
Abstract
Introduction
YLB113 is a biosimilar of the reference product (RP), etanercept, under development for treatment of patients with moderate-to-severe rheumatoid arthritis (RA) and other approved indications. A phase 3 study was conducted in Europe, Japan, and India to compare the efficacy, safety, and immunogenicity of YLB113 with the RP over a treatment period of 52 weeks.
Methods
Overall, 528 patients with moderate-to-severe RA receiving concomitant methotrexate were randomized to receive a once-weekly, subcutaneous dose of 50 mg YLB113 or the RP. The primary endpoint was ACR20 response rate at week 24, with similarity confirmed if the 95% confidence interval (CI) for YLB113 and the RP was within the range of − 15 to 15%. Safety and immunogenicity endpoints were assessed to week 52.
Results
Based on the European analysis, in the full analysis set, ACR20 response at week 24 was 83.3% and 88.5% for YLB113 and the RP, respectively. Responses were within the predefined clinical equivalence margin. The sensitivity analysis in the per protocol set revealed a similar proportion of subjects exhibiting ACR20 response at week 24 between groups, with a difference of − 5.1% (95% CI − 11.07 to 0.81). The incidence of treatment-emergent adverse events was comparable between groups, and the incidence of antidrug antibody development to week 24 favored YLB113 (0.8 vs. 8.3%).
Conclusions
This study demonstrated biosimilarity of YLB113 to the RP regarding efficacy, safety, and immunogenicity in patients with moderate-to-severe RA. Based on the same mechanism of action, biosimilarity could be extrapolated to other therapeutic indications approved for etanercept.
Trial registration
EudraCT Number: 2015-002,809-12.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
YLB113 is being developed as a biosimilar of the reference product (RP) Enbrel® (etanercept). |
Physicochemical characterization and preclinical studies have determined that YLB113 is biosimilar to the RP. |
A phase 3 study was conducted in Europe, Japan, and India to compare the efficacy, safety, and immunogenicity of YLB113 with the RP over a treatment period of 52 weeks. |
What was learned from the study? |
This study demonstrated that YLB113 was biosimilar to the RP in terms of clinical safety, efficacy, and immunogenicity in patients with moderate-to-severe rheumatoid arthritis (RA). |
Based on the same mechanism of action, the biosimilarity of YLB113 can be extrapolated to other therapeutic indications approved for the etanercept RP. |
YLB113 provides another therapeutic option for patients with moderate-to-severe RA, which may improve patient access to etanercept. |
Introduction
Tumor necrosis factor (TNF) is a proinflammatory cytokine secreted by immune cells, such as macrophages, monocytes, neutrophils, and activated natural killer and T-cells [1]. TNF plays an important role in the inflammatory processes of rheumatoid arthritis (RA), polyarticular juvenile idiopathic arthritis (JIA), psoriatic arthritis (PsA), and ankylosing spondylitis (AS), and the resulting joint pathology [1, 2]. In addition, TNF has been shown to play a role in the inflammatory process of psoriasis (PsO) [2]. Increased levels of TNF are found in tissues or serum of patients with immune-mediated chronic inflammatory diseases, including RA, JIA, PsA, AS, and PsO [3].
TNF functions via binding to 2 TNF receptors (TNFRs) that differ based on both signaling mechanisms and cellular localization [1]. TNFR1 is a 55-kDa protein that is ubiquitously expressed, whereas TNFR2 is a 75-kDa protein that is primarily expressed on immune cells. The biological activity of TNF is dependent upon binding to either cell surface TNFR.
Etanercept (Enbrel®; Amgen Inc.; Thousand Oaks, CA, USA) is a TNF inhibitor that is an engineered dimer containing two extracellular portions of TNFR2 fused to a C-terminal human immunoglobulin G1 Fc domain [4, 5]. Etanercept inhibits the binding of TNF to cell-surface TNFRs, which renders TNF biologically inactive. Etanercept is approved for use in adults with moderately to severely active RA, juvenile RA, PsA, and AS [4]. Studies have suggested that patients treated with a combination of biologics and methotrexate exhibit improved responses to treatment compared with patients who only use biologics [6].
