriasis and psoriatic arthritis 2015 treatment recommendations for psoriatic arthritis. Arthritis Rheumatol. 2016;68:1060–71.
Nash P, Ohson K, Walsh J, Delev N, Nguyen D, Teng L, et al. Early and sustained efficacy with apremilast monotherapy in biological-naive patients with psoriatic arthritis: a phase IIIB, randomised controlled trial (ACTIVE). Ann Rheum Dis. 2018;77:690–8.
Kavanaugh A, McInnes IB, Mease PJ, Hall S, Chinoy H, Kivitz AJ, et al. Efficacy of subcutaneous secukinumab in patients with active psoriatic arthritis stratified by prior tumor necrosis factor inhibitor use: results from the randomized placebo-controlled FUTURE 2 study. J Rheumatol. 2016;43:1713–7.
Gladman D, Rigby W, Azevedo VF, Behrens F, Blanco R, Kaszuba A, et al. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N Engl J Med. 2017;377:1525–36.
Ritchlin C, Rahman P, Kavanaugh A, McInnes IB, Puig L, Li S, et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann Rheum Dis. 2014;73:990–9.
Mease PJ, Fleischmann R, Deodhar AA, Wollenhaupt J, Khraishi M, Kielar D, et al. Effect of certolizumab pegol on signs and symptoms in patients with psoriatic arthritis: 24-week results of a phase 3 double-blind randomised placebo-controlled study (RAPID-PsA). Ann Rheum Dis. 2014;73:48–55.
Parmentier JM, Voss J, Graff C, Schwartz A, Argiriadi M, Friedman M, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. 2018;2:23.
Genovese MC, Lertratanakul A, Anderson JK, Papp KA, Tillett W, Van den Bosch F, et al. Efficacy and safety of upadacitinib in patients with active psoriatic arthritis and inadequate response to biologic disease-modifying anti-rheumatic drugs (SELECT-PsA-2): a double-blind, randomized controlled phase 3 trial. Ann Rheum Dis. 2020;79(Suppl 1):139.
Mease P, Lertratanakul A, Anderson JK, Papp KA, Bosch F, Tsuji S, et al. Upadacitinib for psoriatic arthritis refractory to biologics: SELECT-PsA 2. Ann Rheum Dis. 2020;80:312–20.
Mease P, Kavanaugh A, Gladman D, Gossec L, Fitzgerald O, Soriano E, et al. Characterization of remission in patients with psoriatic arthritis treated with upadacitinib: post-hoc analysis from two phase 3 Trials. Arthritis Rheumatol. 2019;71(Suppl 10). https://acrabstracts.org/abstract/characterization-of-remission-in-patients-with-psoriatic-arthritis-treated-with-upadacitinib-post-hoc-analysis-from-two-phase-3-trials/.
Taylor W, Gladman D, Helliwell P, Marchesoni A, Mease P, Mielants H. Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum. 2006;54:2665–73.
Coates L, Fransen J, Helliwell P. Defining minimal disease activity in psoriatic arthritis: a proposed objective target for treatment. Ann Rheum Dis. 2010;69:48–53.
Fredriksson T, Pettersson U. Severe psoriasis—oral therapy with a new retinoid. Dermatologica. 1978;157:238–44.
Langley R, Feldman S, Nyirady J, van de Kerkhof P, Papavassilis C. The 5-point investigator’s global assessment (IGA) scale: a modified tool for evaluating plaque psoriasis severity in clinical trials. J Dermatolog Treat. 2015;26:23–31.
Challa DNV, Crowson CS, Davis JM 3rd. The patient global assessment of disease activity in rheumatoid arthritis: identification of underlying latent factors. Rheumatol Ther. 2017;4:201–8.
Healy P, Helliwell P. Measuring clinical enthesitis in psoriatic arthritis: assessment of existing measures and development of an instrument specific to psoriatic arthritis. Arthritis Rheum. 2008;59:686–91.
Helliwell P, Firth J, Ibrahim G, Melsom R, Shah I, Turner D. Development of an assessment tool for dactylitis in patients with psoriatic arthritis. J Rheumatol. 2005;32:1745–50.
Fries J, Spitz P, Young D. The dimensions of health outcomes: the health assessment questionnaire, disability and pain scales. J Rheumatol. 1982;9:789–93.
Chandran V, Bhella S, Schentag C, Gladman D. Functional assessment of chronic illness therapy-fatigue scale is valid in patients with psoriatic arthritis. Ann Rheum Dis. 2007;66:936–9.
