
Abstract
Introduction
To assess the long-term safety and efficacy of subcutaneous tocilizumab (TCZ-SC) in US patients with rheumatoid arthritis (RA) who rolled over from the two global phase 3 studies, SUMMACTA (NCT01194414) and BREVACTA (NCT1232569), into this open-label, single-arm, phase 3b study.
Methods
Patients continued to receive TCZ-SC 162 mg weekly or every other week or switched from intravenous TCZ to TCZ-SC 162 mg qw for up to 84 weeks. The primary endpoint was the proportion of patients with serious adverse events (SAEs). Secondary endpoints included clinical efficacy, laboratory abnormalities, and immunogenicity.
Results
Of the 217 patients treated, 76.5% were female, and the mean age was 58.4 years. A total of 23 patients (10.6%) had ≥1 SAE. The most common SAEs were infections (3.7%). Alanine aminotransferase elevations (38.2%) were not associated with hepatic injury. Grade 3/4 neutropenia (3%) was transient and not associated with serious infections. Immunogenicity was low (<1%) and not associated with SAEs. No anaphylaxis or deaths occurred. Thirteen patients (6.0%) withdrew due to safety reasons. Mean Clinical Disease Activity Index and Disease Activity Score in 28 joints remained stable throughout the trial.
Conclusions
The long-term safety of TCZ-SC during the long-term extension period was consistent with the safety profiles from SUMMACTA and BREVACTA, with no new safety signals. Efficacy improvements observed from baseline remained stable over time. These results demonstrated the durability of the safety and efficacy responses, and low immunogenicity, with long-term exposure to TCZ-SC in patients with RA.
Funding: F. Hoffmann-La Roche, Ltd.
Trial registration number: ClinicalTrials.gov identifier, NCT01662063.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic, progressive autoimmune disease characterized by inflammation of the joints that, if left untreated, leads to irreversible joint damage and disability. Biologic disease-modifying antirheumatic drugs (bDMARDs) that block inflammatory cytokines, B cells, or T cells are commonly used for the treatment of RA in combination with conventional synthetic DMARDs (csDMARD) or as monotherapy. Due to the long-term treatment of patients with RA with bDMARDs, it is important to establish that these drugs have lasting efficacy and safety.
Tocilizumab (TCZ) is the first drug approved for the treatment of RA that blocks the biological activity of interleukin 6 (IL-6). TCZ is a humanized monoclonal antibody that competitively interferes with the binding of IL-6 to the IL-6 receptor (IL-6R, also known as IL-6Rα or CD126), thereby disrupting receptor association with glycoprotein 130, a co-signal transducer protein (also known as IL-6Rβ or CD130) necessary for initiating intracellular signaling through the JAK-STAT and other pathways [10]. TCZ, in combination with csDMARDs or as monotherapy, has demonstrated efficacy with a well-established safety profile in patients with RA who had an inadequate response to csDMARDs or anti-tumor necrosis factor agents (aTNFs) [2, 4–6]. Intravenous [IV; 4 and 8 mg/kg every 4 weeks (q4w)] and subcutaneous [SC; 162 mg weekly (qw) or every 2 weeks (q2w)] formulations of TCZ are available for the treatment of patients with RA.
The efficacy and safety of TCZ-SC was demonstrated in two recent global phase 3 clinical trials in patients with RA who had an inadequate response to csDMARDs or aTNFs: BREVACTA (NCT1232569) reported the superiority of TCZ-SC q2w plus csDMARDs to placebo SC plus csDMARDs over 24 weeks [8]. SUMMACTA (NCT01194414) reported the noninferiority of TCZ-SC qw to TCZ-IV 8 mg/kg q4w, in combination with a csDMARDs, over 24 weeks [7]. The extensions of SUMMACTA and BREVACTA with TCZ-SC qw and TCZ-SC q2w, respectively, to 96–97 weeks demonstrated that efficacy and safety remained comparable to the known profile of TCZ-IV [9]. Additionally, the MUSASHI (JapicCTI-101117) trial in Japan reported the noninferiority of TCZ-SC q2w as monotherapy to TCZ-IV 8 mg/kg q4w as monotherapy in patients who had an inadequate response to methotrexate or a bDMARD over a 24-week study period; the continued efficacy and safety of TCZ-SC monotherapy in MUSASHI has been reported out to 2 years [1, 11].
