
Abstract
Juvenile idiopathic arthritis is a broad term that describes a clinically heterogeneous group of arthritides of unknown cause, which begin before 16 years of age. This term encompasses several disease categories, each of which has distinct presentation, clinical manifestations, and, presumably, genetic background and etiopathogenesis. Although none of the available drugs has curative potential, prognosis has greatly improved as a result of substantial progresses in disease management. The most important new development has been the introduction of the biologic medications, which constitute a valuable treatment option for patients who are resistant to conventional antirheumatic agents. Further insights into the disease pathogenesis and treatment will be provided by the continuous advances in understanding of the mechanisms related to the immune response and inflammatory process, and by the development of new drugs that are capable of selectively inhibiting single molecules or pathways.
Similar content being viewed by others
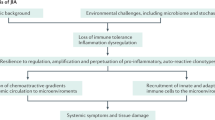
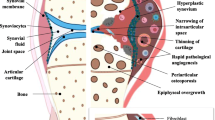

Avoid common mistakes on your manuscript.
Introduction
Juvenile idiopathic arthritis (JIA) is a heterogeneous group of conditions which encompasses all forms of arthritis of unknown etiology lasting for at least 6 weeks and with onset before the age of 16 years [1]. As a result of the lack of pathognomonic features, the diagnosis of JIA is one of exclusion among all possible causes of chronic arthritis in childhood.
The aim of this review is to provide a summary of the epidemiology, clinical features, diagnosis, and treatment of JIA. This article was based on previously conducted studies and did not involve any new studies of human or animal subjects performed by any of the authors.
Epidemiology
JIA is the most common chronic rheumatic disease of childhood and a leading cause of short- and long-term disability. Its reported incidence and prevalence in European and North American populations range from 2 to 20 and from 16 to 150 per 100,000, respectively [1]. However, remarkable disparity in the frequency of JIA subtypes has been noticed in different geographical areas or ethnic groups. In Western countries oligoarthritis is the most common subtype, while polyarthritis predominates in Costa Rica, India, New Zealand, and South Africa [2, 3]. In Asia, systemic arthritis accounts for a greater proportion of childhood arthritis [2, 4]. In India, Mexico, and Canada, a greater incidence of enthesitis-related arthritis (ERA) has been registered, reflecting, at least in part, the high frequency of the human leukocyte antigen (HLA)-B27 in these populations [2]. Rheumatoid factor (RF)-positive polyarthritis is the less common subtype. Distinct distributions of age at onset and sex characterize each onset type. Broader insights into the worldwide variability of JIA phenotypes will come out of the multinational study of the EPidemiology, treatment and Outcome of Childhood Arthritis (EPOCA Study [5]), which has enrolled thus far around 9000 patients from 42 countries in five continents. The potential role of phenotypic variability of JIA across races or ethnic groups in explaining genetic predisposition and pathogenesis has been recently discussed [6].
Classification
Over the last few decades, several classification systems for chronic arthritis in childhood have been proposed [7]. The current scheme, based on the criteria created by the Pediatric Task Force of the International League of Associations for Rheumatology (ILAR) [8], introduced the unifying term of JIA and outlined seven disease categories (Table 1) [7], on the basis of the clinical and laboratory features present in the first 6 months of illness [9]. Although the ILAR classification has served well to harmonize the terminology across Europe and North America and the criteria used to enroll patients in research studies and clinical trials, it has recently been subject to several criticisms [10–19]. In particular, some concerns have been raised about the use of the number of affected joints and the presence of psoriasis as parameters to define homogeneous disease entities [17]. Furthermore, it has been shown that the presence of antinuclear antibodies (ANA) identifies a homogeneous disease subset across various ILAR categories [18, 19]. The rationale underlying a proposal for a new classification of JIA has been recently discussed [20].