Biosimilars are biological agents that are highly similar to reference products (RPs), and are considered for separate marketing approval after the expiration of the patents for the RPs [7]. Because biosimilars are a cost-effective alternative to current marketed products to treat rheumatic diseases, they can help increase patient access to biological therapy and reduce the economic burden to healthcare systems. YLB113 is being developed as a biosimilar of Enbrel (RP), which contains the active ingredient etanercept. Analytical similarity and preclinical studies have determined that YLB113 is biosimilar to the RP (unpublished data).
The purpose of this study is to compare the efficacy and safety at week 24 of YLB113 50 mg and the RP 50 mg given once weekly as a subcutaneous (SC) injection in conjunction with methotrexate in patients with moderate-to-severe RA, and to evaluate the long-term safety and immunogenicity of YLB113 administration compared with the RP.
Methods
Study Design
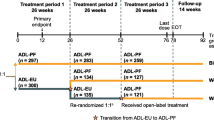
This study was conducted at 101 study centers located in the European Union, Japan, and India. The study allocation was stratified for country and site. Patients were randomized 1:1 in Stage A via the interactive web response system. For each patient, the maximum study duration was 56 weeks, including treatment for 52 weeks and 4 weeks of follow-up (or until study discontinuation).
Stage A consisted of the first 24 weeks of the multicenter, double-blind, randomized, parallel-group, active-control, comparative study, with the objective to compare the efficacy, safety, and immunogenicity of YLB113 and the RP (Fig. 1). Patients received YLB113 50 mg or the RP 50 mg once weekly via SC injection for 24 weeks on a background of stable methotrexate treatment within the range of 6 mg to 25 mg per week. After completing Stage A, subjects reconsented to continue in Stage B (same treatment group as assigned in Stage A) or Stage C (crossover of treatments of Stage A). Stages B and C ran in parallel and both were double-blinded (Fig. 1).

Study Design. ACR20 indicates American College of Rheumatology composite measure defined as a 20% decrease in swollen joint count and tender joint count, as well as a 20% improvement in at least three of the following five measures: subject assessment of pain, subject global assessment of disease activity, physician global assessment of disease activity, C-reactive protein or erythrocyte sedimentation rate, and Health Assessment Questionnaire Disability Index; DB double-blind, R randomized
Stage B consisted of eligible patients who completed evaluations for week 24 in Stage A, who were willing to continue, and who tolerated study medications with no unresolved adverse events (AEs) or serious AEs (SAEs) related to study medications. Patients received the same drug as in Stage A, once weekly via SC injection for an additional 28 weeks. The objective of Stage B was to compare the long-term safety and immunogenicity of YLB113 and the RP.
The objective of Stage C was to compare the sustainability of effects of treatment in patients with RA after crossing over treatments between YLB113 and the RP, following completion of the 24 weeks of treatment in Stage A. Stage C included eligible patients who exhibited reduced baseline Disease Activity Score in 28 joints (DAS28) by at least 0.6 at week 12 and/or week 24, who completed Stage A, and who tolerated study medications with no SAEs or unresolved AEs related to study medications. Patients crossed over to the other treatment arm (YLB113 to RP or RP to YLB113) and received SC injections once weekly for 28 weeks for a total treatment period of 52 weeks plus 4 weeks of follow-up. Stage C was not conducted in Japan, but was performed at centers in Europe and India as an amendment to the original protocol.
The study was conducted in accordance with the protocol, the ethical principles derived from international guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines, applicable International Council for Harmonisation (ICH) Good Clinical Practice (GCP) Guidelines, and applicable laws and regulations. The study was also conducted in accordance with the Japanese GCP in Japan and in accordance with ICH-GCP and/or local regulatory GCP in the European Union and India. An informed consent form (ICF) approved by each study center’s institutional review board/independent ethics committee was signed by the subject or their legally authorized representative (according to the regulatory and legal requirements of the participating country). The ICF contained all relevant information to be conveyed to the prospective subject to assist him/her in making an informed decision about participating in the study.
