
Abstract
Introduction
To evaluate the efficacy and safety of sirukumab in giant cell arteritis (GCA).
Methods
In this multicentre, randomised, double-blind, placebo-controlled, two-part phase 3 trial (NCT02531633; Part A [52-week double-blind treatment]; Part B [104-week follow-up]), patients with GCA were randomised (3:3:2:2:2) to sirukumab 100 mg every 2 weeks plus 6-month or 3-month prednisone taper, sirukumab 50 mg every 4 weeks plus 6-month prednisone taper, or placebo every 2 weeks plus 6-month or 12-month prednisone taper. The primary endpoint was the proportion of patients in sustained remission at week 52. Secondary endpoints included disease flare and safety. The study was terminated early (October 2017; sponsor decision).
Results
Of 161 patients randomised (sirukumab: n = 107; placebo: n = 54), 28 (17.4%) completed week 52 (median treatment duration: 24–30 weeks). In a revised intent-to-treat (ITT) subgroup (completed week 52 or discontinued before study termination [n = 55]); six patients (all receiving sirukumab) achieved the primary endpoint. In the ITT population (n = 161), the proportion of patients with flares (week 2–52) was lower with sirukumab (18.4–30.8%) than placebo (37.0–40.0%). The proportion of patients with flares (week 2–12) was highest with sirukumab 100 mg every 2 weeks plus 3-month prednisone taper (23.1%). In Part A, 94.4% of patients reported ≥ 1 treatment-emergent adverse event (TEAE); 19.3% reported serious TEAEs. The proportions of patients with TEAEs were generally similar across treatment arms. No deaths occurred.
Conclusions
Although data were limited due to early termination and shortened treatment duration, sirukumab treatment resulted in numerically lower proportions of patients with flare by week 52 versus placebo, with no unexpected safety findings.
Trial Registration
Clinicaltrials.gov: NCT02531633.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Giant cell arteritis (GCA) is the most common form of primary systemic vasculitis, predominantly affecting people aged ≥ 50 years. |
Oral glucocorticoids (the mainstay treatment for GCA) require dose adjustment upon disease flare, which can lead to prolonged glucocorticoid tapers, high cumulative glucocorticoid exposure, and substantial toxicity; therefore, there is a need for remission maintenance with glucocorticoid-sparing medications in GCA. |
Sirukumab, a selective, high-affinity human interleukin (IL)-6 monoclonal antibody, may have therapeutic benefit in GCA by interrupting the IL-6 pathway, which plays a major role in GCA pathophysiology; this two-part, phase 3 randomised, placebo-controlled study aimed to evaluate the efficacy and safety of sirukumab (administered subcutaneously every 2 weeks or every 4 weeks, in addition to a prednisone taper) in patients with GCA, over 52 weeks. |
What was learned from the study? |
In a subset of patients who completed the study (or discontinued before the study was terminated), sustained GCA remission at week 52 was achieved by a small number of patients, all receiving sirukumab; in the larger, intent-to-treat population of all randomised patients, the proportion of patients with disease flares up to week 52 was lower with sirukumab than placebo. |
Although data interpretation was limited due to early termination of the study, and thus a shortened treatment duration, the proportion of patients with disease flare by week 52 tended to be lower with sirukumab versus placebo, and overall, safety findings were consistent with the known safety profile of sirukumab. |
Introduction
Giant cell arteritis (GCA) is the most common form of primary systemic vasculitis and predominantly affects people aged ≥ 50 years, with higher prevalence in women than men [1,2,3,4]. Symptoms include headache, jaw claudication, vision impairment, scalp tenderness, and constitutional and polymyalgia rheumatica-related symptoms (e.g., proximal limb girdle stiffness) [5]. The disease may be complicated by blindness, limb claudication, and rarely, stroke and myocardial infarction [1, 3, 4, 6, 7].
The recommended initial GCA treatment is high-dose glucocorticoids [3, 4, 8], tapered gradually over at least 12–18 months [9, 10]. Unfortunately, disease flare requiring adjustment of glucocorticoid dose occurs in 45–80% of patients [11,12,13,14], leading to prolonged glucocorticoid tapers and high glucocorticoid cumulative exposure and toxicity [15,16,17]. Glucocorticoid-related adverse events (AEs) can cause substantial morbidity [4, 17, 18] and are often serious [18]; therefore, there is need for remission maintenance with glucocorticoid-sparing medications in GCA [16, 19, 20].
Interleukin (IL)-6 has a major role in GCA pathophysiology [21,22,23]. It is upregulated in arterial biopsies of patients [24], and serum levels correlate with disease activity [22, 25, 26]; persistent elevation predicts a relapsing disease course [26, 27]. Additionally, patients with active disease demonstrate increased frequencies of IL-17-producing regulatory T cells (‘inflammatory Tregs’) in peripheral blood, which normalise upon IL-6 blockade therapy [28]. In the GiACTA trial, the IL-6 receptor alpha inhibitor tocilizumab, administered subcutaneously weekly or every 2 weeks (q2w) plus a 26-week prednisone taper, was superior to placebo in maintenance of glucocorticoid-free remission in patients with GCA [2, 29].