This open-label long-term extension phase 3b study for up to an additional 84 weeks evaluated the long-term safety, immunogenicity, and efficacy of TCZ-SC in patients in the US who completed the SUMMACTA or BREVACTA studies.
Methods
Participants
The study population consisted of adult patients (≥18 years of age) with RA (≥6 months) in the United States who had moderate-to-high disease activity and completed SUMMACTA [7] or BREVACTA [8] and, on the basis of the investigator’s judgment, might have continued to benefit from TCZ-SC treatment.
Study Design
This was a multicenter phase 3b long-term extension study of SUMMACTA and BREVACTA (Figure S1 in the supplementary material; trial registration number NCT01662063). For enrollment in both SUMMACTA and BREVACTA, patients (≥18 years of age) with RA (≥6 months, revised 1987 American College of Rheumatology criteria) were included and were required to have had an inadequate response to ≥1 DMARD (up to 20% of patients may have had inadequate response to ≥1 aTNF). Patients who completed the 96–97-week BREVACTA and SUMMACTA studies and met the enrollment criteria continued to receive TCZ-SC q2w or TCZ-SC qw, respectively, or switched from TCZ-IV to TCZ-SC 162 mg qw for up to an additional 84 weeks. Thus, all patients in the long-term extension were receiving TCZ-SC. Baseline was defined as the beginning of this long-term extension for all efficacy and safety data. Study treatment at baseline was given at least 6 weeks after the last TCZ-IV dose or at least 4 weeks after the last TCZ-SC dose. The maximum interval allowed between the last dose in the core study and initiation of treatment in the long-term extension study was 3 months.
Concomitant csDMARDs, oral corticosteroids, and non-steroidal anti-inflammatory drugs were permitted up to the maximum dose based on clinical response and the physician’s judgment (Figure S2 in the supplementary material).
The end of the study occurred up to 84 weeks of treatment because the SC formulation of TCZ was approved by the US Food and Drug Administration and commercial supplies became available as planned.
Outcomes and Assessments
The primary study endpoint was the number and percentage of patients with serious adverse events (SAEs) during long-term treatment with TCZ-SC. The secondary safety endpoints were the number and percentage of patients who experienced AEs of special interest, number and percentage of patients with AEs and SAEs causing discontinuation of TCZ, number and percentage of patients who discontinued TCZ for any reason, incidence of clinically significant laboratory abnormalities (hematology, lipid profiles, and liver function), rates of immunogenicity, summary of abnormal vital signs, and patient compliance (drug dispensing logs, patient diary cards, and return records). Clinical efficacy was measured by the Disease Activity Score in 28 joints using the erythrocyte sedimentation rate (DAS28-ESR), Clinical Disease Activity Index (CDAI), Simplified Disease Activity Index (SDAI), total tender joint count (TJC), total swollen joint count (SJC), and achievement of Boolean remission (defined as SJC ≤1, TJC ≤1, patient global assessment visual analog scale score ≤1 cm, and C-reactive protein level ≤1 mg/dL).
AEs were monitored throughout the study, and laboratory measurements were assessed every 12 weeks. Efficacy assessments were made every 12 weeks. Blood samples were collected to evaluate TCZ and soluble IL-6R serum levels and development of anti-TCZ antibodies (Figure S3 in the supplementary material).