Objectives and Endpoints
The primary endpoint of Stage A was the percentage of patients achieving an American College of Rheumatology (ACR)20 response at week 24 of dosing. Secondary endpoints included ACR20 response rate at weeks 4, 8, and 12, ACR50 and ACR70 response rates at weeks 4, 8, 12, and 24 of dosing, and improvement in the DAS28 response rate at weeks 4, 8, 12, and 24. The DAS28 score was calculated based on either erythrocyte sedimentation rate (ESR) or C-reactive protein (CRP). To assess the individual DAS28 score during the course of the study, the same DAS assessment parameters (based on either ESR or CRP) were used throughout the study. Additional endpoints included AEs and immunogenicity assessment at weeks 4, 8, 12, and 24 of dosing. The primary endpoints for stages B and C were safety assessments, including AEs and injection-site assessment, and immunogenicity at weeks 36, 44, and 52 of dosing.
Patients
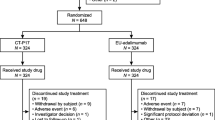
Patients eligible for inclusion were aged 18–75 years at the time of informed consent and were diagnosed with RA according to the 2010 ACR/European League Against Rheumatism (EULAR) classification criteria. Patients were required to exhibit a level of disease activity despite methotrexate treatment. Patients included had been treated with methotrexate for ≥ 3 months at an optimum dose (6 mg–25 mg/week, not exceeding the local approved dose) that remained stable for ≥ 6 weeks prior to screening. Patients were eligible if they had discontinued treatment with disease-modifying antirheumatic drugs other than methotrexate and completed a washout period of ≥ 2 weeks or ≥ 5 half-lives prior to drug administration, whichever was longer. For inclusion, patients were required to have ≥ 6 tender joints and ≥ 6 swollen joints (based on the swollen joint count [SJC] using 66 joints and tender joint count [TJC] using 68 joints) and DAS28 score ≥ 3.2. Exclusion criteria were known hypersensitivity to etanercept, acute or chronic infections, active tuberculosis, and known latex allergies. Of 874 patients screened, 528 were randomized in a 1:1 ratio to the YLB113 or RP arms, and 524 patients were included in the full analysis set (FAS) population, including 261 recruited in Japan, 231 in Europe, and 32 in India. A total of 497 evaluable patients completed Stage A (YLB113, n = 247; RP, n = 250). A total of 471 patients entered Stage B (YLB113, n = 236; RP, n = 235), of which 454 completed the stage (YLB113 and RP, n = 227 each). A total of 18 patients entered Stage C (YLB113 to RP, n = 10; RP to YLB113, n = 8), with only one patient from the YLB113 group not completing stage C.
Efficacy
An ACR20 response is defined as a 20% decrease in SJC and TJC, as well as a 20% improvement in at least three of the following five measures: subject assessment of pain, subject global assessment of disease activity, physician global assessment of disease activity, CRP or ESR, and Health Assessment Questionnaire Disability Index [8].
DAS28 was assessed using a composite score (range, 0–9.4) calculated using results of the 28-joint subset of the 66/68 SJC/TJC, CRP levels or ESR levels, and the subject’s global assessment of disease activity (0–100 scale) [9].
Safety
For the safety assessment, the following parameters were evaluated: AEs, physical examination, vital signs, electrocardiogram, clinical laboratory examination, and injection-site assessment. All outputs for safety outcomes are based on the safety analysis set. No statistical comparisons between the treatment groups were performed for safety data. The safety parameters were reported using descriptive statistics. All AEs were coded by system organ class and preferred term according to the Medical Dictionary for Regulatory Activities version 18.0 or higher. Events were classified as treatment-emergent AEs (TEAEs) if they started on or after the date of the first dose of study drug.
Immunogenicity
Immunogenicity was assessed by measuring the presence of antidrug antibodies (ADAs) against the RP or YLB113 before and after dosing. Immunogenicity was assessed using a validated and highly sensitive Meso Scale Discovery electrochemiluminescence method with acid dissociation and affinity capture as sample pretreatment techniques to enhance drug tolerance. A tiered approach to detection of ADAs was followed, beginning with screening and confirmatory assay followed by test for neutralization. Neutralizing ADAs were evaluated for samples testing positive in the confirmatory ADA assay via a validated method.
