
Abstract
Psoriatic arthritis (PsA) is a chronic, seronegative spondyloarthropathy associated with psoriasis (PsO). Treatment options range from non-pharmacologic measures to NSAIDS, DMARDs, and biologics, depending on patient presentation. Secukinumab (Cosentyx©) is a new biologic treatment option that was approved for use in treating adult patients with PsA in October 2016. Our paper explores the clinical trial evidence available for secukinumab to examine its safety and efficacy as a therapeutic agent for the treatment of PsA. While indirect comparisons of indicate that secukinumab is as effective as other treatment options, further studies directly comparing available treatments will be necessary to establish its place in treatment guidelines. As these and other trials are conducted, the evidence produced will further elucidate the clinical potential of secukinumab as a treatment option for patients with rheumatologic disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Psoriatic arthritis (PsA) is a chronic, seronegative spondyloarthropathy associated with psoriasis (PsO). Psoriasis affects as many as 7.5 million Americans, with up to 30% having psoriatic arthritis. Psoriasis occurs in all races with higher rates reported among Caucasians. The onset of disease most often occurs during young adulthood [1]. Meanwhile, psoriatic arthritis tends to appear between the ages of 30 and 50 years [2]. Comorbidities associated with psoriasis and psoriatic arthritis were studied in the large multicenter, non-interventional PREPARE trial (Prevalence of Psoriatic Arthritis in Adults with Psoriasis: An Estimate From Dermatology Practice). Of 949 patients with PsO evaluated, 285 received a diagnosis of PsA by the study rheumatologist through medical history, physical examination, and laboratory findings [3].
There are multiple treatment options for PsA. Treatment is based on the severity and response of disease. Treatment options range from non-pharmacological measures to NSAIDs, DMARDs, and biologics depending on the patient presentation. A newer treatment option that has been approved for use in PsO and PsA is the biologic medication secukinumab (Cosentyx©). Secukinumab is a human IgG1-kappa monoclonal antibody that binds to interleukin (IL) 17A. There has been extensive clinical trial experience with secukinumab in patients with psoriasis, and trials have also been conducted to examine its safety and efficacy in treating PsA patients [4]. Secukinumab is approved for use in treating adult patients with moderate-to-severe plaque PsO in the United States since January 2015 and for PsA in October 2016. This paper explores the clinical trials of secukinumab and examines its role as a therapeutic agent for PsA.
A PubMed search was conducted, using the keywords and phrases “psoriatic arthritis”, “secukinumab”, “efficacy”, “safety”, and “FDA approval”. Original articles published within the last 5 years as well as recent review articles were given preference. Priority was placed on utilizing data from secukinumab prescribing information and the American College of Rheumatology presentation entitled “Expert Insights on the approved uses of Cosentyx”, as this included data on file for secukinumab. Author opinion was utilized to provide insight in the discussion. This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
IL-17 in PsA
Conventional therapy for PsA includes disease-modifying anti-rheumatic drugs such as methotrexate and the use of non-steroidal anti-inflammatory drugs (NSAID) [5]. However, in recent years, targeted therapies—anti-tumor necrosis factor (TNF) agents, interleukin (IL)-12/23 inhibitors, and phosphodiesterase 4 (PDE4) inhibitors—have been studied and approved for use in PsA [5, 6]. A variety of targeted immunotherapies are approved for use in treating PsO, and these same immune pathways are being studied for their potential use in treating PsA. One of the pathways identified as a therapeutic target for PsA is that of the inflammatory cytokine interlukin-17A (IL-17A).
IL-17A is produced by a number of immune cells and can affect the function of cells such as neutrophils, endothelial cells, fibroblast-like synoviocytes, chondrocytes, and osteoblasts [5, 7,8,9]. The activation of these cells furthers the inflammatory cascade through the release of pro-inflammatory cytokines, which not only promote inflammation but also signal pathological processes such as hyperproliferation, vessel activation, matrix destruction cartilage damage, and bone erosion [5, 7,8,9]. The IL-17 pathway plays an important role in the pathogenesis of PsA [5]. An enrichment of IL-17A and IL-17RA can be found in the synovial fluid of PsA [10]. Furthermore, synovial fluid IL-17-producing CD4+ T cells are present in greater numbers in PsA than in osteoarthritis [10]. CD8+ T-cells also produce IL-17A and are more abundant in the synovial fluid of PsA patients than in healthy individuals. The level of these cells positively correlates to joint damage progression and measures of disease activity [5, 11]. Furthermore, in preclinical animal studies, local over-expression of IL-17 during collagen-induced arthritis is associated with joint destruction and increased synovial inflammation [5, 12]. As a result, multiple biologic therapies targeting IL-17 have been studied in patients with PsA, PsO, and other spondyloarthropathies, such as ankylosing spondylitis [5, 8].