Ware J, Kosinski M, Dewey J. How to score version two of the SF-36 health survey. Lincoln: Quality Metric; 2000.
Armstrong A, Banderas B, Foley C, Stokes J, Sundaram M, Shields A. Development and psychometric evaluation of the self-assessment of psoriasis symptoms (SAPS)–clinical trial and the SAPS–real world patient-reported outcomes. J Dermatolog Treat. 2017;28:505–14.
Garrett S, Jenkinson T, Kennedy LG, Whitelock H, Gaisford P, Calin A. A new approach to defining disease status in ankylosing spondylitis: the Bath Ankylosing Spondylitis Disease Activity Index. J Rheumatol. 1994;21:2286–91.
Clegg DO, Reda DJ, Mejias E, Cannon GW, Weisman MH, Taylor T, et al. Comparison of sulfasalazine and placebo in the treatment of psoriatic arthritis. A Department of Veterans Affairs cooperative study. Arthritis Rheum. 1996;39:2013–20.
Mease PJ, Woolley JM, Bitman B, Wang BC, Globe DR, Singh A. Minimally important difference of Health Assessment Questionnaire in psoriatic arthritis: relating thresholds of improvement in functional ability to patient-rated importance and satisfaction. J Rheumatol. 2011;38:2461–5.
Burmester GR, Kremer JM, Van den Bosch F, Kivitz A, Bessette L, Li Y, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391:2503–12.
Fleischmann R, Pangan AL, Song IH, Mysler E, Bessette L, Peterfy C, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III, double-blind, randomized controlled trial. Arthritis Rheumatol. 2019;71:1788–800.
Fleischmann RM, Genovese MC, Enejosa JV, Mysler E, Bessette L, Peterfy C, et al. Safety and effectiveness of upadacitinib or adalimumab plus methotrexate in patients with rheumatoid arthritis over 48 weeks with switch to alternate therapy in patients with insufficient response. Ann Rheum Dis. 2019;78:1454–62.
Genovese MC, Fleischmann R, Combe B, Hall S, Rubbert-Roth A, Zhang Y, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet. 2018;391:2513–24.
Smolen JS, Pangan AL, Emery P, Rigby W, Tanaka Y, Vargas JI, et al. Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet. 2019;393:2303–11.
van Vollenhoven R, Takeuchi T, Pangan AL, Friedman A, Mohamed MF, Chen S, et al. Efficacy and safety of upadacitinib monotherapy in methotrexate-naive patients with moderately to severely active rheumatoid arthritis (SELECT-EARLY): a randomized, double-blind, active-comparator, multi-center, multi-country trial. Arthritis Rheumatol. 2020;72:1607–20.
McInnes IB, Anderson JK, Magrey M, Merola JF, Liu Y, Kishimoto M, et al. Efficacy and safety of upadacitinib versus placebo and adalimumab in patients with active psoriatic arthritis and inadequate response to non-biologic disease-modifying anti-rheumatic drugs (SELECT-PsA-1): a double-blind, randomized controlled phase 3 trial. Ann Rheum Dis. 2020;79(Suppl 1):12. https://doi.org/10.1136/annrheumdis-2020-eular.6727.
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70.
Cameron MC, Lee E, Hibler BP, Barker CA, Mori S, Cordova M, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303–17.
Gross RL, Schwartzman-Morris JS, Krathen M, Reed G, Chang H, Saunders KC, et al. A comparison of the malignancy incidence among patients with psoriatic arthritis and patients with rheumatoid arthritis in a large US cohort. Arthritis Rheumatol. 2014;66:1472–81.
Nash P, Coates LC, Kivitz AJ, Mease PJ, Gladman DD, Covarrubias-Cobos JA, et al. Safety and efficacy of tofacitinib in patients with active psoriatic arthritis: interim analysis of OPAL balance, an open-label, long-term extension study. Rheumatol Ther. 2020;7:553–80.
Smolen JS, Genovese MC, Takeuchi T, Hyslop DL, Macias WL, Rooney T, et al. Safety profile of baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J Rheumatol. 2019;46:7–18.
Wollenhaupt J, Lee EB, Curtis JR, Silverfield J, Terry K, Soma K, et al. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: final results of a global, open-label, long-term extension study. Arthritis Res Ther. 2019;21:89.