Statistics
Statistical data analysis was performed using SAS Version 9.3 or higher (SAS Institute, Inc; Cary, NC; 2000). Summary statistics and statistical analyses were performed for subjects included in the relevant analysis population. All statistical tests were two-sided and evaluated at a 5% level of significance. Missing ACR20 data were imputed using either a single mutation technique or a combination of nonresponder imputation and multiple imputation methods. Missing safety data were not imputed. However, because this was an international study that included patients with RA from Japan, Europe, and India, the outcome analysis for the primary and the most important secondary endpoint, ACR20 and DAS28, respectively, has been performed based on different statistical methodology, including imputation techniques requested by the Pharmaceuticals and Medical Devices Agency (PMDA) and European Medicines Agency (EMA). The results therefore include, in addition to the EMA-requested analysis of ACR20 and DAS28, the last observation carried forward, which is the PMDA-accepted analysis of missing data. An equivalence margin of 15% was estimated by carrying out meta-analysis of relevant studies and this approach was accepted by the EMA. The DAS28 reductions were analyzed using the least square means.
Results
Patient Characteristics
Baseline demographics were similar across treatment groups. The median age was 53.0 years, the majority were female (76.1%), and the average body mass index was 24.9 kg/m2 (Table 1). Baseline RA characteristics were similar across treatment groups. The majority (68.1%) of patients had a Class II ACR global functional status. Overall, 70.2% of subjects tested positive for rheumatoid factor, whereas anti-cyclic citrullinated peptide was detected in 73.5% of subjects.
ACR Response Rates
Primary Endpoint
The primary endpoint, the percentage of patients achieving an ACR20 response at week 24 using the EMA-requested imputation methods, was similar between groups, with a slightly higher rate in the RP arm (87.1%) compared with the YLB113 arm (average, 81.3%) in the FAS population (Fig. 2a). The difference was − 5.8% (95% confidence interval [CI], − 11.8 to 0.2), which was within the predetermined clinical equivalence margin of ± 15%. The results were confirmed in the per protocol set population: 90.6 and 86.0% for the RP and YLB113, respectively, with a difference of − 4.6 (95% CI − 10.1 to 0.8). The primary endpoint, ACR20 at week 24 in the FAS population, using PMDA-requested imputation methods, was 88.5 and 83.3% for the RP and YLB113, respectively, with a difference of − 5.1 (95% CI − 11.07 to 0.81). The percentage of patients who achieved ACR20 response at week 24 was similar between male and female patients.

ACR20/50/70 response rates over time. ACR20/50/70 indicates American College of Rheumatology composite measure defined as a 20, 50, or 70% decrease in swollen joint count and tender joint count, as well as a 20, 50, or 70% improvement in at least three of the following five measures: subject assessment of pain, subject global assessment of disease activity, physician global assessment of disease activity, C-reactive protein or erythrocyte sedimentation rate, and Health Assessment Questionnaire Disability Index; RP etanercept reference product
Secondary Endpoints
The following secondary endpoints are described using the nonresponder/multiple imputation technique, with the exception of DAS28. At week 4, 56.0% of patients in the YLB113 arm and 54.6% in the RP arm achieved ACR20 responses; these increased over time to week 12, when 77.6% in the YLB113 arm and 81.4% in the RP arm achieved ACR20 responses. No significant differences were observed between treatment groups.
ACR50 (the same instrument as ACR20, with improvement levels defined as 50% instead of 20%) response rates increased over time, with 20.1% of patients in the YLB113 arm and 23.2% in the RP arm achieving ACR50 responses at week 4, and 56.5 and 67.5% in the YLB113 and RP arms, respectively, achieving ACR50 responses at week 24 (Fig. 2b). A significant difference was observed between treatment groups only at week 24 but was not confirmed in subsequent weeks of treatment.
ACR70 (the same instrument as ACR20, with improvement levels defined as 70% instead of 20%) response rates also increased over time, with 8.8% of patients in the YLB113 arm and 6.8% in the RP arm achieving ACR70 responses at week 4, and 35.2 and 36.2% in the YLB113 and RP arms, respectively, achieving ACR70 responses at week 24 (Fig. 2c). No significant differences were observed between treatment groups.