Secukinumab
Secukinumab is a fully human IgG1κ anti-IL-17A monoclonal antibody that acts by selectively binding and neutralizing IL-17A, thus inhibiting its interaction with IL-17 receptors. In this way, activation of the downstream inflammatory cascade is halted, while other immune functions are not disturbed [5, 8]. In PsO patients, serum levels of total IL-17A increase to plateau concentrations after administration of secukinumab [5, 13]. In PsO patients, neutrophil markers and infiltrating epidermal neutrophils were reduced in skin lesions after 1–2 weeks of treatment compared to baseline [5, 13].
The pharmacokinetic profile of secukinumab in PsA patients is similar to that of PsO patients and is typical for an IgG1 monoclonal antibody [5]. In patients with PsA, the bioavailability of secukinumab is 85%, and clearance is independent of age, although both clearance and volume of distribution increase with body weight [5, 13]. In PsO patients, the bioavailability of secukinumab is 73%, with time to reach maximum concentration of 31–34 days after initial weekly dosing for first month [13, 14]. Although not examined in human subjects, there is no evidence to suggest that IL-17A influences the expression of CYP450 enzymes. Additionally, in PsA studies there was no interaction seen in the administration of secukinumab together with methotrexate and/or corticosteroids [13, 14].
In patients with PsA, secukinumab provides greater improvement than placebo in clinical response, C-reactive protein (CRP) levels, erythrocyte sedimentation rate (ESR), and quality of life (QoL) measures [5, 15]. Sustained reductions in clinical manifestations of PsA, inhibition of radiographic progression, and QoL measures were subsequently achieved across two phase 3 studies with 1003 total patients (703 secukinumab patients and 300 placebo patients) [5, 14, 16]. In these studies, FUTURE 1 and FUTURE 2, patients with active PsA were randomized to one of multiple treatment arms consisting of variable dosing regimens of secukinumab or placebo. In both studies, placebo patients switched to SC secukinumab based on clinical response. Both studies remain ongoing to collect long-term efficacy and safety data.
FUTURE-1 and FUTURE-2 Trials
The FUTURE 1 study is an ongoing, randomized, double-blind, placebo-controlled trial that evaluated 606 patients with active PsA. Patients were randomized (1:1:1) and those in the treatment group received an IV loading dose of secukinumab 10 mg/kg at weeks 0, 2, and 4, followed by either 75 or 150 mg SC injection secukinumab every 4 weeks [4, 14]. Patients receiving placebo were then re-randomized and switched to SC secukinumab 150 or 75 mg at week 16 or 24, depending upon their clinical response. Clinical status was defined as responder or non-responder (<20% improvement from baseline in tender and swollen joint counts).
FUTURE 2 is a multicenter, randomized, double-blind, placebo-controlled trial that evaluated 397 adult patients with active PsA who received SC secukinumab 75, 150, or 300 mg, or placebo, at weeks 0, 1, 2, 3, and 4, followed by the same dose every 4 weeks. Patients receiving placebo were re-randomized to receive secukinumab at week 16 or week 24 based on responder status [4, 16]. As in FUTURE1, patients were re-randomized based on clinical response at week 16 and were classified as responders or non-responders, receiving secukinumab (either 150 or 300 mg every 4 weeks) from week 16 (non-responders) or week 24 (responders).
In both studies, the primary end point was percentage of patients achieving an ACR20 response at week 24 [5, 16]. Achieving an ACR20 response required a ≥20% improvement in the number of tender and swollen joint counts, as well as in three of five other parameters: patient assessment of pain, physician and patient global assessment of disease activity, disability [Health Assessment Questionnaire Disability Index (HAQ-DI) score], and acute-phase reactant (CRP or ESR) [14]. To achieve an ACR50 response, patients needed to experience ≥50% improvement in the parameters listed above. Finally, ACR70 indicates ≥70% improvement in the above parameters. Multiple secondary endpoints were measured, and the presence of dactylitis and enthesitis were measured as accompanying symptoms of the disease (Table 1).