Statistical Methods
No formal statistical hypothesis testing was performed. Patients were grouped by their initial dose frequency. Safety analyses included all patients who received ≥1 dose of study medication and had ≥1 postdose safety assessment. Efficacy analyses included all patients who received ≥1 dose of study medication and had ≥1 efficacy measurement performed. Due to the noncomparative nature of the study, descriptive statistics were produced for all endpoints.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional review boards: Southern Arizona VA Health Care System, St. Vincent Hospital/Reliant Medical Group/Fallon Community Health Plan, Western, Oklahoma Medical Research Foundation, Via Christi Hospitals Wichita, Inc. and Dartmouth Hitchcock Medical Center Quorum) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients included in the study.
Results
Patient Disposition and Baseline Characteristics
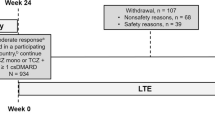
A total of 217 patients who completed BREVACTA or SUMMACTA were enrolled and received treatment, including 44 patients who received TCZ-SC q2w and 173 patients who received TCZ-SC qw (Fig. 1). Of the 217 patients, 183 (84.3%) completed the study at the time of TCZ-SC formulation approval. The mean length of TCZ-SC additional exposure was 0.93 years past the original 96-week 2-year studies. The integrated mean duration of TCZ-IV or TCZ-SC exposure (the duration in the core studies combined with the duration in this study) was 2.6 years.

Overview of patient disposition. aOne patient was not treated. bFourteen patients did not meet the screening criteria including unable and unwilling to provide written informed consent and to comply with the requirements of the study protocol; any major episode of infection requiring hospitalization or treatment with intravenous antibiotics ≤4 weeks of screening or oral antibiotics ≤2 weeks of screening; receiving intraarticular or parenteral corticosteroids ≤4 weeks prior to baseline; diagnosed with neuropathies or other conditions that might interfere with pain evaluation; and meeting any lab exclusion criteria at screening (i.e., alanine aminotransferase or aspartate aminotransferase >3 times upper limit of normal (ULN), total bilirubin >ULN). Fifteen patients did not enroll in the long-term extension due to either the physicians’ recommendations or personal decisions not to participate. AE adverse event; qw every week, q2w every 2 weeks, SC subcutaneous, TCZ tocilizumab
Baseline demographics and clinical characteristics were balanced across the dosing frequency groups (Table 1). The majority of patients (76.5%) were female, and the mean [standard deviation (SD)] age was 58.4 (10.3) years. The mean (SD) duration of RA was 12.8 (9.0) years, the mean (SD) CDAI was 23.1 (16.2), the mean (SD) DAS28 was 4.6 (1.9), and the mean (SD) SDAI was 24.4 (16.8).
Safety
The rates of AEs and SAEs were stable over time in both TCZ-SC qw and TCZ-SC q2w dosing groups (Table 2). A total of 179 patients (82.5%) experienced 954 AEs (133 AEs in the TCZ-SC q2w group and 821 AEs in the TCZ-SC qw group). The most common AEs for all patients were upper respiratory tract infection (19.8%), sinusitis (11.1%), and nasopharyngitis (10.1%). A total of 23 patients (10.6%) experienced 37 SAEs (7 SAEs in the TCZ-SC q2w group and 30 SAEs in the TCZ-SC qw group). The rate of SAEs was 14.72 events per 100 patient-years (PY), and the most common SAEs for all patients were infections (3.7%), nervous system disorders (2.3%), gastrointestinal (GI) disorders (1.8%), and musculoskeletal and connective tissue disorders (1.8%). There were no deaths.
There were 85 patients (49.1%) in the TCZ-SC qw group and 12 patients (27.3%) in the TCZ-SC q2w group who experienced AEs leading to dose modification or interruption. Seven patients (3.2%) had their dosing frequency reduced from TCZ-SC qw to TCZ-SC q2w due to AEs. Withdrawals occurred in 15.7% of patients, with 6.0% of patients withdrawing due to safety reasons (2.3% in the TCZ-SC q2w group and 6.9% in the TCZ-SC qw group).