Sirukumab, a selective, high-affinity human IL-6 monoclonal antibody [30], may have therapeutic benefit in GCA treatment by interrupting the IL-6 pathogenic pathway. Sirukumab reduced rheumatoid arthritis (RA) disease activity and improved physical function and quality of life in previous trials, in which patients with RA received sirukumab 100 mg every 2 weeks (q2w), 50 mg every 4 weeks (q4w) or placebo q2w for 52 weeks [31, 32].
This two-part study aimed to evaluate efficacy and safety of sirukumab in GCA treatment. The study was terminated prematurely due to the sponsor’s decision to discontinue sirukumab development in autoimmune disease treatment.
Methods
Study Design and Treatment
This was a multicentre, randomised, double-blind, placebo-controlled, two-part phase 3 trial: Part A was a 52-week double-blind treatment phase to establish efficacy and safety of sirukumab; Part B was a 104-week long-term extension phase with optional open-label sirukumab treatment (up to 52 weeks) for patients with active disease at the discretion of the investigator. The study was conducted at 56 hospitals and clinics in Australia, Belgium, Bulgaria, France, Germany, Hungary, Italy, the Netherlands, New Zealand, Spain, the UK, and the USA.
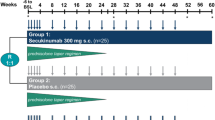
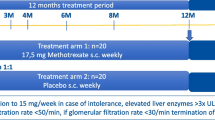
Following a ≤ 6-week screening phase, eligible patients were randomised (3:3:2:2:2) at baseline (week 0) to receive: sirukumab 100 mg q2w for 12 months plus a 6-month prednisone taper; sirukumab 100 mg q2w for 12 months plus a 3-month prednisone taper; sirukumab 50 mg q4w for 12 months plus a 6-month prednisone taper; placebo q2w for 12 months plus a 6-month prednisone taper; or placebo q2w for 12 months plus a 12-month prednisone taper (Fig. 1).

Study design (as originally planned). aRescue glucocorticoid permitted without the requirement to withdraw. bOptional (investigator discretion). q2w every 2 weeks, q4w every 4 weeks
Randomisation was performed centrally via an Interactive Response Technology System according to a computer-generated schedule (generated prior to study commencement), stratified by baseline prednisone dose (< 30 mg/day; ≥ 30 mg/day). Enrolment of patients with relapsed/refractory disease would be capped at approximately 50% to ensure enough patients with new-onset disease were included.
To maintain blinding, patients in the sirukumab 50 mg q4w arm received placebo injections q2w (alternating between sirukumab and placebo), both administered subcutaneously in visually matched syringes using prefilled SmartJect® autoinjectors. All patients received prednisone (≥ 20 mg/day) at the start of screening. At baseline, prednisone dose was 20–60 mg/day, with the starting dose chosen by investigators based on their best clinical judgement. Patients remained on this prednisone dose for the first week following baseline; subsequently, the prednisone taper was administered open-label for doses ≥ 20 mg/day and blinded for doses < 20 mg/day by providing prednisone and matching placebo in blister packs. Patients experiencing disease flare were treated with an investigator-defined open-label prednisone rescue regimen while continuing the double-blind injections of sirukumab/placebo. Investigators, patients, and the sponsor/study team were blinded to treatment throughout the 52-week treatment period (detailed in Supplementary Appendix 1). During Part B, open-label sirukumab treatment was administered as per investigator discretion.
Patients completing Part A were eligible to enter Part B. Patients in remission at week 52 discontinued blinded sirukumab/placebo and were observed for maintenance of remission; in the event of disease flare, patients could receive open-label sirukumab 100 mg q2w for up to 52 weeks (per investigator discretion). Patients not in remission at week 52 or unable to tolerate prednisone taper could also receive open-label sirukumab 100 mg q2w for up to 52 weeks.
Ethics
This study (GSK study number: 201677; NCT02531633) was reviewed and approved by institutional review boards and local research ethics committees before commencement (listed in Supplementary Appendix 2). The study was conducted in accordance with International Council on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Good Clinical Practice ethical principles and the Declaration of Helsinki [33]. Patients provided written informed consent prior to study commencement. The full study protocol is available at https://www.clinicaltrials.gov/ProvidedDocs/33/NCT02531633/Prot_000.pdf.