Patients included in the studies were individuals of at least 18 years of age with active PsA and met the Classification Criteria for Psoriatic Arthritis (CASPAR). Active disease was defined as ≥3 tender joints and ≥3 swollen unresponsive to previous treatment with nonsteroidal anti-inflammatory drugs (NSAIDs), disease-modifying anti-rheumatic drugs (DMARDs), or TNF inhibitors (Fig. 1) [14, 16]. Patients who were on a TNF inhibitor must have experienced an inadequate response to previous or current treatment with a TNF inhibitor given at an approved dose for ≥3 months, or have stopped treatment due to safety/tolerability problems after ≥1 administration of a TNFα inhibitor.

Inclusion and exclusion criteria of FUTURE-1 and FUTURE-2 trials
Efficacy in FUTURE 1 and FUTURE 2 Trials
Secukinumab therapy provided clinically significant improvement in both skin and joint symptoms that was observed in both FUTURE 1 and FUTURE 2. All primary endpoints were met in both studies at both treatment dosing regimens. In FUTURE 1, the ACR20 response rates were 50.0% with intravenous (IV) secukinumab 150 mg, 50.5% with IV secukinumab 75 mg, and 17.3% with placebo (P < 0.001 for both) at week 24 [14]. ACR50/70 responses at week 24 were 34.7%/18.8%, 34.7%/16.8%, and 7.4%/2.0%, respectively (all P < 0.001) (Table 2). Two-year follow-up data from FUTURE 1 sustained clinical improvement with long-term secukinumab therapy [5].
In FUTURE 2, ACR20 response was 54.0% with secukinumab 300 mg (P < 0.0001), 51.0% with 150 mg (P < 0.0001), and 29.3% with 75 mg (P = 0.0399) compared with 15.3% with placebo [16]. ACR50 responses were achieved by 35.0, 35.0, and 18.2% of patients in the secukinumab 300, 150, and 75 mg groups, respectively, compared with 7.1% in the placebo group (Table 3).
Compared with placebo, secukinumab 300 mg also improved ACR50 and HAQ-DI scores, however improvements in the other endpoints were not statistically significant. Clinical improvements were sustained through week 52, with observed response rates of 64.0% in the secukinumab 300 mg group, 64.0% in the secukinumab 150 mg group, and 50.5% in the secukinumab 75 mg groups [16]. At week 52, observed ACR20 responses were 73, 73, and 67% for the 300, 150, and 75 mg groups, respectively.
Furthermore, responses were similar in patients regardless of concomitant MTX treatment and prior anti-TNFα therapy. Patients who initially received placebo and who were assessed as non-responders at week 16 were re-randomized to receive secukinumab without a loading regimen; they achieved similar ACR20 responses over time. In patients with concomitant PsO, those receiving secukinumab (n = 99) had greater improvement in the skin lesions of PsO than did placebo treated subjects as measured by the PASI [4].
Enthesitis and Dactylitis Improvement
Improvements in enthesitis and dactylitis scores were observed in each secukinumab group compared to placebo at week 24 in the FUTURE 1 trial [4]. In data using non-responder imputation for missing values, patients with enthesitis or dactylitis at baseline had complete resolution of symptoms at week 24 in 46 and 48.8% of the 150-mg treatment group, and 48.8 and 56.7% in the 75-mg group, compared with 12.8 and 15.5% in the placebo group (all comparisons P < 0.0001) [5] (Table 4).
At week 52 using observed data, 81.6 and 79.4% of subjects in the 150-mg and 75-mg groups, respectively, were free from enthesitis (versus 37.6 and 36.1% at baseline), while 87.7% in the 150 mg group and 89.7% of the 75 mg group were free from dactylitis (compared with 48.5 and 48.5% at baseline) [5] (Fig. 2).

Comparison of percentage of patients without symptoms of enthesitis and dactylitis in the FUTURE-1 trial at week 52 versus baseline using observed data (Mease et al. [5])
While in the FUTURE 2 trial there were improvements in the number of patients with complete resolution of enthesitis and dactylitis, hierarchical analysis determined the comparisons between treatment and placebo to be not statistically significant [5, 16]. However, exploratory analysis of enthesitis and dactylitis resolution by individual doses suggested that secukinumab 300 mg and 150 mg could have clinically meaningful improvement in resolving these symptoms [5, 17]. At week 24, 56.5% in the 300 mg group and 50% in the 150 mg group had complete resolution of dactylitis compared with 14.8% of the placebo group (P < 0.01 for all comparisons). For enthesitis, 48.2% of the 300-mg group (P < 0.01) and 42.2% (P < 0.05) in the 150-mg group had complete resolution compared with 21.5% with placebo [5, 17] (Table 4). At week 52, 88.2% of patients in the 300-mg group and 90.9% of patients in the 150-mg group were free of dactylitis symptoms, compared with 54.0 and 68.0% at baseline (Fig. 3). For enthesitis, 72.0 and 69.3% were free from symptoms at week 52, compared with 44.0 and 36.0% at baseline in the 300- and 150-mg groups, respectively. At week 24, secukinumab also reduced the number of enthesitis site dactylitic digits, as determined by multiple methods of assessment [5].