There were 11 serious infections in eight patients (4.38 per 100 PY), and there were no opportunistic infections (Table 2). The serious infections reported in the TCZ-SC qw group included cellulitis (3 events), septic arthritis (1 event), endocarditis (1 event), staphylococcal bacteremia (2 events), and bronchopneumonia (1 event). In the TCZ-SC q2w group, endophthalmitis (1 event) and pyelonephritis (2 events) were reported. There were two GI perforation events in the TCZ-SC qw group and no GI perforation events in the TCZ-SC q2w group. One GI perforation event was a diverticular perforation considered related to study treatment and the other was a gastric ulcer perforation complicated by peritonitis. There were no myocardial infarctions, demyelinating disorders, or serious hepatic events. There were a total of five strokes, all in the TCZ-SC qw group. One patient in the TCZ-SC q2w group experienced a serious bleeding event (hematuria). There were two malignancies, one in the TCZ-SC q2w group (basal cell carcinoma) and one in the TCZ-SC qw group (endometrial stromal sarcoma).
Hypersensitivity Reactions, Injection Site Reactions and Immunogenicity
There were six non-serious hypersensitivity reactions defined as all AEs [except for injection site reactions (ISRs)] that occurred during or within 24 h of injection and which were not judged ‘unrelated’ to treatment by the investigator, regardless of whether or not they were consistent with hypersensitivity. There were 29 ISRs in six patients during the study (one patient in the TCZ-SC q2w group and five patients in the TCZ-SC qw group; Table 3). None of the ISRs in any patient was serious, and none of the patients discontinued study treatment due to an ISR. One patient had 24 ISRs. In this patient, ISRs occurred with every TCZ-SC qw injection up to 216 study days, and there were no reports of ISRs ≈4 months after reducing the dose frequency to TCZ-SC q2w; the patient did not discontinue treatment and no clinical sequelae were associated with the ISRs. This patient experienced ISRs with every injection in the core trial as well.
One patient developed antidrug antibodies in the TCZ-SC qw arm (Table 3). No patients experienced anaphylaxis, serious hypersensitivity, or clinically significant hypersensitivity reactions, including the one who developed anti-TCZ antibodies. There was no association between lack of efficacy or loss of therapeutic response and development of anti-TCZ antibodies. None of the patients who withdrew due to insufficient therapeutic response had a positive anti-TCZ antibody screening assay.
Laboratory Abnormalities
The overall incidence of markedly abnormal laboratory values was ≤5%. Elevations >3 × the upper limit of normal (ULN) at any point in alanine aminotransferase (ALT) levels were observed in two patients and in aspartate aminotransferase levels were observed in one patient (Table 4). Two patients in the TCZ-SC qw group had ≥2 consecutive observations of ALT >3 × ULN. No serious hepatic events were observed in any patients. There were six cases of grade 3 neutropenia (≥500 to <1000 cells/mm3) reported as AEs (one in the TCZ-SC q2w group and five in the TCZ-SC qw group). No patients who experienced neutropenia developed serious or opportunistic infections. One patient in the TCZ-SC qw group experienced grade 3 thrombocytopenia (≥50,000 to <25,000 cells/mm3) that was reported as an SAE. Among the patients with normal cholesterol levels at baseline, there were nine grade 1 levels (>ULN to ≤7.75 mmol/L; 2 in the TCZ-SC q2w group and 7 in the TCZ-SC qw group) and 18 grade 2 levels (>7.75 to ≤10.34 mmol/L; all in the TCZ-SC qw group). Among patients with normal triglyceride levels as baseline, there were 28 grade 1 (>1.71 to ≤3.42 mmol/L), 1 grade 2 (>3.42 to ≤5.7 mmol/L) and 1 grade 3 (>5.7 to ≤11.4 mmol/L) levels. Seven patients (3%) had markedly high (≥5.4 mmol/L) low density lipoprotein cholesterol levels, all of whom were in the TCZ-SC qw group.