Patient Population
Patients aged ≥ 50 years with active GCA were included. GCA was diagnosed according to the following criteria: history of erythrocyte sedimentation rate (ESR) ≥ 50 mm/h and/or C-reactive protein (CRP) ≥ 2.45 mg/dl; plus unequivocal cranial GCA symptoms and/or unequivocal polymyalgia rheumatica symptoms; plus features of GCA by temporal artery biopsy or imaging (e.g., ultrasound, magnetic resonance angiography, computed tomography angiography, positron emission tomography-computed tomography). Active GCA was defined as the presence of unequivocal cranial GCA symptoms and/or unequivocal polymyalgia rheumatica symptoms and ESR or CRP elevation (ESR ≥ 30 mm/h or CRP ≥ 1 mg/dl) within 6 weeks of baseline. Patients with a major ischemic event (unrelated to GCA) within 12 weeks of screening, marked prolongation of QTc interval, or receiving certain medications were excluded. Major inclusion/exclusion criteria are detailed in Supplementary Appendix 1.
Endpoints and Assessments
The primary endpoint was the proportion of patients in sustained remission at week 52. Sustained remission was defined as achievement of all the following: remission (absence of clinical signs and symptoms of GCA and normalisation of ESR [< 30 mm/h] and CRP [< 1 mg/dl]) by week 12; absence of disease flare (recurrence of symptoms attributable to active GCA with or without elevations in ESR/CRP) following remission at week 12 through week 52; completion of assigned prednisone taper protocol; no requirement for rescue therapy at any time through week 52.
Secondary endpoints in Part A included the proportion of patients with disease flare, cumulative prednisone-equivalent dose at week 52, patient- and physician-reported outcomes (Supplementary Appendix 1), change in serum ESR and CRP from baseline over time (collected at every visit), and safety of sirukumab compared with placebo.
Safety assessments included incidence of treatment-emergent AEs (TEAEs; from first dose to 16 weeks post last dose), treatment-emergent serious AEs (TESAEs), AEs of special interest (AESIs; listed in Supplementary Appendix 1), and changes in clinical chemistry parameters. The National Cancer Institute Common Terminology Criteria for AEs version 4.03 [34] were used to grade laboratory abnormalities. Additional secondary endpoints are listed in Supplementary Appendix 1.
Key endpoints in Part B were maintenance of disease remission (proportion of patients remaining in remission without requiring rescue therapy or treatment change over time) at week 24 (Part B) and after cessation of 12-month sirukumab treatment, and safety (as per Part A).
Statistical Analyses
Due to early study termination, sample size was the number of patients randomly assigned to treatment at termination. The original study sample size was calculated assuming a 30% sustained remission rate in the placebo plus 6-month prednisone taper arm, versus a 70% sustained remission rate in the sirukumab plus 6-month prednisone taper arms at week 52. In order to detect a difference with a 5% significance level, the following sample sizes provided > 91% power: sirukumab 100 mg q2w, n = 51; sirukumab 50 mg q4w, n = 34; placebo, n = 34; all arms in combination with a 6-month prednisone taper. However, given early study termination, the actual sample size was the number of patients randomly assigned to treatment at study termination. A planned interim futility analysis was not performed due to early study termination.
No statistical analyses were performed due to limited patient numbers as a result of early study termination. All data interpretations were based on listed data summarised by treatment group. The intent-to-treat (ITT) population included all randomised patients who received ≥ 1 dose of sirukumab/placebo, analysed according to treatment allocated at randomisation. The primary endpoint was summarised in the revised ITT population: patients who received ≥ 1 dose of sirukumab/placebo and completed 52 weeks or discontinued prior to sponsor announcement of study termination. For the revised primary endpoint, patients who withdrew early were considered treatment failures; missing components, including absence of disease flare, were imputed with the assumption that they have not been met. The safety population included all patients who received ≥ 1 dose of sirukumab/placebo, analysed according to treatment received. The randomised population included all randomised patients. No patients were excluded from ITT or safety populations; therefore, these three populations comprised the same patients.
Post hoc analyses were performed as described in Supplementary Appendix 1. For patients in the revised primary analysis, patient-level data review was performed after study completion to evaluate presence of investigator-reported disease flare, without imputation. Patient-level data review after study completion was also performed to evaluate disease flare rate up to week 12 in all randomised patients.
Results
Patient Demographics and Clinical Characteristics
In total, 246 patients were screened and 161 (of 204 patients originally planned) were randomised (sirukumab: n = 107; placebo: n = 54) at the time of study termination. The first patient was randomised 18 November 2015, study termination was 10 October 2017, and the last patient last visit was 21 March 2018. Overall, 28/161 (17.4%) patients completed week 52 and 133/161 (82.6%) discontinued early, largely due to sponsor request (early study termination) (Fig. 2). Demographic characteristics were similar across treatment arms (Table 1). Proportions of patients with baseline prednisone doses < 30 mg/day and ≥ 30 mg/day were similarly balanced across treatment arms.

