
Abstract
Introduction
This study assessed if concomitant use of conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) or corticosteroids altered the response or safety outcomes to baricitinib in rheumatoid arthritis (RA) patients.
Methods
Patients with ≥ 6 swollen/tender joints and no prior biologic DMARD were eligible for study inclusion. In RA-BUILD, csDMARD-inadequate responder (IR) patients were randomized to placebo or baricitinib (2 or 4 mg) once daily (QD). In RA-BEAM, methotrexate (MTX)-IR patients were randomized to placebo QD, baricitinib 4-mg QD, or adalimumab 40-mg biweekly. Patients continued background csDMARD (including MTX) therapy. This post hoc analysis of placebo and baricitinib 4-mg patients assessed the number and type of concomitant csDMARDS and concurrent corticosteroid use.
Results
From 716 placebo patients, 71, 21, and 6% were taking MTX alone, MTX + ≥ 1 csDMARD, and non-MTX csDMARDs, respectively; from 714 baricitinib patients, the rates were 74, 18, and 6%; 56% of placebo and 55% of baricitinib patients used corticosteroids at baseline (mean dose, 6.0 mg/day for both groups); patients continued use throughout the studies. The odds ratios for achieving American College of Rheumatology response at the 20% improvement level (ACR20) and Clinical Disease Activity Index (CDAI) ≤ 10 at week 12 favored baricitinib for most subgroups; no significant interactions were observed. Rates of adverse events were similar regardless of csDMARD group or corticosteroid use. There were numerically more serious adverse events in placebo patients taking corticosteroids (4.2 vs. 1.6%) and a higher rate of discontinuations in baricitinib patients taking corticosteroids (4.1 vs. 1.2%).
Conclusions
Baricitinib was efficacious regardless of concomitant use of csDMARDs or corticosteroids; the incidence of adverse events was similar across all groups of patients.
Funding
Eli Lilly and Company and Incyte Corporation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Current American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR) rheumatoid arthritis (RA) guidelines suggest that methotrexate (MTX) should be considered the initial drug of choice for most patients diagnosed with RA [1, 2]. For patients not able to tolerate MTX, the physician may change the patient to another conventional synthetic disease-modifying antirheumatic drug (csDMARD) such as leflunomide, sulfasalazine, or hydroxychloroquine. If the patient simply fails MTX, another csDMARD may be added to the MTX regimen. Regardless of treatment program, physicians may add corticosteroids when patients are having an RA flare and keep them on low-dose corticosteroids to prevent future flares. In this manuscript, we examined the safety and efficacy of baricitinib in patients whose concomitant csDMARDs were: (1) MTX only (with and without corticosteroids), (2) MTX plus other csDMARDs including sulfasalazine and hydroxychloroquine (with and without corticosteroids); and (3) non-MTX csDMARDS, including leflunomide, sulfasalazine, and hydroxychloroquine (with and without corticosteroids).
These groups may be distinctly different. Patients in the “MTX plus” group may be a more refractory group than the “MTX only” group because they are likely to have failed MTX monotherapy leading to the addition of other csDMARDs to the treatment regimen to achieve relief of symptoms. Thus, patients receiving non-MTX csDMARDs are likely to have failed or could not tolerate the side effects associated with MTX. Patients receiving non-MTX therapy could also reflect different prescribing patterns in various parts of the world, thus creating a unique group based on geography. Corticosteroid use may reflect the need to suppress chronic disease or prevent flares from occurring despite csDMARD therapy, although their use in clinical trials tend to be limited or restricted not reflecting full cumulative doses in clinical settings. From a safety standpoint, adverse reactions to therapy may occur for each of the drugs, and thus, the more medications patients are on, the more likely they are to experience adverse reactions.
The objectives of this post hoc analysis were to determine if these subgroups, based on csDMARDs or corticosteroids, were similar or dissimilar with respect to the following: (1) baseline characteristics; (2) adverse reactions to therapy; and (3) efficacy of baricitinib. Baricitinib, an oral selective inhibitor of Janus kinase (JAK)1 and JAK 2, is approved for the treatment of moderately to severely active RA in adults in over 50 countries, including European countries, the United States, and Japan.