Efficacy
The efficacy of TCZ remained stable over time. Mean DAS28-ESR, CDAI, and SDAI scores decreased from baseline and then remained stable over time (Fig. 2a and Figure S4 in the supplementary material). At the time the study ended due to the SC formulation of TCZ becoming commercially available, only two and one patients from the TCZ-SC q2w group remained up to 60 and 72 weeks, respectively; therefore, the efficacy for this group beyond 60 weeks cannot be accurately evaluated. Overall, the proportion of patients who achieved low disease activity (LDA) and remission based on DAS28-ESR (defined as DAS28-ESR ≤ 2.6 and DAS28-ESR ≤3.2, respectively) at 36 weeks in the extension period, after which there was not a sufficient number of patients for analysis, was 48.5% and 36.4%, respectively (Fig. 2b; results by group are shown in Fig. 2c, d). A total of 62.0% of patients achieved an ACR20 response at week 36 of the extension period. Based on CDAI, LDA and remission at 36 week of the extension period were achieved in 40.9% and 11.1% of patients, respectively, and based on SDAI, LDA and remission at 36 weeks in the extension period were achieved in 40.9% and 14.1% of patients, respectively. Four patients (1.8%) had their dosing frequency reduced from TCZ-SC qw to TCZ-SC q2w due to sustained clinical remission.

Efficacy over time. Time is shown in weeks from the beginning of the extension study; total TCZ exposure time is the indicated extension study week plus 96 or 97 weeks from the core trial. Mean (95% CI) DAS28-ESR in patients who received TCZ-SC qw or TCZ-SC q2w (total patients) is shown for each 12 week interval during the study (a). The percentage of patients in DAS28 LDA or remission is shown over the course of the study for the total population (b) and those who received TCZ-SC q2w (c) or TCZ-SC q2w (d). Remission was defined as DAS28 ≤2.6 and LDA was defined as DAS28 ≤3.2. CI confidence interval, LDA low disease activity, SC subcutaneous, TCZ tocilizumab, qw weekly, q2w every other week, DAS28-ESR disease activity score in 28 joints using the erythrocyte sedimentation rate
Pharmacokinetics and Pharmacodynamics
The mean concentration of serum TCZ in patients in the TCZ-SC qw treatment group increased from baseline (the beginning of the extension period) to week 12, then remained constant until week 72, ranging between 36.6 and 40.4 µg/mL. The mean concentration of serum TCZ in patients in the TCZ-SC q2w treatment group increased from baseline to week 48, ranging between 8.4 and 15.9 µg/mL.
The mean soluble IL-6R concentration in patients receiving TCZ-SC qw increased from baseline to week 12, then stayed constant until week 72, and post baseline, the concentration ranged between 523.2 and 579.4 ng/mL. The mean soluble IL-6R concentration in patients receiving TCZ-SC q2w steadily increased from baseline to week 48, and from week 12 to week 48, the concentration ranged between 398.7 and 518.9 ng/mL.
Discussion
This long-term extension of the SUMMACTA and BREVACTA studies assessed the safety, immunogenicity, and efficacy of the extended use of TCZ-SC qw and TCZ-SC q2w in US patients with RA. The long-term use of TCZ-SC was well tolerated and consistent with the known safety profile of TCZ. The rate of SAEs (events/100 PY) in this study was 14.7, consistent with the rates observed for TCZ-SC at 96 or 97 weeks in BREVACTA and SUMMACTA [11.2 events/100 PY (TCZ-SC q2w) and 14.6 events/100 PY (TCZ-SC qw), respectively] [9]. Serious infections were the most common SAE, and the overall incidence of serious infections in this study (4.38/100 PY) was similar to that in previous TCZ studies, including the 97-week SUMMACTA trial (3.95/100 PY for TCZ-SC qw and 3.92/100 PY for TCZ-IV q4w), the long-term cumulative safety analyses of the phase 3 TCZ-IV clinical trials (4.5/100 PY), and the 3-year postmarketing report of TCZ-IV in Japan (3.67/100 PY) [3, 9, 12]. No new safety signals were identified.