Comparison of percentage of patients without symptoms of enthesitis and dactylitis in the FUTURE-2 trial at week 52 versus baseline using observed data (Mease et al. [5])
Adverse Reactions
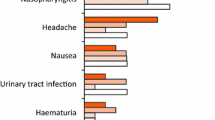
During the 16-week, placebo-controlled period of the trials in patients with PsA, the overall proportion of patients with adverse events (AEs) was similar in the secukinumab groups and placebo groups (59 and 58%, respectively). Overall, upper respiratory tract infections, nasopharyngitis, and headaches were the most common infections, occurring at similar rates in across the secukinumab treatment groups [16]. Other adverse effects varied between the FUTURE-1 and FUTURE-2 trials with some overlap (Fig. 4).

Adverse events in FUTURE-1 and FUTURE-2 trials
Similar to clinical trials of PsO, an increased proportion of patients contracted infections in the secukinumab groups compared with the placebo groups [4, 18, 19]. The rates of infections and serious infections across indications for PsA patients in secukinumab clinical trials were 29 and 1.3%, respectively (n = 703), compared with 25 and 0.3% in the placebo groups (n = 300). The infections seen included Candida infections, herpes viral infections, and staphylococcal infections [18].
In the PsA trials, Candida infections were more commonly seen with secukinumab treatment versus placebo [16]. The potential for Candida infections is to be expected with IL17 blockade, because the deficiency in the IL-17 pathway results in mucocutaneous candidiasis [20]. In FUTURE-2, Candida infections were reported in 11 patients on secukinumab, or 3.7% of exposed patients [16] (Table 5).
Six cases of oral candidiasis (two patients in the 300-mg group, three patients in the 150-mg group, and one patient in the 75-mg group); four cases of vulvovaginal candidiasis (one patient in the 300-mg group and three patients in the 150-mg group), one case of esophageal candidiasis (300-mg group), and one unspecified Candida infection (300-mg group) [16]. In FUTURE-1, oral candidiasis was reported in four patients, each in the secukinumab 150- and 75-mg groups; in the 150-mg group, there were reports of esophageal candidiasis in one patient and a Candida infection of the skin in another [14]. All cases of candidiasis, including one serious case, responded to oral therapy, and patients continued in the study.
There were no reports of serious opportunistic infections or cases of active tuberculosis. Overall, adverse events leading to discontinuations of the drug were noted in less than 5% of the patients for FUTURE-1 and less than 2% of patients in FUTURE-2, and were similar among the three groups [14, 16].
A concern with IL17 blockade is possible exacerbation of inflammatory bowel disease (IBD). A history of IBD was recorded for two patients (1.0%) in the secukinumab 75-mg dose group and for none in the other secukinumab or placebo groups; a history of Crohn’s disease was recorded for three (0.2%) secukinumab 150-mg patients, one (0.1%) secukinumab 300-mg patient, one (0.3%) placebo patient, and no patients in the secukinumab 75-mg group [19]. Among the 1003 patients with PsA in the clinical trials, there were three cases of new-onset IBD [4]. Two cases occurred in patients who received the drug, while one occurred in a patient who received placebo. The development of IBD in secukinumab-treated patients is consistent with the findings of a phase II placebo-controlled study in which patients with Crohn’s disease were treated with secukinumab; those treated with secukinumab had higher rates of adverse events and had worse IBD than those treated with placebo [21].
Comparing Safety Profiles
In comparing the safety of secukinumab use across clinical trials of PsA, PsO, and ankylosing spondylitis (AS), similar rates of infection were seen [18, 19, 22] (Table 6). The rates of infections and serious infections across indications for PsA patients in secukinumab clinical trials were 29 and 1.3%, respectively (n = 703), compared with 25 and 0.3% in the placebo groups (n = 300). At 12 weeks in PsO trials, the rate of infection with secukinumab was 28.7% (n = 1382) compared with 18.9% in the placebo group (n = 694) [4]. Only 0.14% of secukinumab subjects developed serious infections, compared with 0.3% of placebo. The most common infections observed were viral illnesses rather than those anticipated by blockage of IL-17 such as superficial Candida or staphylococcal infection.