Patient disposition. aApplies to patients who withdrew early from the study or who attended with week 52 visit of Part A and did not enter Part B. bViolation of inclusion/exclusion criteria. cOther: sponsor request. dPatients who received ≥ 1 dose SC sirukumab/placebo. ePatients who received ≥ 1 dose of SC sirukumab/placebo and completed 52 weeks (n = 28) or discontinued prior to study termination (n = 27). All patients who were randomised received their allocated treatment. A total of 26 patients continued to Part B; 8/26 received ≥ 1 dose of open-label sirukumab. No patients completed the 104-week extension phase; this was due to early withdrawal or study termination (25 patients withdrawn at sponsor request; one lost to follow-up). ITT intent-to-treat, q2w every 2 weeks, q4w every 4 weeks, SC subcutaneous
Twenty-six patients continued to Part B; 8/26 (30.8%) received ≥ 1 dose of open-label sirukumab and 22/26 (84.6%) completed the 16-week safety follow-up visit. No patients had completed Part B at study termination. One patient was lost to follow-up.
Treatment duration was shorter than planned across all treatment arms (Table 2).
Proportion of Patients in Sustained Remission at Week 52 (Revised Primary Endpoint)
The primary analysis was revised due to early study termination and included only patients who had completed or discontinued the study prior to the study termination announcement to avoid potential bias. In the revised ITT population (N = 55), 28 patients completed week 52 and 27 patients discontinued prior to study termination. The sirukumab 100 mg q2w plus 3-month prednisone arm had the highest proportion of withdrawals (8/13 [61.5%]) (Table 3). A high proportion of patients failed to achieve sustained remission at week 52 across all treatment arms; the most common reason was non-completion of prednisone taper due to early study termination. Six patients, all in sirukumab arms, fulfilled sustained remission criteria at week 52 (Table 3). Sustained remission was not achieved by 82.4–88.9% patients in sirukumab arms and all patients in placebo arms due to: not being in remission at week 12; disease flares from week 12 to week 52; receiving glucocorticoid rescue therapy.
Using the imputation rule for primary endpoint components (where patients who had withdrawn from the study early were counted as having had a flare), 52.9–69.2% and 71.4–88.9% patients in sirukumab and placebo arms, respectively, were considered to have experienced disease flare from week 12–52 due to early study withdrawal (Table 3). An ad hoc review of patient-level data (without imputation) showed the proportion of patients experiencing disease flare from week 12–52 was 5.9–11.1% in sirukumab arms and 28.6–55.6% in placebo arms, lower than in the primary analysis in all treatment groups (Table 3). Glucocorticoid rescue therapy use was consistent with the non-imputed flare results from week 12 to week 52 (Table 3).
Proportion of Randomised Patients with Disease Flare
In randomised patients (n = 161) with ≥ 1 flare assessment from week 2–52, the observed proportion of patients with disease flare was lower in sirukumab (100 mg plus 6-month prednisone: 7/38 [18.4%]; 100 mg plus 3-month prednisone: 11/39 [28.2%]: 50 mg plus 6-month prednisone: 8/26 [30.8%]) than placebo arms (placebo plus 6-month prednisone: 10/25 [40.0%]; placebo plus 12-month prednisone 10/27 [37.0%]). An ad hoc patient-level data review showed 11.9–23.1% and 14.8–18.5% patients in sirukumab and placebo arms, respectively, experienced ≥ 1 flare between week 2 and week 12; this was highest in the sirukumab 100 mg q2w plus 3-month prednisone taper arm (23.1%) (Table 4). In an ad hoc patient-level data review of patients completing week 52 in Part A (completer analysis; N = 28), 11.1–20.0% and 50.0–80.0% patients in sirukumab and placebo arms, respectively, experienced disease flare between week 12 and week 52 (Table 5).
Mean cumulative prednisone-equivalent dose for patients completing week 52 was 2418–2974 mg and 3157–3603 mg in sirukumab and placebo arms, respectively (Supplementary Appendix 3; Supplementary Table 1).
Change in Inflammatory Markers
In sirukumab arms, mean CRP and ESR decreased at week 2 compared with baseline; sustained suppression was observed throughout the study. In placebo arms, mean ESR and CRP increased or remained stable over time (Fig. 3).

Change in inflammatory markers from baseline to week 52: a C-reactive protein and b erythrocyte sedimentation rate (safety population). q2w every 2 weeks, q4w every 4 weeks, SC subcutaneous
Safety
Treatment-Emergent Adverse Events
In Part A, 152/161 (94.4%) patients experienced ≥ 1 TEAE; similar proportions of TEAEs were observed across treatment arms (Table 6). The most common TEAEs (by preferred term) were: headache (43/161 [26.7%]); arthralgia (19/161 [11.8%]); back pain (19/161 [11.8%]). Seventy-seven (47.8%) patients experienced a TEAE considered related to sirukumab/placebo ± prednisone. Thirty-four (21.1%) patients experienced a TEAE related to both sirukumab/placebo and prednisone, most commonly (by system organ class) infections and infestations (23/161 [14.3%]).