Immunogenicity is a concern with biologic therapies. Antidrug antibodies can lead to loss of efficacy, hypersensitivity reactions, and/or allergic responses [13, 14]. In this long-term extension study of TCZ-SC, immunogenicity was very low, which was consistent with previous studies of TCZ-SC and TCZ-IV. [7, 9, 11, 15] Only one patient (0.5%) developed anti-TCZ antibodies, and this patient did not experience anaphylaxis, serious hypersensitivity reactions, or ISRs. Moreover, this patient did not experience a lack of efficacy or loss of therapeutic response associated with the development of anti-TCZ antibodies. Similarly, in the 97-week SUMMACTA study in which patients received TCZ-SC or TCZ-IV, 0.5–1.6% of patients developed anti-TCZ antibodies, with no correlation between anti-TCZ antibody development and clinical response or AEs [9]. This and other studies have shown that TCZ has one of the lowest incidences of antidrug antibody development among bDMARDs [16, 17], even lower than what has recently been reported for a fully human monoclonal antibody against the IL-6R [18].
Overall, the efficacy data in this study showed benefit of the extended use of TCZ-SC, given that patients who remained in the study and continued to receive TCZ-SC remained stable from baseline in mean DAS28-ESR, CDAI, and SDAI. The proportion of patients who achieved LDA increased or remained stable with the continuation of TCZ-SC treatment. Improvements in patient’s and physician’s global assessment of disease activity and pain were also maintained over time with the extended use of TCZ-SC.
There were some limitations to this analysis. Whereas SUMMACTA and BREVACTA were global studies, this long-term extension of SUMMACTA and BREVACTA was conducted only in US patients. Despite this difference, the safety and efficacy profiles in this study were comparable with those in the global studies. Overall, the number of patients was low (N = 217), with only 44 patients in the TCZ-SC q2w group. In addition, this extension study was limited to patients who completed SUMMACTA and BREVACTA; those who discontinued for any reason, including due to safety or efficacy, were not included. Only 14 patients (6.5%) reduced dosing frequency from TCZ-SC qw to TCZ-SC q2w (7 patients reduced due to AEs and four patients reduced due to sustained remission); therefore, this study provided limited additional information concerning the patients who received less frequent dosing.
Conclusion
In this long-term extension analysis to the SUMMACTA and BREVACTA trials, the long-term use of TCZ-SC showed no increased risk of safety events and efficacy was maintained or improved. Overall, the results of this study supported the long-term safety and efficacy of TCZ-SC qw or TCZ-SC q2w therapy in patients with RA.
References
Ogata A, Tanimura K, Sugimoto T, et al. Phase III study of the efficacy and safety of subcutaneous versus intravenous tocilizumab monotherapy in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66:344–54.
Smolen JS, Beaulieu A, Rubbert-Roth A, et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371:987–97.
Yamamoto K, Goto H, Hirao K, et al. Longterm safety of tocilizumab: results from 3 years of followup postmarketing surveillance of 5573 patients with rheumatoid arthritis in Japan. J Rheumatol. 2015;42:1368–75.
Genovese MC, McKay JD, Nasonov EL, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheum. 2008;58:2968–80.
Emery P, Keystone E, Tony HP, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67:1516–23.
Jones G, Sebba A, Gu J, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis. 2010;69:88–96.
Burmester GR, Rubbert-Roth A, Cantagrel A, et al. A randomised, double-blind, parallel-group study of the safety and efficacy of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional disease-modifying antirheumatic drugs in patients with moderate to severe rheumatoid arthritis (SUMMACTA study). Ann Rheum Dis. 2014;73:69–74.
Kivitz A, Olech E, Borofsky M, et al. Subcutaneous tocilizumab versus placebo in combination with disease-modifying antirheumatic drugs in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66:1653–61.