The lower rate in the placebo group in the PsO studies may also be attributed to the socially debilitating nature of severe psoriasis, limiting patients’ contact with other people, and therefore, their exposure to infectious disease. At 16 weeks in AS clinical trials, 31% of secukinumab patients developed infections and 0.3% developed serious infections (n = 394), compared to 18 and 0% of the placebo group (n = 196). Most cases of secukinumab-associated neutropenia were both transient and reversible, and not associated with any serious infection [4, 19, 22].
Among the 1003 patients with PsA in the clinical trials, there were three cases of new-onset IBD. In the plaque PsO program, 3430 patients were exposed to secukinumab over the entire treatment period, for up to 52 weeks (2725 patient-years) [4]. There were three cases (0.11 per 100 patient-years) of exacerbation of Crohn’s disease about 39 subjects with known Crohn’s disease, two cases (0.08 per 100 patient-years) of exacerbation of ulcerative colitis, and two apparently new cases (0.08 per 100 patient-years) of ulcerative colitis. There were no cases in placebo patients (N = 793, 176 patient-years) during the 12-week placebo-controlled period [4]. Only one case of exacerbation of Crohn’s disease was reported from long-term non-controlled portions of ongoing clinical trials in plaque PsO. In the 571 patients exposed to secukinumab in the AS clinical study, there were eight cases of IBD reported during the entire treatment period. During the placebo-controlled 16-week period, there were two Crohn’s disease exacerbations and one new-onset ulcerative colitis case that was considered a serious AE [22]. During the remainder of the study, when all patients received secukinumab, there were two cases of new-onset IBD, and three cases of exacerbation. A history of IBD was reported by 2.3% of patients receiving any dose of secukinumab and in 2% of patients on placebo [22].
Overall Safety Profile of Secukinumab
Safety data from multiple studies confirms an overall favorable safety profile for secukinumab. Aside from a pre-treatment test for latent tuberculosis, there are no screening tests indicated before starting secukinumab. Patients should be made aware of the risk of mucocutaneous candidiasis. Candida infections with secukinumab are mucocutaneous, mild to moderate in severity, and respond well to standard oral or topical therapies. Standard treatment for candidiasis is sufficient without evidence to support discontinuation of secukinumab or prophylaxis [23]. Hematologic adverse effects of any biologic are very rare. Secukinumab trials saw a grade 3 neutropenia in 1.0% of patients and was not associated with serious infections. With regard to malignancy, in a pooled analysis from phase II/III secukinumab studies, IR of malignant or unspecified tumors (excluding NMSC) over 52 weeks of secukinumab were consistent with the expected rate of malignancy in the general population. This analysis also failed to reveal an increased risk for cardiovascular events specific to secukinumab, with overall risk comparable to etanercept [23]. No specific monitoring is required beyond regular screening based on risk factors and comorbid conditions. Evidence is limited with respect to the safety profile of secukinumab in pregnant patients [24]. There are no human studies, however studies in mice and monkeys show no embryofetal toxicity [24].
Immunogenicity
As with all therapeutic proteins, there is the potential for immunogenicity when using secukinumab. In the clinical trials of secukinumab, neutralizing antibodies were not associated with loss of efficacy in patients treated with secukinumab. Using an electrochemiluminescence-based bridging immunoassay, <1% of patients treated with secukinumab developed antibodies to secukinumab in up to 52 weeks of treatment [4]. Approximately 0.5% of total patients, or half of those who had antibodies, were classified as having neutralizing antibodies. These neutralizing antibodies were not associated with loss of efficacy. This assay has limitations in detecting anti-secukinumab antibodies in the presence of secukinumab; therefore, the incidence of antibody development might not have been reliably determined. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay [4]. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to secukinumab with the incidences of antibodies to other products may be misleading. Among patients who were anti-drug antibody (ADA)-negative prior to initiating treatment with secukinumab, secukinumab-specific treatment-emergent ADAs were detected in 0.1% of patients with PsA and 0.3% of patients with AS [19, 22]. In the PsO clinical development program, less than 1% of subjects treated with secukinumab developed antibodies to secukinumab in up to 52 weeks of treatment [4].