Methods
Patients and Study Design
The study design and patient inclusion/exclusion criteria for each study have been described previously [3, 4]. Briefly, data were pooled from two phase 3 studies in which patients with inadequate response to csDMARDs with ≥ 6 swollen and tender joints and no prior biologic DMARD (bDMARD) use were eligible for study inclusion. RA-BEAM (NCT01710358) was a 52-week study of patients with inadequate response to MTX. Patients were randomized 3:3:2 to placebo once daily (QD), baricitinib 4-mg QD, or adalimumab 40-mg biweekly. Use of stable dose (7.5–25 mg/week) of concomitant oral MTX for ≥ 8 weeks was required prior to study entry and patients remained on the same dose of MTX throughout the study. Patients could also have been on concomitant hydroxychloroquine (up to 400 mg/day) or sulfasalazine (up to 3000 mg/day) and must have been receiving a stable dose for ≥ 8 weeks prior to entry into the study and remained on that dose throughout the study. Concomitant leflunomide was not permitted in RA-BEAM. RA-BUILD (NCT01721057) was a 24-week study of patients with inadequate response to csDMARDs. Patients were randomized 1:1:1 to placebo or baricitinib 2-mg or 4-mg QD. Use of stable doses of concomitant csDMARD for ≥ 8 weeks prior to study entry was permitted (7.4–25 mg/day MTX, up to 400 mg/day hydroxychloroquine, up to 3000 mg/day sulfasalazine, up to 20 mg/day leflunomide, and/or up to 150 mg/day or 2 mg/kg/day azathioprine) and patients remained on that dose throughout the study. In both studies, doses of concomitant csDMARDs could be reduced for safety reasons.
In both studies, corticosteroids of doses ≤ 10 mg/day of prednisone (or equivalent) were allowed but must have been maintained at stable levels from 6 weeks prior to randomization and through the treatment phase of the study, unless a patient received rescue therapy. After rescue, new corticosteroids or increases in doses of ongoing concomitant corticosteroids were permitted. Patients who were not on corticosteroids prior to randomization were not to initiate corticosteroid therapy during the study and were not to receive other systemic corticosteroids during the study including intra-muscular or intra-articular corticosteroids. Topical, intranasal, intra-ocular, and inhaled corticosteroids were permitted.
The primary endpoint in the studies was the ACR response at the 20% improvement level (ACR20) at week 12. Secondary endpoints included ACR responses at the 50 and 70% levels (ACR50/70), improvement from baseline in the Disease Activity Score based on 28 joints (DAS28) using the high-sensitivity C-reactive protein (hsCRP), and the Health Assessment Questionnaire-Disability Index (HAQ-DI), as well as the percentage of patients who achieved low disease activity (LDA) or remission based on the Simplified Disease Activity Index (SDAI) and the Clinical Disease Activity Index (CDAI).
Each study was conducted in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practice Guidelines and was approved by the Quorum Review institutional review board (IRB) #27257 for RA-BEAM and Quorum Review IRB #27258 for RA-BUILD. Ethics approvals were also obtained for all 281 sites for the RA-BEAM trial and all 182 sites for the RA-BUILD trial. All patients provided written informed consent. The studies were designed by the sponsor, Eli Lilly and Company and Incyte Corporation, with input obtained from an academic advisory board in which the non-Lilly author of this manuscript participated. All authors participated in the preparation and review of this manuscript and approved the final version.
Efficacy Endpoints for This Analysis
Efficacy for this post hoc analysis was examined based on concomitant csDMARD and concomitant corticosteroid status of patients during the placebo-controlled periods of the studies. The primary endpoints were percent of patients with ACR20 response and percent of patients with LDA as measured by CDAI ≤ 10 at week 12. Other endpoints included ACR50/70 response rates, percent of patients with SDAI ≤ 11 and ≤ 3.3, CDAI ≤ 2.8, DAS28-hsCRP ≤ 3.2 and < 2.6, HAQ-DI improvement ≥ 0.3 and ≥ 0.22, and EULAR responses of good and good + moderate. Good EULAR Response was defined as DAS28-hsCRP improvement from baseline > 1.2 with post-baseline DAS28-hsCRP level of ≤ 3.2; good + moderate response was defined as DAS28-hsCRP improvement from baseline > 1.2 or improvement > 0.6 with post-baseline DAS28-hsCRP level of ≤ 5.1. Change from baseline to week 12 for HAQ-DI, DAS28-hsCRP, CDAI, and SDAI were also evaluated.
Safety Assessments
Safety results at 12 weeks were assessed based on concomitant csDMARD and concomitant corticosteroid status during the placebo-controlled periods of the studies. Outcomes included adverse events (AE), serious adverse events (SAE), discontinuation due to AE, deaths, serious infections, herpes zoster, and malignancies.