Thirty-one (19.3%) patients experienced a TESAE (Table 6); the most common (by preferred term) were: syncope (3/161 [1.9%]); pneumonia (2/161 [1.2%]); temporal arteritis (considered disease flare; 2/161 [1.2%]). TESAE incidence per 100 patient-years of exposure was similar across treatment arms (Table 7). The proportion of patients who discontinued treatment due to a TEAE was higher in sirukumab than placebo arms (Table 6; Supplementary Appendix 3). No deaths occurred during the study.
Adverse Events of Special Interest
Overall, 111/161 (68.9%) patients experienced ≥ 1 AESI; 81/107 (75.7%) and 30/54 (55.6%) in sirukumab and placebo arms, respectively. AESI incidence per 100 patient-years of exposure was higher in sirukumab than placebo arms. The most common AESIs by category were infections and infestations, occurring more frequently with sirukumab than placebo (Table 7); incidence of serious and/or opportunistic infections was similar across treatment arms (Table 7).
The number of patients experiencing ≥ 1 injection site reaction was higher in sirukumab than placebo arms; proportions were similar with sirukumab 100 mg and 50 mg (Table 7). The proportion of patients experiencing glucocorticoid-related events was higher with sirukumab than placebo. No cases of anaphylaxis, tuberculosis activation, or gastrointestinal perforation were reported.
Four patients experienced a malignancy (Table 6). Three patients (all receiving sirukumab) experienced visual disturbances. One patient receiving sirukumab 100 mg q2w plus 3-month prednisone developed retinal artery occlusion with visual disturbance on day 51 (unresolved at study termination). Ophthalmology evaluation revealed a cholesterol embolus of the retinal artery consistent with a transient ischaemic attack and GCA-related optic ischaemia. One patient receiving sirukumab 50 mg q4w plus 6-month prednisone developed a visual field defect with other symptoms of disease flare on study day 142 (unresolved at study termination). Prednisone was discontinued on study day 114 (no further glucocorticoid use noted), but the patient continued sirukumab. Finally, one patient receiving sirukumab 100 mg q2w plus 6-month prednisone developed mild temporary vision loss on day 435 (85 days after the most recent dose of sirukumab) associated with a transient ischaemic attack, secondary to blood loss associated with a fall, considered unrelated to GCA.
AESIs associated with liver function test abnormalities occurred in 6/161 (3.7%) patients in sirukumab arms (no patients in placebo arms). Two patients receiving sirukumab experienced a liver monitoring or stopping event (Table 6; Supplementary Table 2). Nine patients (all in sirukumab arms) experienced a total of 12 cytopenia events (thrombocytopenia [six events]; decreased platelet count [three events]; neutropenia, decreased neutrophil count and decreased white blood cell count [each one event]). No meaningful difference across sirukumab arms and no dose-dependent relationship was observed.
Laboratory Abnormalities
Excluding decreased lymphocytes, grade 3 or 4 laboratory abnormalities in haematology, lipid and liver function tests were reported only in sirukumab arms (Table 8).
Part B
Five patients in sustained remission at week 52 (Part A) entered Part B; 3/5 (60%) maintained remission at week 4 and did not receive open-label sirukumab, and 2/5 (40%) withdrew prior to week 4. None of these patients experienced disease flare.
Overall, 34 AEs were reported in 8 patients who received open-label sirukumab. The only AE experienced by ≥ 2 patients was headache (one patient in the sirukumab 50 mg q4w plus 6-month prednisone arm and 1 in the placebo plus 6-month prednisone arm). One patient (in the placebo plus 12-month prednisone arm) permanently discontinued open-label sirukumab due to abnormal liver function tests. No TESAEs or AESIs were reported in Part B.
Discussion
This study aimed to investigate the safety and efficacy of sirukumab in GCA; however, due to early study termination, a high proportion of patients failed to complete week 52 to achieve the pre-defined sustained remission criteria, limiting interpretation of results from the primary analysis. In a subgroup of patients qualifying for the revised primary analysis (N = 55), sustained remission at week 52 was achieved by only 6 patients, all in sirukumab treatment arms.
As the number of patients in the revised ITT population was limited, secondary endpoints were examined for all randomised patients to increase the robustness of the findings. The proportion of patients in the revised ITT population who completed week 52 and experienced disease flare was lower in sirukumab than placebo arms, in line with observations in the GiACTA trial, although definition of flare differed slightly between studies [2]. Although the majority of randomised patients did not flare prior to week 12, based on patient-level data, the highest proportion of patients with disease flare from week 2 to week 12 was in the sirukumab 100 mg plus 3-month prednisone taper arm. This may reflect the short prednisone taper compared with other treatment arms. Glucocorticoid rescue therapy use was consistent with these flare results.