The mean reductions in DAS28 scores were similar between treatment groups at weeks 4, 8, and 12 (Fig. 3). No significant differences in DAS28 score reduction were observed between treatment groups. At week 24, no significant differences in the DAS28-CRP or DAS28-ESR score reduction were observed between treatment groups. The analysis of DAS28 score reduction based on the single imputation technique, as requested by the PMDA, revealed similar results in both groups.

Mean change from baseline in DAS28 scores. DAS28 indicates disease activity score in 28 joints
Safety
During Stage A, 60.3% of subjects experienced at least one TEAE (YLB113, 55.3%; RP, 65.4%; Table 2). TEAEs were defined as AEs that started or worsened on or after the first dose of study medication and prior to the last date of study medication. The incidence of TEAEs related to study medication was greater in the RP arm compared with the YLB113 arm (35.8 vs. 22.0%). The overall incidence of AEs leading to discontinuation was comparable between arms (YLB113, 0.8%; RP, 1.9%). No new safety signals were reported. The most common TEAEs were nasopharyngitis (11.4 and 10.0%), injection-site reaction (ISR; 3.8 and 13.5%), and injection-site erythema (1.9 and 9.6%) for YLB113 and RP, respectively.
In Stage B, 57.3% of subjects experienced at least one TEAE (YLB113, 52.5%; RP, 62.1%; Table 2). The incidence of TEAEs related to study medication was greater in the RP arm compared with the YLB113 arm (26.4 vs. 11.9%). The overall incidence of AEs leading to discontinuation was comparable between arms (YLB113, 1.7%; RP, 1.7%). No new safety signals were reported. The most common TEAEs were similar to the ones in Stage A: nasopharyngitis (14.8 and 18.7%), ISR (1.3 and 7.2%), and injection-site erythema (0 and 4.3%) for YLB113 and the RP, respectively.
In Stage C, 33.3% of subjects experienced at least 1 TEAE (YLB113, 30.0%; RP, 37.5%; Table 2). The incidence of TEAEs related to study medication was greater in the RP arm compared with the YLB113 arm (25.0 vs. 10.0%). No AEs led to discontinuation in either arm. ISRs were observed in three patients in the RP arm, but were no longer observed in Stage C after switching to YLB113. In contrast, ISRs were newly reported in patients who switched to the RP in Stage C.
Immunogenicity
The incidence of ADA binding in stages A and B was lower with YLB113 compared with the RP. The long-term immunogenicity (subjects who tested positive at any time point during 52 weeks of treatment, pooled from stages A and B) with YLB113 was lower (0.8%) compared with the RP (9.4%; Table 3). Most of the reported ADAs were of low titer. Overall, two subjects with binding ADAs in the RP group and no subjects in the YLB113 group tested positive for neutralizing antibodies (NAbs).
Among 85 evaluated patients in the YLB113 arm with at least one AE of special interest (AESI), 1 (0.4%) patient had transient positive ADAs. Among 117 evaluated patients in the RP arm with at least one AESI, eight (3.1%) subjects had transient positive ADAs. Among patients evaluated for ADA status, none had positive ADAs for any of the AESIs at weeks 36, 44, or 52. Of subjects with transient positive ADAs, 0.4% of patients in the YLB113 arm and no patients in the RP arm reported allergic/hypersensitivity events. No patients with transient positive ADAs treated with YLB113 and 2.3% of patients treated with the RP reported ISRs. Infections were reported in 0.4 and 1.2% of patients with transient positive ADAs in the YLB113 arm and the RP arm, respectively. Overall, the incidence of transient AESIs with positive ADA status was not appreciably different in the RP group compared with the YLB113 group. Among all patients evaluated for ADA status, allergic/hypersensitivity events, ISRs, and infections were reported in 6.4, 0.4, and 23.9% of patients in the YLB113 arm and in 11.5, 23.5, and 27.7% of patients in the RP arm, respectively.