Statistical Analysis
This post hoc analysis combined data from both trials providing overall samples for placebo (n = 716) and baricitinib 4-mg (n = 714). Analyses were performed for patients in three sub-populations based on concomitant csDMARD status: patients randomized to placebo or baricitinib 4-mg whose background csDMARD was MTX alone, MTX plus ≥ 1 other csDMARD, or one or more non-MTX csDMARD. Analyses were also performed based on use of concomitant corticosteroids. Descriptive summary tables are presented for baseline and safety data as well as descriptive graphs for response rates of efficacy outcomes.
For the subgroup efficacy analyses, comparisons between each baricitinib 4-mg and placebo group were performed across subgroups at 12 weeks using the modified intent-to-treat population, which was defined as all randomized patients who received ≥ 1 dose of the study drug. For the categorical measurements, nonresponder imputation was used in the analysis of patients who received either rescue therapy or discontinued from the study or study treatment. To assess the consistency of baricitinib treatment effect across the subgroups, the interaction between treatment and subgroups was evaluated using the following logistic regression model: treatment group + subgroup + treatment-by-subgroup + study. When the logistic regression sample size requirements were not met (< 5 responders in any of the factors in the model), interaction p value was not assessed.
An interaction p value ≤ 0.10 was considered to be statistically significant. Within a subgroup, odds ratios and 95% confidence intervals were calculated from a logistic regression model as follows: treatment group + study. When the logistic regression sample-size requirements were not met (< 5 responders in any study or treatment), the Cochran–Mantel–Haenszel (CMH) test stratified by study, was applied to generate p values, odds ratios, and 95% confidence intervals of odds ratios. Results are presented for those outcome measures where ≤ 1 subgroup required the CMH test. Response rate 95% confidence intervals are from exact (Clopper–Pearson) method. Interpretation of subgroup interaction analyses that had a p value of ≤ 0.10 began with an examination of the direction (same as or opposite to overall treatment effect) and then the magnitude of the treatment effect across the strata.
For the continuous outcomes (change from baseline to week 12), the following analysis of covariance (ANCOVA) model was used to evaluate interaction p values: baseline + treatment group + subgroup + study + treatment-by-subgroup. For least-squares mean change from baseline, the following ANCOVA model was used: baseline + treatment group + study. Modified last observation carried forward imputation was applied for missing data for continuous measurements.
Results
The pooled data from both studies included 1989 patients, of which 229 were randomized to baricitinib 2-mg in RA-BUILD and 330 to adalimumab in RA-BEAM; this analysis focused on patients in the placebo (n = 716) and baricitinib 4-mg (n = 714) arms. Of these, 71, 21, and 6% randomized to placebo were taking MTX alone, MTX + ≥ 1 csDMARD, and non-MTX csDMARDs, respectively; the rates for patients randomized to baricitinib 4-mg were 74, 18, and 6%, respectively. Oral corticosteroids were used in 56% of placebo and 55% of baricitinib 4-mg patients at baseline (mean dose of 6.0 mg/day for both groups). Patients continued stable levels of csDMARD and corticosteroid use through week 12 of the studies. Overall, the frequency of corticosteroid use was similar across the csDMARD groups (56, 56, and 58% in the MTX only, MTX + ≥ 1 csDMARD, and non-MTX groups, respectively). These rates were similar in the placebo and baricitinib groups (Table 1). Because corticosteroid use was similar regardless of csDMARD group, and the sample sizes of the subgroups was small when broken into treatment group by csDMARD group and corticosteroid group, analyses were performed on csDMARD subgroups and corticosteroid subgroups separately.
Baseline demographics and clinical characteristics are presented by treatment group and concomitant csDMARD status, and were generally similar across groups (Table 1). In the non-MTX group for patients treated with both placebo and baricitinib, a lower percentage of patients were anti-citrullinated protein antibodies (ACPA) and rheumatoid factor (RF) positive compared to the other csDMARD groups. A higher percentage of patients in Asia (including Japan) were in the MTX + ≥ 1 csDMARD group whereas the highest percentage of patients in the non-MTX group were from the US and Canada. The regions in which the separate RA-BEAM and RA-BUILD studies were carried out were similar (Supplementary Table 1), thereby allowing the combined regions to be examined in this analysis. However, per protocol, patients in RA-BEAM were required to be taking MTX; therefore all patients in the non-MTX subgroup are from the RA-BUILD study.