Burmester GR, Rubbert-Roth A, Cantagrel A, et al. Efficacy and safety of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional DMARDs in patients with RA at week 97 (SUMMACTA). Ann Rheum Dis. 2016;75:68–74.
Eulenfeld R, Dittrich A, Khouri C, et al. Interleukin-6 signalling: more than Jaks and STATs. Eur J Cell Biol. 2012;91:486–95.
Ogata A, Amano K, Dobashi H, et al. Longterm safety and efficacy of subcutaneous tocilizumab monotherapy: results from the 2-year open-label extension of the MUSASHI study. J Rheumatol. 2015;42:799–809.
Genovese MC, Rubbert-Roth A, Smolen JS, et al. Longterm safety and efficacy of tocilizumab in patients with rheumatoid arthritis: a cumulative analysis of up to 4.6 years of exposure. J Rheumatol. 2013;40:768–80.
van Schouwenburg PA, Rispens T, Wolbink GJ. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat Rev Rheumatol. 2013;9:164–72.
Maggi E, Vultaggio A, Matucci A. Acute infusion reactions induced by monoclonal antibody therapy. Expert Rev Clin Immunol. 2011;7:55–63.
Keiserman M, Codreanu C, Handa R, et al. The effect of antidrug antibodies on the sustainable efficacy of biologic therapies in rheumatoid arthritis: practical consequences. Expert Rev Clin Immunol. 2014;10:1049–57.
Mok CC, van der Kleij D, Wolbink GJ. Drug levels, anti-drug antibodies, and clinical efficacy of the anti-TNFalpha biologics in rheumatic diseases. Clin Rheumatol. 2013;32:1429–35.
Schaeverbeke T, Truchetet ME, Kostine M, Barnetche T, Bannwarth B, Richez C. Immunogenicity of biologic agents in rheumatoid arthritis patients: lessons for clinical practice. Rheumatology (Oxford). 2016;55:210–20.
Genovese MC, Fleischmann R, Kivitz AJ, et al. Sarilumab Plus Methotrexate in Patients With Active Rheumatoid Arthritis and Inadequate Response to Methotrexate: Results of a Phase III Study. Arthritis Rheumatol. 2015;67:1424–37.
Acknowledgments
The authors wish to thank all investigators and patients who participated in the study and all members of the ML28338 study team. Sponsorship and article processing charges for this study were funded by F. Hoffmann-La Roche, Ltd. Support for third-party writing assistance for this manuscript, furnished by Ellen Mercado, PhD, of Health Interactions, was provided by F. Hoffmann-La Roche, Ltd. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published.
Disclosures
AKivitz has received research grants and/or consulting fees from AbbVie, Pfizer, Genentech, UCB, Amgen, Celgene, Bristol-Myers Squibb, AstraZeneca and Janssen. E Olech has received research grants and/or consulting fees from AbbVie, Amgen, Celgene, Genentech, Janssen, Regeneron, Sanofi-Aventis, UCB and Vertex Pharmaceuticals. M Borofsky has no conflict of interest. T Wallace is an employee of Genentech. J Devenport is an employee of Genentech. J Pei is an employee of Genentech. M Michalska is an employee of Genentech.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional review boards: Southern Arizona VA Health Care System, St. Vincent Hospital/Reliant Medical Group/Fallon Community Health Plan, Western, Oklahoma Medical Research Foundation via Christi Hospitals Wichita, Inc. and Dartmouth Hitchcock Medical Center Quorum) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients for being included in the study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/A0E6F0602E4A01A1.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kivitz, A., Wallace, T., Olech, E. et al. Long-Term Safety and Efficacy of Subcutaneously Administered Tocilizumab for Adult Rheumatoid Arthritis: A Multicenter Phase 3b Long-term Extension Study. Rheumatol Ther 3, 291–304 (2016). https://doi.org/10.1007/s40744-016-0043-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-016-0043-1