Discussion
Based on available evidence, secukinumab offers an effective new addition to the PsA treatment repository. The recommended use of secukinumab for moderate-to-severe PsA with coexistent moderate-to-severe PsO is 300 mg by subcutaneous injection every 4 weeks, following initial loading doses of 300 mg for 4 weeks. Each 300-mg dose is given as two injections of 150 mg. For some patients, a dosage of 150 mg may be acceptable. For other patients with PsA, it is recommended to administer secukinumab with or without a loading dosage by subcutaneous injection [4]. The recommended dosage with a loading dosage is 150 mg at weeks 0, 1, 2, 3, and 4 and every 4 weeks thereafter. Without a loading dosage, the recommended regimen is 150 mg every 4 weeks. If a patient continues to have active PsA, physicians can consider a dosage of 300 mg. Secukinumab appears to have a broad therapeutic window, as doses of up to 30 mg/kg have been administered to patients without signs of dose-limiting toxicity [4].
The recommended dosage of secukinumab for moderate-to-severe plaque PsO as per the most recent guidelines is 300 mg by SC injection at weeks 0, 1, 2, 3, and 4 followed by 300 mg every 4 weeks. Each 300-mg dosage is delivered as two SC injections of 150 mg. A dose of 150 mg may be acceptable for some patients [4].
Secukinumab may be administered with or without methotrexate. For all injectable medications, the first self-injection should be performed under the supervision of a qualified healthcare professional. Patients should be trained on proper injection techniques prior to self-administration by either physician or appropriately trained health-care staff.
However, some potential weaknesses of the FUTURE trials lie within their study design of utilizing uncontrolled extensions during which time ACR measures were exploratory. Following week 24, patients were made aware they were taking active treatment, but remained blind to the dose. The use of an uncontrolled extension phase creates limitations, as placebo comparisons cannot be made, and inherent bias, as patients remaining in the extension phase may be those who experienced better results.
As secukinumab enters the market for rheumatologic diseases, treatment guidelines may need revision, as this first-in-class drug must be given a place among currently available treatments [25]. Direct comparison trials are needed to assess secukinumab against other available therapies, but indirect comparisons indicate that the drug is as effective as other treatment options in treating PsA [5]. Multiple biologic therapies targeting TNF-alpha, IL-12/23, and now IL-17 are available and recommended for patients who fail NSAID treatment or conventional disease-modifying anti-rheumatic drugs [5]. The use of anti-IL-17 therapy has been added to recently updated treatment guidelines for PsA in the by the European League Against Rheumatism (EULAR) and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) [26, 27]. However, these guidelines differ in their recommendations based on their different evaluation processes, as the focus of the EULAR is primarily rheumatological while the GRAPPA is balanced between the rheumatological and dermatologic aspects of PsA [28]. In both publications, data on secukinumab are reviewed, including its effects on progression of radiographic disease [28, 29]. However, in the EULAR recommendations, TNF inhibitors are given preference as first-line biologics based on the larger quantity of available efficacy and safety data as well as longer experience with these biologic agents compared with newer drugs [27, 28]. In the GRAPPA recommendations, TNF inhibitors and other biologic agents are placed at the same “step”, allowing all biologic agents to be at the same level of first-line therapy if deemed clinically appropriate [28, 29]. For some of the PsA domains in particular, all biologic agents are placed at the same level [28, 29]. Based on these recommendations in conjunction with the positive results seen in clinical trials of secukinumab, the drug should be given consideration as a first-line biologic therapy for patients with severe skin disease in addition to their PsA.
While these guidelines provide a place for secukinumab in the current treatment paradigm for PsA, further studies are being conducted that will help establish its role in treating the disease. Currently, three phase 3 clinical trials for secukinumab in PSA are ongoing and seek to examine safety and efficacy of different doses versus placebo (FUTURE 3; NCT01989468), safety and efficacy of the secukinumab versus placebo with and without loading dose (FUTURE 4; NCT02294227), and the effect of secukinumab on the progression of structural damage (FUTURE 5; NCT02404350) [5]. Other clinical trials, one examining secukinumab in treating juvenile psoriatic arthritis and enthesitis-related arthritis (NCT03031782), and one comparing secukinumab monotherapy with the biologic adalimumab in patients with active PsA are both registered but not yet recruiting (EXCEED 1; NCT02745080).
Although these studies have been ongoing, it remains to be seen if future compliance issues will arise. Especially because secukinumab is given as an injection, patients may have inherent fears and bias towards the medication [30]. These fears may prevent patients from being fully adherent to their medication due to a reluctance to give themselves a weekly injection. There are different strategies that might be used to assuage this reluctance, such as anchoring. This technique resets patients’ frame of reference to become more amenable to the frequency of biologic injections [30]. Future studies might examine more techniques to help patients become more comfortable with injectable medications. Further studies might also examine the adherence rates to injectable biologic medications to assess the potential problem of nonadherence in patients taking them.