Efficacy by csDMARD and Corticosteroid Group
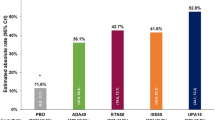
Clinical efficacy outcomes as measured by percent achieving ACR20 and CDAI ≤ 10 at week 12 are presented in Fig. 1. For the csDMARD subgroups, small sample sizes precluded the calculation of interaction p values; however, the odds ratios favored baricitinib over placebo. There were no significant interactions for the corticosteroid subgroup between baricitinib 4-mg and placebo, and the odds ratios favored baricitinib over placebo for both ACR20 and CDAI ≤ 10. For other outcome measures numerically higher response rates were observed for baricitinib regardless of the csDMARD (Supplementary Fig. 1) or corticosteroid (Supplementary Fig. 2) group. For those outcomes that met the requirement of ≤ 1 subgroup requiring the CMH test, odds ratios predominately favored baricitinib (Supplementary Fig. 3). There were no significant interactions for change from baseline to week 12 for HAQ-DI, DAS28-hsCRP, CDAI, and SDAI based on csDMARD (Table 2) and corticosteroid (Table 3) use.

Percentage of patients achieving ACR20 and CDAI ≤ 10 at week 12 by concomitant csDMARD and corticosteroid subgroups. Data (non-responder imputation) are presented as n/N (%). ACR20 American College of Rheumatology 20% response rate; CDAI Clinical Disease Activity Index, csDMARD conventional synthetic disease-modifying antirheumatic drug, MTX methotrexate. *Interaction p value not calculated because sample size requirement was not met †Odds ratio and 95% confidence interval calculated from Cochran–Mantel–Haenszel test due to sample size requirement not being met for logistic regression
Safety
Overall, there were few differences between randomized treatment groups or the concomitant csDMARD or corticosteroid subgroups with regard to safety outcomes. As has been previously reported [3, 4], there were more cases of herpes zoster in patients treated with baricitinib (Table 4). There was a numerically higher rate of serious adverse events in patients treated with placebo in the non-MTX csDMARD group (7.0%) compared to the other csDMARD groups for both placebo and baricitinib (range, 0.8–2.8%) (Table 2). Numerically, a higher rate of patients treated with placebo who were taking corticosteroids had serious adverse events (4.2%) relative to other subgroups (range, 1.6–2.6%), and more baricitinib patients on corticosteroids discontinued due to adverse events (4.1%) relative to other subgroups (range 1.2–2.7%) (Table 5). There were two deaths, both in patients treated with placebo who were using corticosteroids; one was due to subarachnoid hemorrhage, and the other due to renal failure. There were no events of tuberculosis in any patients treated with placebo or baricitinib through 12 weeks of treatment.
Discussion
This post hoc analysis of pooled data of over 1400 patients from two phase III studies of baricitinib 4-mg in csDMARD-inadequate responder (IR) patients with RA indicated that baricitinib was favored over placebo regardless of the number or type of concomitant csDMARDs or corticosteroid use. The quantitative differences observed in patients in the non-MTX subgroup should be evaluated with caution. The small number of patients in the non-MTX subgroup resulted in wide confidence intervals with the point estimates favoring baricitinib. Baricitinib as monotherapy has been shown to be superior to placebo; [3] therefore, it is less likely that baricitinib would be ineffective in a population treated with—generally weaker—csDMARDs. However, it is also possible that this patient population may be different than the patient population in the other two subgroups (MTX alone, MTX + ≥ 1 csDMARD) and because the non-MTX group was the smallest, with approximately 40 patients in each treatment group, this may reflect a type 2 error.
Baseline characteristics were generally similar in patients across the three csDMARD groups; clinical parameters such as DAS28-hsCRP were almost identical and duration of disease was similar across groups. However, it is possible that patients in the MTX + ≥ 1 csDMARD subgroup are more refractory as they needed at least two drugs to get to the same baseline state with similar durations of RA disease, as patients in the MTX only group. The data in this study do not, however, address when the second or third csDMARD was added in the MTX + ≥ 1 csDMARD subgroup. Additionally, lower proportions of patients in the non-MTX subgroup, receiving both placebo and baricitinib, were ACPA and RF positive. The literature is not consistent on whether seronegative patients may have higher disease activity [5, 6] and therefore be more difficult to treat with standard first-line therapy such as MTX. While the non-MTX subgroup in this study is small, this subgroup appears to differ from the other two subgroups. The distribution of geographic regions across the csDMARD subgroups was also interesting. While the percentages of patients in the MTX only group were, in general, evenly spread across the regions, between 42 and 47% of patients in the MTX + ≥ 1 csDMARD group were from Asia (including Japan), whereas approximately one-third of patients in the non-MTX group were from the United States and Canada.