As reported in other studies of anti-IL-6 therapy in GCA [2, 35] and in line with the known pharmacodynamic effects of IL-6 pathway inhibition [23], levels of acute-phase reactant proteins decreased with sirukumab treatment.
The nature of TEAEs reported in this study was consistent with that reported previously in patients with RA treated with sirukumab [31, 32, 36, 37], and in patients with GCA treated with tocilizumab [2, 38]. The safety profile was similar across sirukumab arms, with no meaningful differences in TEAEs or TESAE incidence. The nature of laboratory abnormalities was consistent with that reported previously for anti-IL-6 therapies [2, 31, 36,37,38].
Overall, the proportion of patients with AESIs was higher in sirukumab than placebo arms. The proportion of patients with glucocorticoid-related AESIs was not lower in patients receiving sirukumab compared with placebo, possibly due to the short study duration. Therefore, it is not possible to draw conclusions regarding the full benefits of glucocorticoid-sparing therapy. The proportion of patents with injection site reactions was higher in sirukumab than placebo arms; these were higher than reported in previous sirukumab studies in RA and tocilizumab studies in GCA [2, 31, 32, 36, 37]. However, consistent with sirukumab phase 3 RA study findings, no meaningful difference in incidence was noted between sirukumab 50 mg and 100 mg arms [31, 36].
Unlike previous sirukumab trials in RA [31, 32, 37], no gastrointestinal perforations, a known consequence of anti-IL-6 therapies [39], were reported in the current study; and there were no reported deaths or tuberculosis re-activations. Two patients receiving sirukumab had visual disturbances considered potentially GCA related. One patient receiving tocilizumab in the GiACTA trial experienced vision loss, which resolved with glucocorticoid treatment [2]. Additionally, in a prospective study evaluating 3-month intravenous tocilizumab with prednisone taper in patients with GCA, one of the ten disease flares occurring during the 1-year follow-up period included transient vision loss, which resolved with glucocorticoid treatment [40]. Therefore, it is important to remain vigilant for ocular ischaemia, even in patients receiving IL-6 blockade therapy in addition to glucocorticoids [2].
The main limitation of this study was early termination, decreasing the number of treated patients and duration of exposure to sirukumab, and therefore limiting interpretation of results. Additionally, the study population may not be truly representative of the real-world GCA patient population, limiting result generalisability; in one study site, only 12/95 (13%) of patients pre-screened for study participation were eligible for randomisation, largely due to concomitant diseases [41]. However, this is a well-known limitation in randomised trials [42, 43]. Finally, consistent with the GiACTA trial [2], the ‘sustained remission’ definition may have favoured patients in IL-6 inhibitor groups, since normalisation of ESR and CRP, a hallmark of anti-IL-6 therapy, was part of the definition. To mitigate this, a sensitivity analysis omitting CRP from the definition of sustained remission could have been performed.
Conclusions
Early study termination and short treatment duration limited interpretation of results. In a primary analysis subset of patients, sustained remission at week 52 was achieved by a small number of patients, all in sirukumab arms. Based on listed data summarised by treatment group, the proportion of patients with disease flare-up to week 52 tended to be lower in the sirukumab arms compared with placebo. Overall, safety findings were consistent with the known safety profile of sirukumab.
References
Salvarani C, Crowson CS, O'Fallon WM, Hunder GG, Gabriel SE. Reappraisal of the epidemiology of giant cell arteritis in Olmsted County, Minnesota, over a fifty-year period. Arthritis Rheum. 2004;51:264–8.
Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. 2017;377:317–28.
Dasgupta B, Borg FA, Hassan N, et al. BSR and BHPR guidelines for the management of giant cell arteritis. Rheumatology. 2010;49:1594–7.
Liddle J, Bartlam R, Mallen CD, et al. What is the impact of giant cell arteritis on patients’ lives? A UK qualitative study. BMJ Open. 2017;7:e017073.
Pineles SL. Giant cell arteritis. 2019. https://eyewiki.aao.org/Giant_Cell_Arteritis. Accessed 9 Mar 2020.
Villa-Forte A. Giant cell arteritis: suspect it, treat it promptly. Cleve Clin J Med. 2011;78:265–70.
Amiri N, De Vera M, Choi HK, Sayre EC, Avina-Zubieta JA. Increased risk of cardiovascular disease in giant cell arteritis: a general population-based study. Rheumatology. 2016;55:33–40.
Hellmich B, Agueda A, Monti S, et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis. 2020;79:19–30.
Docken WP. Treatment of giant cell arteritis. 2019. https://www.uptodate.com/contents/treatment-of-giant-cell-arteritis/print. Accessed 1 July 2019.
National Institute for Health and Care Excellence. Giant cell arteritis. 2014. https://www.cks.nice.org.uk/giant-cell-arteritis#!scenario. [Accessed 3 June 2019].