Clinical Manifestations
Systemic arthritis accounts for 5–15% of children with JIA in North America and Europe [21]. The ILAR criteria for systemic arthritis require the presence of arthritis accompanied or preceded by a documented quotidian fever of at least 2 weeks’ duration, plus at least one of the following: characteristic rash (Fig. 1), generalized symmetrical lymphadenopathy, enlargement of liver or spleen, or serositis (pericarditis, pleural or pericardial effusion, rarely peritonitis). The fever has a typical intermittent pattern, with one or two daily spikes, up to 39 °C or higher, followed by rapid return to baseline. The erythematous, salmon pink, evanescent macular rash usually appears with fever [2]. Arthritis is often symmetrical and polyarticular, but may be absent at onset and develop much later. In these cases, diagnosis cannot be considered definite until arthritis is present. There are always signs of systemic inflammation, but no specific laboratory abnormalities. A sharp rise of ferritin, together with a drop in platelet count, an increase in serum transaminases, and a decrease of fibrinogen level, in conjunction with a change in the fever pattern from intermittent to continuous, may herald the occurrence of macrophage activation syndrome (MAS). In 2005, preliminary diagnostic guidelines for MAS in patients with systemic JIA were published [22, 23]. A multinational collaborative effort aimed at developing new classification criteria for the syndrome has been recently accomplished [24–27] (Table 2).

Salmon-macular rash in systemic juvenile idiopathic arthritis
Rheumatoid factor (RF)-negative polyarthritis is defined as an arthritis that affects five or more joints during the first 6 months of disease in the absence of immunoglobulin (IgM) RF. This is a heterogeneous category that may manifest with at least three different phenotypes [2, 3]. The first is very similar to early-onset oligoarthritis; the second subtype is more similar to RF-negative rheumatoid arthritis (RA) of the adults, with symmetric arthritis of large and small joints, later onset, and negative ANA; the third subset, known as “dry synovitis”, exhibits negligible joint swelling but prominent stiffness and flexion contractures. This subset is often poorly responsive to treatment and may pursue a destructive course [28].
Oligoarthritis accounts for 50–80% of all children with chronic arthritis in North American and European white populations [2] and affects four or fewer joints during the first 6 months of disease (Fig. 2). The ILAR classification distinguishes two further subsets: persistent, if arthritis remains confined to four or fewer joints during the whole disease course; or extended, if arthritis spreads to more than four joints after the initial 6 months of illness. Most of these children display a characteristic phenotype, with asymmetric arthritis, early disease onset (<6 years), female predilection, high frequency of positive ANA, and high risk of iridocyclitis [2, 3]. Oligoarthritis predominantly affects the joints of the lower extremities, with the knee being most frequently involved (30–50% of the cases), followed by the ankle. The main complication is chronic uveitis (20–30% of patients), with ANA positivity (70–80% of patients) representing the most important risk factors for its occurrence [18, 19]. Wrists and ankle arthritis and high erythrocyte sedimentation rate (ESR) at onset have been identified as predictors for an extended course [29–31].

Arthritis of the right knee in a child with oligoarticular juvenile idiopathic arthritis
Enthesitis-related arthritis (ERA) mainly affects male patients older than 6 years and is characterized by the association of enthesitis and arthritis [2, 3]. Most of the patients are HLA-B27 positive and have negative RF and ANA. The onset of ERA may be insidious, as intermittent osteoarticular pain and stiffness may be present, with or without objective inflammation of peripheral joints. The presence of enthesitis, especially at the calcaneal insertions of the Achilles tendon, the plantar fascia, and the tarsal area, is the most helpful diagnostic feature. The joints of the lower extremities and the hip are predominantly affected. Sacroiliitis, mono- or bilateral, may be a clinical feature of ERA, as a part of the axial skeleton involvement. A plain radiograph does not exclude the diagnosis of sacroiliitis, and in that case magnetic resonance imaging (MRI) is very helpful. In some cases the disease progresses to the clinical picture of ankylosing spondylitis (AS) [2]. ERA is often remitting and mild. Nevertheless, limitation in the expansion of thorax or back may occur and for this reason should be documented early. Patients with ERA may also develop cardiopulmonary and cerebrovascular complications, which are also a leading cause of shorter life expectancy. Amyloidosis and renal sequelae more frequently occur in adult-onset AS, but little information is actually available in children. The diagnosis of juvenile psoriatic arthritis (JPsA) by the ILAR criteria requires the coexistence of arthritis and a typical psoriatic rash or, when the rash is missing, the presence of arthritis and any two of the following: family history of psoriasis in a first-degree relative, dactylitis (sausage-like swelling of individual digits that extends beyond the joint margins), and nail pitting or onycholysis. There is increasing evidence that JPsA is not a homogeneous disease entity, but includes at least two distinct subgroups: one shares the same characteristics as early-onset ANA-positive JIA, the other belongs to the spectrum of spondyloarthropathies [32].