The percentage of patients receiving concomitant corticosteroids in our study, approximately 55%, was similar to that of patients with RA in a study that examined the effects of glucocorticoids on clinical outcomes when taken with tofacitinib, another JAK inhibitor [5]. As in our study, in patients who used glucocorticoids through baseline and then with tofacitinib during the phase 3 studies, their post hoc analysis found that the concomitant use of the glucocorticoids did not appear to affect clinical efficacy [7]. A study of glucocorticoids taken with tocilizumab during RA trials also found no evidence that concomitant glucocorticoid therapy affected efficacy and safety [8].
In general, no major differences were seen with regard to safety outcomes. There was a numerically higher percentage of patients with serious adverse events in placebo patients in the non-MTX group (7.0 vs. 2.8% and 2.7% in the MTX alone and MTX + ≥ 1 csDMARD subgroups, respectively), however, the 7.0% represents three events. Similarly, a higher percentage of baricitinib patients in the non-MTX subgroup discontinued due to adverse events (7.1 vs. 2.1% and 3.8%), but again, this represented three cases. Placebo patients who were using corticosteroids had a numerically higher percentage of serious adverse events and there was a higher percentage of baricitinib patients using corticosteroids who discontinued the study. Aside from these exceptions, safety was similar in patients who were and who were not using corticosteroids. Prior studies have revealed concomitant corticosteroids increase the risk of infections with biological treatment [9, 10]. The current study did not share this observation. However, the noted numerical increase in discontinuations due to adverse events might nonetheless indicate a subtle shift in the pattern or the experienced severity of adverse events.
There were several limitations in this study, including those inherent to post hoc analyses, such as the lack of a clear a priori hypothesis for testing and the smaller numbers in each of the subgroups. Additionally, we did not include data on patients who received baricitinib 2-mg, which was evaluated in two trials in the baricitinib phase III development program. This analysis was focused on csDMARD-IR patients, the patient populations of RA-BEAM and RA-BUILD.
RA-BEAM, the larger trial, did not include a 2-mg dose. RA-BUILD did include baricitinib 2-mg. RA-BEACON, the other phase III trial that included baricitinib 2-mg, enrolled patients who were tumor necrosis factor inhibitor IR. These patients who, on average, were older, with longer RA disease duration, more extensive treatment experience and demonstrated refractoriness are dissimilar from the patients enrolled in RA-BEAM and RA-BUILD. Based on the different characteristics of patients enrolled in the two studies with baricitinib 2-mg, different efficacy responses are expected for csDMARD-IR versus bDMARD-IR patients. Therefore we do not believe it is appropriate to combine the populations from RA-BUILD and RA-BEACON for analysis. There was a limitation with the studies we did choose to combine. While both studies enrolled csDMARD-IR patients, the one difference in our subgroups was that MTX was required in RA-BEAM and therefore all patients in the non-MTX subgroup came from RA-BUILD. However, the patient demographics and corticosteroid use were similar for these individual studies, supporting their combination for this analysis. The clinical trial data reported here may not be representative of patients in clinical settings due to eligibility criteria of the trials. Finally, the observation period is short, especially for the placebo group, limiting the controlled-period assessment of safety.
Conclusions
In conclusion, patients that require multiple csDMARDs and those who need corticosteroids to help control their symptoms may differ in their disease burden from patients who use only one csDMARD and do not require corticosteroids. Yet, baricitinib was shown to be efficacious when added to MTX only or a combination of concomitant csDMARD therapy and regardless of corticosteroids use. Overall the incidence of adverse events was similar across all these groups of patients. Knowing that baricitinib has a similar efficacy and safety profile for MTX-IR, non-MTX-IR, and multiple csDMARD-IR patients, even during the short observation period of this study, is valuable information when considering adding baricitinib to the treatment regimen of patients with RA who are not responding to current therapy.
References
Singh JA, Saag KG, Bridges SL Jr, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res. 2016;68:1–25. https://doi.org/10.1002/acr.22865.
Smolen J, Landwew R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;2017(76):960–77. https://doi.org/10.1136/annrheumdis-2016-210715 (Published online first 6 Mar).