Discussion
In the current study, YLB113 demonstrated biosimilarity to the RP based on the percentage of patients achieving ACR20 after 24 weeks of treatment, the primary endpoint. Safety and tolerability were also similar through 52 weeks of treatment. The long-term safety and immunogenicity profile based on ADA analysis was comparable between treatment groups. In addition, a small subgroup of patients who crossed over to the other treatment after 24 weeks demonstrated sustained efficacy for the next 28 weeks. Although the number of patients in the crossover portion of the study was small, it showed a favorable trend of sustainable safety. Overall, YLB113 tolerability was marginally better, with a lesser immunogenic potential compared with the RP.
In this study, ACR20 was chosen as the primary endpoint based on the ability to compare results with recently published data. Because previous studies have evaluated both ACR and DAS28 responses [10,11,12], DAS28 responses were also determined to provide a more complete clinical efficacy assessment and comparison of the current results with historical data. At week 24, the ACR20 responses in the RP and YLB113 groups (90.6 vs. 86.0%) in the per protocol population were comparable with ACR20 rates reported in a previous biosimilar study [13].
Using a highly selective method with acid dissociation and affinity capture, a higher incidence of ADA formation was observed in patients treated with the RP compared with those treated with YLB113 in this study. The ADAs were transient and non-neutralizing, and most of them disappeared after 24 weeks of treatment. In the treatment group of Stage A that received the RP, two patients developed NAbs, which were no longer detectable in Stage B. Interchangeability assessment conducted in 18 patients (Stage C) revealed that one patient who switched from YLB113 in Stage A to the RP in Stage C expressed both ISR and transient ADA formation at week 36, but these did not persist through week 52. These results indicate that ISRs and ADAs may be dependent on product-specific variables (e.g., product aggregation or impurities) or other factors, and does not seem to be associated with the assay method [14, 15].
In the current study, a higher incidence of ISRs was reported in the RP group compared with the YLB113 group. Emery and colleagues reported a similar observation with the etanercept biosimilar, Benepali®, which does not involve l-arginine in its formulation and uses syringe needles without latex coating [16]. The YLB113 formulation and syringe needles have similar features to Benepali®. l-arginine has not been shown to be associated with an increased risk for ISR, but it is possible that this formulation difference contributes to the observed ISRs. Latex is associated with skin allergic reactions, and may contribute to the higher rate of ISRs in the RP group, whereas the YLB113 group did not use latex-coated syringes [17].
Limitations of the current study include small differences in product formulation and syringe coating between YLB113 and the RP. These differences may be contributing to the observed difference in ISRs reported between the two groups. Based on the importance of safety to the development process of biosimilars, the evaluation of ISRs and other AEs is needed to assess biosimilarity [18]. Another limitation is the small sample size in the subgroup of patients who switched from one treatment to the other in the current study. Although there was a trend toward sustained efficacy over the subsequent 28 weeks, the sample was too small to determine statistical significance and additional studies are warranted.
Clinical trials are designed mainly to show clinical equivalence with the reference medicine using sensitive endpoints in a population where product-related differences in clinical performance can be detected. Therefore, the equivalence design specifically rules out clinically relevant differences in safety or efficacy between the biosimilar and the reference medicine, and thus, confirms biosimilarity. The equivalence approach, therefore, is the preferred design to demonstrate biosimilarity.
Having demonstrated biosimilarity, YLB113 presents an alternative therapeutic option for patients with RA, because branded anti-TNF agents are starting to lose patent protections. The patents on Enbrel expired in Europe in August 2015 and are set to expire in the United States in November 2028, after Amgen was granted a new patent [19]. Furthermore, as more biosimilars enter the market and physician experience grows, the overall availability of biologic medications will increase. This greater availability may be of benefit for patients and healthcare systems by providing lower pricing while demonstrating similar efficacy and safety.
Currently, five TNF inhibitors are approved for the treatment of RA, including adalimumab, certolizumab pegol, etanercept, golimumab, and infliximab [20]. Available biosimilars on the market include etanercept (four biosimilars), adalimumab (three biosimilars), infliximab (three biosimilars), and rituximab (one biosimilar), with additional biosimilars under investigation [21, 22]. YLB113, which received approval for use as a biosimilar in Japan early in 2019, provides an additional alternative for the treatment of RA.