Based on the success of secukinumab in treating symptoms of PsA such as dactylitis and enthesitis, the medication has the potential for use in treating other rheumatologic diseases. The drug is approved for the treatment of ankylosing spondylitis after completing phase 2 and 3 trials with positive results. These trials have pointed to the efficacy of secukinumab in addressing the key clinical domains of ankylosing spondylitis and suggest that IL17A plays a role in the pathogenesis of this disease in addition to that of PsA and PsO [31]. However, in the case of rheumatoid arthritis, phase II/III randomized controlled trials failed to demonstrate resounding evidence that secukinumab is effective in treating this disease [25]. There are various phase 3 trials that have been either recently completed or are still in progress regarding the use of secukinumab in rheumatoid arthritis. As this data becomes available, it will further define the potential this drug has for the treatment of rheumatoid arthritis patients.
Conclusions
Although the drug has been FDA approved for PsO, PsA, and AS, the full potential of secukinumab in treating the various inflammatory rheumatic diseases has not been fully explored. Likewise, ongoing studies will help delineate therapeutic strategies to optimize secukinumab for its already approved uses. As further research is completed in the coming years, clinical trial evidence will further assess the potential clinical utility of this drug in treating patients with rheumatologic disease.
References
National Psoriasis Foundation. Fact Sheet Library | National Psoriasis Foundation. https://www.psoriasis.org/publications/patient-education/fact-sheets. Accessed 30 May 2017.
Gottlieb A, Korman NJ, Gordon KB, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 2. Psoriatic arthritis: overview and guidelines of care for treatment with an emphasis on the biologics. J Am Acad Dermatol. 2008;58(5):851–64. doi:10.1016/j.jaad.2008.02.040.
Mease PJ, Gladman DD, Helliwell P, et al. Comparative performance of psoriatic arthritis screening tools in patients with psoriasis in European/North American dermatology clinics. J Am Acad Dermatol. 2014;71(4):649–55. doi:10.1016/j.jaad.2014.05.010.
Cosentyx (secukinumab) [prescribing information]. East Hanover: Novartis Pharmaceuticals Corporation. 2016.
Mease P, McInnes IB. Secukinumab: a new treatment option for psoriatic arthritis. Rheumatol Ther. 2016;3(1):5–29. doi:10.1007/s40744-016-0031-5.
Kavanaugh A, Mease PJ, Gomez-Reino JJ, et al. Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis. 2014;73(6):1020–6. doi:10.1136/annrheumdis-2013-205056.
Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361(9):888–98. doi:10.1056/NEJMra0707449.
Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11(10):763–76. doi:10.1038/nrd3794.
Smith JA, Colbert RA. Review: the interleukin-23/interleukin-17 axis in spondyloarthritis pathogenesis: th17 and beyond. Arthritis Rheumatol. 2014;66(2):231–41. doi:10.1002/art.38291.
Raychaudhuri SP, Raychaudhuri SK, Genovese MC. IL-17 receptor and its functional significance in psoriatic arthritis. Mol Cell Biochem. 2012;359(1–2):419–29. doi:10.1007/s11010-011-1036-6.
Menon B, Gullick NJ, Walter GJ, et al. Interleukin-17+ CD8+ T cells are enriched in the joints of patients with psoriatic arthritis and correlate with disease activity and joint damage progression. Arthritis Rheumatol. 2014;66(5):1272–81. doi:10.1002/art.38376.
Lubberts E, Joosten LA, Oppers B, et al. IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001;167(2):1004–1013. http://www.ncbi.nlm.nih.gov/pubmed/11441109. Accessed 6 Apr 2017.
Novartis. Cosentyx summary of product characteristics. http://ec.europa.eu/health/documents/community-register/2015/20150115130444/anx_130444_en.pdf. Accessed 30 July 2017.
Mease PJ, McInnes IB, Kirkham B, et al. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. N Engl J Med. 2015;373(14):1329–39. doi:10.1056/NEJMoa1412679.
McInnes IB, Sieper J, Braun J, et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis. 2014;73(2):349–56. doi:10.1136/annrheumdis-2012-202646.
McInnes IB, Mease PJ, Kirkham B, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2015;386(9999):1137–46. doi:10.1016/S0140-6736(15)61134-5.