Olivera PA, Lasa JS, Bonovas S, Danese S, Peyrin-Biroulet L. Safety of Janus kinase inhibitors in patients with inflammatory bowel diseases or other immune-mediated diseases: a systematic review and meta-analysis. Gastroenterology. 2020;158(1554–73):e12.
Acknowledgments
We thank the participants of the study.
Funding
AbbVie funded this study and participated in the study design, research, analysis, data collection, interpretation of data, reviewing, and approval of the publication. No honoraria or payments were made for authorship. AbbVie also funded the journal’s rapid service fee.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article; all authors had access to relevant data and participated in drafting the article and revising it critically for important intellectual content and take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Medical Writing Assistance
Medical writing assistance was provided by John Ewbank, PhD, Frances Smith, PhD, and Russell Craddock, PhD, of 2 the Nth (Cheshire, UK), and was funded by AbbVie.
Disclosures
Philip Mease has received research grants, consulting fees, and/or speaker’s fees from AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squib, Celgene, Galapagos, Genentech, Gilead, GlaxoSmithKline, Janssen, Lilly, Novartis, Pfizer, Sun Pharma, and UCB. Kim Papp has served as a scientific adviser and/or clinical study investigator for AbbVie, Akros, Allergan, Almirall, Amgen, Arcutis, Avillion, Bausch Health, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Dermavant, Dermira, Eli Lilly and Company, Galderma, Genentech/Roche, GlaxoSmithKline, Janssen, Kyowa Kirin, LEO, Meiji, Merck Sharp & Dohme, Novartis, Pfizer, Regeneron, Sanofi Genzyme, Sienna Pharmaceuticals, Sun Pharma, Takeda, UCB, and Valeant; and as a paid speaker for AbbVie, Akros, Allergan, Almirall, Amgen, Bausch Health, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Dermavant, Dermira, Eli Lilly and Company, Galderma, Genentech/Roche, Janssen, Kyowa Kirin, LEO, Meiji, Merck Sharp & Dohme, Novartis, Pfizer, Regeneron, Sanofi Genzyme, Sienna Pharmaceuticals, Sun Pharma, Takeda, UCB, and Valeant. Filip van den Bosch has received speaker and/or consulting fees from AbbVie, Bristol Myers Squibb, Celgene, Eli Lilly, Galapagos, Janssen, Merck, Novartis, Pfizer, Sanofi, and UCB. Shigeyoshi Tsuji has received research grants and speaker and/or consulting fees from AbbVie, Eli Lilly, Janssen, Novartis, and UCB. Eva Dokoupilova has received research grants from AbbVie, Affibody AB, Eli Lilly, Galapagos, Gilead, GlaxoSmithKline, Hexal AG, Merck, Novartis, Pfizer, R-Pharm, Sanofi-Aventis, and UCB Biopharma SPRL. Apinya Lertratanakul, Xianwei Bu, Liang Chen, Reva M. McCaskill, Patrick Zueger, Erin L. McDearmon-Blondell, and Aileen L. Pangan are employees of AbbVie and may own stock or options. Mauro Keiserman has received research/speaker/advisory board grants/fees from AbbVie, Celgene, Lilly, Bristol, Roche, Novartis, Pfizer, Janssen, and UCB. William Tillett has received grant/research support from AbbVie, Celgene, Janssen, and Eli Lilly, has acted as a consultant for AbbVie, Amgen, Celgene, Eli Lilly, MSD, Novartis, Pfizer, and UCB, and has received speaker bureau fees from AbbVie, Amgen, Celgene, Eli Lilly, Janssen, Novartis, UCB, and Pfizer.
Compliance with Ethics Guidelines
The study was conducted according to the International Conference on Harmonisation guidelines, applicable regulations and guidelines governing clinical trial conduct, and the Declaration of Helsinki 1964 and its later amendments. The trial protocol was approved by independent ethics committees and institutional review boards provided in supplementary materials. All patients provided written informed consent.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis datasets), as well as other information (e.g., protocols and Clinical Study Reports), provided the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and statistical analysis plan, and execution of a Data Sharing Agreement. Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Mease, P.J., Lertratanakul, A., Papp, K.A. et al. Upadacitinib in Patients with Psoriatic Arthritis and Inadequate Response to Biologics: 56-Week Data from the Randomized Controlled Phase 3 SELECT-PsA 2 Study. Rheumatol Ther 8, 903–919 (2021). https://doi.org/10.1007/s40744-021-00305-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-021-00305-z