Dougados M, van der Heijde D, Chen YC, et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA-BUILD study. Ann Rheum Dis. 2016;2017(76):88–95. https://doi.org/10.1136/annrheumdis-2016-210094 (Published Online First 29 September).
Taylor PC, Keystone EC, van der Heijde D, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. 2017;2017(376):652–62. https://doi.org/10.1056/NEJMoa1608345 (Published Online First 16 February).
Nordberg LB, Lillegraven S, Lie E, et al. Patients with seronegative RA have more inflammatory activity compared with patients with seropositive RA in an inception cohort of DMARD-naïve patients classified according to the 2010 ACR/EULAR criteria. Ann Rheum Dis. 2016;2017(76):341–5. https://doi.org/10.1136/annrheumdis-2015-208873 (Published online first 19 Apr).
Smolen JS, Van Der Heijde DM, St Clair EW, et al. Predictors of joint damage in patients with early rheumatoid arthritis treated with high-dose methotrexate without or with concomitant infliximab: results from the ASPIRE trial. Arthritis Rheum. 2006;54:702–10. https://doi.org/10.1002/art.21678.
Charles-Schoeman C, van der Heijde D, Burmester GR, et al. Effect of glucocorticoids on the clinical and radiographic efficacy of tofacitinib in patients with rheumatoid arthritis: a posthoc analysis of data from 6 phase III studies. J Rheum. 2018;45:177–87.
Safy M, Jacobs JWG, Edwardes M, et al. No evidence that concomitant glucocorticoid therapy affects efficacy and safety of tocilizumab monotherapy in rheumatoid arthritis clinical trials. Ann Rheum Dis. 2018;77:945.
Haraoui B, Jovaisas A, Bensen WG, et al. Use of corticosteroids in patients with rheumatoid arthritis treated with infliximab: treatment implications based on a real-world Canadian population. RMD Open. 2015;1:e000078. https://doi.org/10.1136/rmdopen-2015-000078.
Strangfeld A, Eveslage M, Schneider M, et al. Treatment benefit or survival of the fittest: what drives the time-dependent decrease in serious infection rates under TNF inhibition and what does this imply for the individual patient? Ann Rheum Dis. 2011;2011(70):1914–20. https://doi.org/10.1136/ard.2011.151043 (Published online first 25 Jul).
Acknowledgements
Funding
This study and article processing charges were funded by Eli Lilly and Company and Incyte Corporation. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Medical Writing and/or Editorial Assistance
The authors thank Kathy Oneacre, MA, of Syneos Health for medical writing support and assistance with preparation and submission of this manuscript, which was funded by Eli Lilly and Company. The authors wish to thank the patients and their families and study personnel who participated in these clinical trials.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Ronald van Vollenhoven has received grants from AbbVie, Amgen, BMS, GSK, Pfizer, Roche, UCB; received consulting fees from AbbVie, Biotest, BMS, Celgene, Crescendo, GSK, Janssen, Lilly, Merck, Novartis, Pfizer, Roche, UCB, Vertex. Cameron Helt is an employee and stockholder of Eli Lilly and Company. Vipin Arora is an employee and stockholder of Eli Lilly and Company. Ana Pinto Correia is an employee and stockholder of Eli Lilly and Company. Inma de la Torre is an employee and stockholder of Eli Lilly and Company. David Muram was an employee of Eli Lilly and Company at the time of this study. Jinglin Zhong has nothing to disclose.
Compliance with Ethics Guidelines
Each study was conducted in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practice Guidelines and was approved by the Quorum Review IRB #27257 for RA-BEAM and Quorum Review IRB #27258 for RA-BUILD. Ethics approvals were also obtained for all 281 sites for the RA-BEAM trial and all 182 sites for the RA-BUILD trial. All patients provided written informed consent.
Data Availability
Lilly provides access to relevant anonymized patient level data from studies on approved medicines and indications as defined by the sponsor specific information on clinicalstudydatarequest.com. For details on submitting a request see the instructions provided at clinicalstudydatarequest.com.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced digital features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.7151969.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
van Vollenhoven, R., Helt, C., Arora, V. et al. Safety and Efficacy of Baricitinib in Patients Receiving Conventional Synthetic Disease-Modifying Antirheumatic Drugs or Corticosteroids. Rheumatol Ther 5, 525–536 (2018). https://doi.org/10.1007/s40744-018-0128-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-018-0128-0