Kirkham B, McInnes I, Mease P, et al. THU0421 secukinumab is effective in reducing dactylitis and enthesitis using multiple measures in patients with psoriatic arthritis: data from a phase 3 randomized, multicenter, double-blind, placebo-controlled study (future 2). Ann Rheum Dis. 2015;74:351. doi:10.1136/annrheumdis-2015-eular.2276.
Novartis. Expert insights on the approved uses of COSENTYX (secukinumab). In: American College of Rheumatology annual 2016 meeting. Washington, DC. 2016.
Data on file. AIN457F summary of clinical safety in psoriatic arthritis. Novartis Pharmaceuticals Corp. 2015.
Gaffen SL, Hernández-Santos N, Peterson AC. IL-17 signaling in host defense against Candida albicans. Immunol Res. 2011;50(2–3):181–7. doi:10.1007/s12026-011-8226-x.
Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61(12):1693–700. doi:10.1136/gutjnl-2011-301668.
Data on file. AIN457H summary of clinical efficacy in ankylosing spondylitis. Novartis Pharmaceuticals Corp. 2015.
van de Kerkhof PCM, Griffiths CEM, Reich K, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75(1):83–98.e4. doi:10.1016/j.jaad.2016.03.024.
Porter ML, Lockwood SJ, Kimball AB. Update on biologic safety for patients with psoriasis during pregnancy. Int J Womens Dermatol. 2017;3(1):21–5. doi:10.1016/j.ijwd.2016.12.003.
Koenders MI, van den Berg WB. Secukinumab for rheumatology: development and its potential place in therapy. Drug Des Dev Ther. 2016;10:2069–80. doi:10.2147/DDDT.S105263.
Coates LC, Kavanaugh A, Mease PJ, et al. Group for research and assessment of psoriasis and psoriatic arthritis 2015 treatment recommendations for psoriatic arthritis. Arthritis Rheumatol. 2016;. doi:10.1002/art.39573.
Gossec L, Smolen JS, Ramiro S, et al. European League Against Rheumatism (EULAR) recommendations for the management of psoriatic arthritis with pharmacological therapies: 2015 update. Ann Rheum Dis. 2016;75(3):499–510. doi:10.1136/annrheumdis-2015-208337.
Gossec L, Coates LC, de Wit M, et al. Management of psoriatic arthritis in 2016: a comparison of EULAR and GRAPPA recommendations. Nat Rev Rheumatol. 2016;12(12):743–50. doi:10.1038/nrrheum.2016.183.
Coates LC, Murphy R, Helliwell PS. New GRAPPA recommendations for the management of psoriasis and psoriatic arthritis: process, challenges and implementation. Br J Dermatol. 2016;174(6):1174–8. doi:10.1111/bjd.14667.
Oussedik E, Cardwell LA, Patel NU, Onikoyi O, Feldman SR. An anchoring-based intervention to increase patient willingness to use injectable medication in psoriasis. JAMA Dermatol. 2017. doi:10.1001/jamadermatol.2017.1271.
Baeten D, Sieper J, Braun J, et al. Secukinumab, an interleukin-17a inhibitor, in ankylosing spondylitis. N Engl J Med. 2015;373(26):2534–48. doi:10.1056/NEJMoa1505066.
Acknowledgements
No funding or sponsorship was received for this study or publication of this article. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Dr. Feldman is a speaker for Janssen and Taro. He is a consultant and speaker for Novartis Pharmaceuticals, Galderma, Stiefel/GlaxoSmithKline, Abbott Labs, Leo and Pharma Inc. Dr. Feldman has received grants from Galderma, Janssen, Abbott Labs, Amgen, Stiefel/GlaxoSmithKline, Celgene, and Anacor. He is a consultant for Amgen, Baxter, Caremark, Gerson Lehrman Group, Guidepoint Global, Hanall Pharmaceutical Co Ltd, Kikaku, Lilly, Merck & Co Inc, Merz Pharmaceuticals, Mylan, Novartis Pharmaceuticals, Pfizer Inc, Qurient, Suncare Research and Xenoport. He is on an advisory board for Pfizer Inc. Dr. Feldman is the founder and holds stock in Causa Research and holds stock and is majority owner in Medical Quality Enhancement Corporation. He receives Royalties from UpToDate and Xlibris. Nupur Patel, Nora Vera, Emily Rose Shealy, and Margaret Wetzel have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/4FEBF0606BE719EF.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Patel, N.U., Vera, N.C., Shealy, E.R. et al. A Review of the Use of Secukinumab for Psoriatic Arthritis. Rheumatol Ther 4, 233–246 (2017). https://doi.org/10.1007/s40744-017-0076-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-017-0076-0