Hachulla E, Boivin V, Pasturel-Michon U, et al. Prognostic factors and long-term evolution in a cohort of 133 patients with giant cell arteritis. Clin Exp Rheumatol. 2001;19:171–6.
Alba MA, García-Martínez A, Prieto-González S, et al. Relapses in patients with giant cell arteritis: prevalence, characteristics, and associated clinical findings in a longitudinally followed cohort of 106 patients. Medicine. 2014;93:194–201.
Labarca C, Koster MJ, Crowson CS, et al. Predictors of relapse and treatment outcomes in biopsy-proven giant cell arteritis: a retrospective cohort study. Rheumatology. 2016;55:347–56.
Mainbourg S, Addario A, Samson M, et al. Prevalence of giant cell arteritis relapse in patients treated with glucocorticoids: a meta-analysis. Arthritis Care Res. 2020;72:838–49.
Meskimen S, Cook TD, Blake RL Jr. Management of giant cell arteritis and polymyalgia rheumatica. Am Fam Physician. 2000;61(2061–8):73.
Fraser JA, Weyand CM, Newman NJ, Biousse V. The treatment of giant cell arteritis. Rev Neurol Dis. 2008;5:140–52.
Proven A, Gabriel SE, Orces C, O'Fallon WM, Hunder GG. Glucocorticoid therapy in giant cell arteritis: duration and adverse outcomes. Arthritis Rheum. 2003;49:703–8.
Low C, Conway R. Current advances in the treatment of giant cell arteritis: the role of biologics. Ther Adv Musculoskelet Dis. 2019;11:1759720X19827222.
Chandran A, Udayakumar PD, Kermani TA, Warrington KJ, Crowson CS, Matteson EL. Glucocorticoid usage in giant cell arteritis over six decades (1950 to 2009). Clin Exp Rheumatol. 2015;33:S-98–S-102.
Evans J, Steel L, Borg F, Dasgupta B. Long-term efficacy and safety of tocilizumab in giant cell arteritis and large vessel vasculitis. RMD Open. 2016;2:e000137.
Terrades-Garcia N, Cid MC. Pathogenesis of giant-cell arteritis: how targeted therapies are influencing our understanding of the mechanisms involved. Rheumatology. 2018;57:ii51–ii62.
Dasgupta B, Panayi GS. Interleukin-6 in serum of patients with polymyalgia rheumatica and giant cell arteritis. Br J Rheumatol. 1990;29:456–8.
Unizony S, Kermani TA. IL-6 blockade and its therapeutic success in giant cell arteritis. J Neuroophthalmol. 2018;38:551–8.
Hernandez-Rodriguez J, Segarra M, Vilardell C, et al. Tissue production of pro-inflammatory cytokines (IL-1beta, TNFalpha and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatology. 2004;43:294–301.
Emilie D, Liozon E, Crevon MC, et al. Production of interleukin 6 by granulomas of giant cell arteritis. Hum Immunol. 1994;39:17–24.
van der Geest KS, Abdulahad WH, Rutgers A, et al. Serum markers associated with disease activity in giant cell arteritis and polymyalgia rheumatica. Rheumatology. 2015;54:1397–402.
Garcia-Martinez A, Hernandez-Rodriguez J, Espigol-Frigole G, et al. Clinical relevance of persistently elevated circulating cytokines (tumor necrosis factor alpha and interleukin-6) in the long-term follow-up of patients with giant cell arteritis. Arthritis Care Res. 2010;62:835–41.
Miyabe C, Miyabe Y, Strle K, et al. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann Rheum Dis. 2017;76:898–905.
Stone JH, Tuckwell K, Dimonaco S, et al. Glucocorticoid doses and acute-phase reactants at giant cell arteritis flare in a randomized trial of tocilizumab. Arthritis Rheumatol. 2019;71:1329–38.
Xu Z, Bouman-Thio E, Comisar C, et al. Pharmacokinetics, pharmacodynamics and safety of a human anti-IL-6 monoclonal antibody (sirukumab) in healthy subjects in a first-in-human study. Br J Clin Pharmacol. 2011;72:270–81.
Takeuchi T, Thorne C, Karpouzas G, et al. Sirukumab for rheumatoid arthritis: the phase III SIRROUND-D study. Ann Rheum Dis. 2017;76:2001–8.
Aletaha D, Bingham CO 3rd, Tanaka Y, et al. Efficacy and safety of sirukumab in patients with active rheumatoid arthritis refractory to anti-TNF therapy (SIRROUND-T): a randomised, double-blind, placebo-controlled, parallel-group, multinational, phase 3 study. Lancet. 2017;389:1206–17.
International Council for Harmoisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). ICH E6 good clinical practice. 2016. https://www.ich.org/page/efficacy-guidelines. Accessed 9 Mar 2020.
National Institutes for Health (NIH) National Cancer Institute. National Cancer Institute common terminology criteria for adverse events (NCI CTCAE v4.03). 2010. https://www.ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_40. Accessed 9 Mar 2020.