According to EULAR recommendations, RA incurs high individual, medical, and societal costs, all of which should be considered in its management by the treating rheumatologist [23]. Biosimilar treatment approaches should be preferred, as long as safety and clinical outcomes, particularly the immunogenicity profiles, are similar to those of branded therapies. The high cost of treatment is an important factor that limits the availability of modern therapies in some countries; this should be considered when choosing a treatment strategy. Therefore, the development and approval of biosimilars provides an opportunity to reduce the pressure on healthcare budgets.
Conclusions
The lower immunogenicity of YLB113 does not affect its classification as a biosimilar based on clinical equivalency and safety results. Overall, the biosimilarity of YLB113 to the RP was demonstrated based on safety, efficacy, and immunogenicity in patients with moderate-to-severe RA. This can be extrapolated to other therapeutic indications that have been approved for treatment with etanercept.
References
Menegatti S, Bianchi E, Rogge L. Anti-TNF therapy in spondyloarthritis and related diseases, impact on the immune system and prediction of treatment responses. Front Immunol. 2019;10:382.
Coates LC, FitzGerald O, Helliwell PS, Paul C. Psoriasis, psoriatic arthritis, and rheumatoid arthritis: Is all inflammation the same? Semin Arthritis Rheum. 2016;46(3):291–304.
Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. 2002;2(5):364–71.
Nestorov I. Clinical pharmacokinetics of TNF antagonists: how do they differ? Semin Arthritis Rheum. 2005;34(5):12–8.
Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor: Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340(4):253–9.
Gabay C, Riek M, Scherer A, Finckh A, Physicians SC. Effectiveness of biologic DMARDs in monotherapy versus in combination with synthetic DMARDs in rheumatoid arthritis: data from the Swiss Clinical Quality Management Registry. Rheumatology (Oxford). 2015;54(9):1664–72.
Combe C, Tredree RL, Schellekens H. Biosimilar epoetins: an analysis based on recently implemented European medicines evaluation agency guidelines on comparability of biopharmaceutical proteins. Pharmacotherapy. 2005;25(7):954–62.
Felson DT, Anderson JJ, Boers M, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38(6):727–35.
Fransen JSG, van Riel PLCM. Rheumatoid arthritis measures. Disease activity score (DAS), disease activity score-28 (DAS28), rapid assessment of disease activity in rheumatology (RADAR), and rheumatoid arthritis disease activity index (RADAI). Arthritis Care Res (Hoboken). 2003;49(5S):S214–24.
Bathon JM, Martin RW, Fleischmann RM, et al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N Engl J Med. 2000;343(22):1586–93.
Genovese MC, Bathon JM, Martin RW, et al. Etanercept versus methotrexate in patients with early rheumatoid arthritis: two-year radiographic and clinical outcomes. Arthritis Rheum. 2002;46(6):1443–50.
Genovese MC, Bathon JM, Fleischmann RM, et al. Long-term safety, efficacy, and radiographic outcome with etanercept treatment in patients with early rheumatoid arthritis. J Rheumatol. 2005;32(7):1232–42.
Matucci-Cerinic M, Allanore Y, Kavanaugh A, et al. Efficacy, safety and immunogenicity of GP2015, an etanercept biosimilar, compared with the reference etanercept in patients with moderate-to-severe rheumatoid arthritis: 24-week results from the comparative phase III, randomised, double-blind EQUIRA study. RMD Open. 2018;4(2):e000757.
Singh SK. Impact of product-related factors on immunogenicity of biotherapeutics. J Pharm Sci. 2011;100(2):354–87.
Rosenberg AS. Effects of protein aggregates: an immunologic perspective. AAPS J. 2006;8(3):E501–7.
Emery P, Vencovsky J, Sylwestrzak A, et al. A phase III randomised, double-blind, parallel-group study comparing SB4 with etanercept reference product in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2017;76(1):51–7.
Levy DA, Charpin D, Pecquet C, Leynadier F, Vervloet D. Allergy to latex. Allergy. 1992;47(6):579–87.
Agency EM. Guideline on similar biological medicinal products containing monoclonal antibodies–non-clinical and clinical issues. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf Accessed 19 April 2019.
Initiative GO-GaB. Biosimilars of etanercept. 2018. http://gabionline.net/Biosimilars/General/Biosimilars-of-etanercept.
Rein P, Mueller RB. Treatment with biologicals in rheumatoid arthritis: an overview. Rheumatol Ther. 2017;4(2):247–61.
Dörner T, Isaacs J, Gonçalves J, et al. Biosimilars already approved and in development. Consid Med. 2017;1(1):7–12.
Dorner T, Strand V, Cornes P, et al. The changing landscape of biosimilars in rheumatology. Ann Rheum Dis. 2016;75(6):974–82.
Smolen JS, Landewe R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–77.
Acknowledgements
We would like to thank the subjects who participated in the trial.
Funding
Funding for the phase 3 study was provided by Yoshindo Lupin Biologics Ltd., Tokyo, Japan. Funding support for the Rapid Service Fee was provided by Mylan Inc. All authors had full access to all the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Editorial Assistance
Editorial assistance was provided under the direction of the authors by Jessica D’Amico of The Lynx Group LLC. Editorial Assistance was funded by Mylan Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Dhananjay Bakhle is an employee and shareholder of Lupin Pharmaceuticals Ltd. Hisashi Yamanaka received grants and personal fees from YL Biologics Limited, and grants from AbbVie and Eisai. He received additional grants from Bristol-Myers, Novartis, Boehringer, Astellas, Kaken, Nippon Shinyaku, Pfizer, UCB, Ayumi, Ono, Daiichi-Sankyo, Taisho-Toyama, Takeda, Tanabe-Mitsubishi, Chugai, Teijin Pharma, and Torii, along with personal fees from Bristol-Myers, Pfizer, and Teijin Pharma. Naoyuki Kamatani has nothing to disclose. Yoshiya Tanaka has received speaking fees and/or honoraria from Daiichi-Sankyo, Astellas, Chugai, Eli Lilly, Pfizer, AbbVie, YL Biologics, Bristol-Myers, Takeda, Mitsubishi-Tanabe, Novartis, Eisai, Janssen, and Teijin, and has received research grants from Asahi Kasei, Mitsubishi-Tanabe, Chugai, Takeda, Sanofi, Bristol-Myers, UCB, Daiichi-Sankyo, Eisai, and Ono. Toshihiko Hibino is an employee of YL Biologics Limited. Edit Drescher has nothing to disclose. Juan Sánchez-Bursón has nothing to disclose. Manfred Rettenbacher is an employee of Lupin Pharmaceuticals Ltd. and was during the conduct of the study. Girish Bhatia has nothing to disclose. Snehal Gadve was an employee of Lupin Pharmaceuticals Ltd. at the time the study was conducted. Chirag Shah is an employee of Lupin Pharmaceuticals Ltd.
Compliance with Ethics Guidelines
The study was conducted in accordance with the protocol, the ethical principles derived from international guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines, applicable International Council for Harmonisation (ICH) Good Clinical Practice (GCP) Guidelines, and applicable laws and regulations. The study was also conducted in accordance with the Japanese GCP in Japan and in accordance with ICH-GCP and/or local regulatory GCP in the European Union and India. An informed consent form (ICF) approved by each study center’s institutional review board/independent ethics committee was signed by the subject or their legally authorized representative (according to the regulatory and legal requirements of the participating country). The ICF contained all relevant information to be conveyed to the prospective subject to assist him/her in making an informed decision about participating in the study. The full list of institutional review boards/independent ethics committees that approved this study can be found in the Supplementary Material.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to: https://doi.org/10.6084/m9.figshare.11113196.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any non-commercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yamanaka, H., Kamatani, N., Tanaka, Y. et al. A Comparative Study to Assess the Efficacy, Safety, and Immunogenicity of YLB113 and the Etanercept Reference Product for the Treatment of Patients with Rheumatoid Arthritis. Rheumatol Ther 7, 149–163 (2020). https://doi.org/10.1007/s40744-019-00186-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-019-00186-3