Calderon-Goercke M, Loricera J, Aldasoro V, et al. Tocilizumab in giant cell arteritis. Observational, open-label multicenter study of 134 patients in clinical practice. Semin Arthritis Rheum. 2019;49(1):126–35.
Thorne C, Takeuchi T, Karpouzas GA, et al. Investigating sirukumab for rheumatoid arthritis: 2-year results from the phase III SIRROUND-D study. RMD Open. 2018;4:e000731.
Taylor PC, Schiff MH, Wang Q, et al. Efficacy and safety of monotherapy with sirukumab compared with adalimumab monotherapy in biologic-naive patients with active rheumatoid arthritis (SIRROUND-H): a randomised, double-blind, parallel-group, multinational, 52-week, phase 3 study. Ann Rheum Dis. 2018;77:658–66.
Villiger PM, Adler S, Kuchen S, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387:1921–7.
Jagpal A, Curtis JR. Gastrointestinal perforations with biologics in patients with rheumatoid arthritis: implications for clinicians. Drug Saf. 2018;41:545–53.
Samson M, Devilliers H, Ly KH, et al. Tocilizumab as an add-on therapy to glucocorticoids during the first 3 months of treatment of giant cell arteritis: a prospective study. Eur J Intern Med. 2018;57:96–104.
Schmidt WA, Hofheinz K, Burger S, Schafer VS, Juche A. Eligibility of patients with giant cell arteritis for entry into a prospective randomised controlled trial: A single-centre experience. Ann Rheum Dis. 2018;77(Suppl):A1496.
Rothwell PM. Factors that can affect the external validity of randomised controlled trials. PLoS Clin Trials. 2006;1:e9.
Murad MH, Katabi A, Benkhadra R, Montori VM. External validity, generalisability, applicability and directness: a brief primer. BMJ Evid Based Med. 2018;23:17–9.
Acknowledgements
Our thanks to the patients who participated in this study, and to the staff and principal investigators at the study sites (see Supplementary Appendix 4). The authors acknowledge Janssen Research & Development, LLC, for supply of the investigative product.
Funding
This study (GSK study number: 201677; NCT02531633) and Rapid Service Fee were funded by GlaxoSmithKline (GSK).
Medical Writing and Editorial Assistance
Medical writing and editorial support were provided by Gillian Wallace, MSc, at Fishawack Indicia Ltd, UK, and were funded by GSK.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Prior Presentation
The results of this study were published in part in abstract form (Schmidt et al. Ann Rheum Dis. 2019;78:827) and were presented in part as a poster presentation at the annual European League Against Rheumatism (EULAR) meeting, Madrid, 12–15 June 2019.
Disclosures
Wolfgang A. Schmidt has received consulting fees and grants from GSK, Novartis, Roche, and Sanofi; has participated in speaker bureaus for Chugai, GSK, Novartis, Roche, and Sanofi; and has been an investigator on clinical trials for GSK, Novartis, Roche, and Sanofi. Bhaskar Dasgupta has received consultancy fees from GSK, Roche, Sanofi, Bristol-Myers Squibb, and AbbVie, and has received speaker fees from Roche. Raashid Luqmani has received grants from Celgene, GSK, Roche, and Vifor. Sebastian H. Unizony has received consulting fees from GSK, Kiniksa, and Sanofi, and has received grants from Genentech. Daniel Blockmans has received consultancy fees from Roche. Zhihong Lai, Regina H. Kurrasch, and Ivana Lazic are employees of GSK and hold shareholder status in the company. Kurt Brown and Ravi Rao were employees of GSK and held shareholder status at the time of study conduct. Current affiliations: Kurt Brown, Incyte (Delaware, USA); Ravi Rao, Aeglea BioTherapeutics (Texas, USA).
Compliance with Ethics Guidelines
This study (GSK study number: 201677; NCT02531633) was reviewed and approved by institutional review boards and local research ethics committees before commencement (listed in Supplementary Appendix 2). The study was conducted in accordance with International Council on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Good Clinical Practice ethical principles and the Declaration of Helsinki [33]. Patients provided written informed consent prior to study commencement. The full study protocol is available at https://www.clinicaltrials.gov/ProvidedDocs/33/NCT02531633/Prot_000.pdf.
Data Availability
The datasets generated during and/or analysed during the current study are available in the ClinicalStudyDataRequest.com repository upon request, from https://www.clinicalstudydatarequest.com.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features
To view digital features for this article go to: https://doi.org/10.6084/m9.figshare.12746993.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Schmidt, W.A., Dasgupta, B., Luqmani, R. et al. A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate the Efficacy and Safety of Sirukumab in the Treatment of Giant Cell Arteritis. Rheumatol Ther 7, 793–810 (2020). https://doi.org/10.1007/s40744-020-00227-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-020-00227-2