Undifferentiated arthritis includes patients who do not meet the criteria for any category, or who meet the criteria for more than one. Several proposals for revision of this category have been put forward [16, 31].
Diagnosis
Juvenile idiopathic arthritis (JIA) is a diagnosis of exclusion that, when suspected, requires a complete clinical evaluation, including family to personal history and recent pathologic events, and specific attention to pain and morning stiffness. A detailed physical examination should always be performed to examine all body joints at both first evaluation and follow-up visits [33]. At the end of the visit, the physician is asked to provide his/her global rating of the overall level of disease activity on a visual analog scale (VAS), ranging from 0 (no activity) to 10 (maximum activity) [33, 34]. The differential diagnosis of JIA is wide (Table 3). The identification of systemic JIA may be challenging as arthritis is often not present at onset.
Updates on Outcome Measures and Future Outcomes
The incorporation of patient-reported or parent-reported outcomes (PRCOs), when measuring the health state of patients with pediatric rheumatic diseases, has become crucial in the last few years [35–38]. These tools may help the physician to improve the patient management through the identification of the most salient clinical issues and to focus the attention on the most relevant matters for the patient management. Conversely, this may improve adherence of the patient to treatment by actively participating in shared decision-making [35, 38]. PCROs in JIA may be assessed by different tools, including a VAS for rating a child’s overall well-being and intensity of pain, and questionnaires for the evaluation of functional ability and health-related quality of life (HRQoL) [34–37, 39]. Recently, the Juvenile Arthritis Multidimensional Assessment Report (JAMAR) was created with this aim [34], introducing a new approach to clinical care for children with JIA, through quantitative data collected at each visit as a standardized procedure in order to guide the physician in monitoring the patient over time [40].
A recent physician-centered outcome measure in JIA is the Juvenile Arthritis Disease Activity Score (JADAS). JADAS is a composite disease activity index that is made up by pooling four individual measures: physician’s global assessment of disease activity (PGA), parent’s/patient’s assessment of child’s well-being (PPGA), count of joints with active arthritis (assessed in 71, 27, or 10 joints, depending on the version), and ESR [33]. Recent studies have shown that the ESR can be replaced by the C-reactive protein without altering the performance of the instrument [41]. In addition, a three-item version (clinical JADAS, cJADAS), which excludes the acute phase reactant, was found to correlate closely with the original tool [42]. The cutoff values of JADAS that correspond to the main disease activity states of JIA have been recently established [36, 37, 39]. The care of JIA patients cannot be possible without appropriate and validated outcome measures, for which further work is required [41].
Imaging
Conventional radiography remains the gold standard for the detection of structural joint damage and growth and maturation disturbances of bones in JIA patients [43, 44]. In recent years, a great deal of effort has been made to develop new radiographic scoring systems or to adapt adult methods for use in JIA [45–51]. However, the poor sensitivity of plain radiographs in identifying active synovitis and its limited ability to disclose erosive changes early in the disease course has raised interest in alternative imaging methodologies.
MRI is the only tool that has the ability to simultaneously assess all features of synovial disease and is exquisitely suited for the evaluation of disease activity in the temporomandibular, hip, sacroiliac, and vertebral joints [52–55] (Fig. 3). The main advantage of MRI over conventional radiography is the direct visualization of synovitis, cartilage, and early erosive lesions. Dynamic contrast-enhanced MRI (DCE-MRI) enables the analysis of the time course of signal changes following gadolinium administration [56]. A peculiar lesion detectable by MRI is periarticular bone marrow edema. This abnormality represents a key predictor of erosive joint damage in adults with arthritis [57], but its meaning is still debated in JIA, as some studies have shown that bone abnormalities on MRI resembling bone marrow edema can be seen in healthy subjects [58]. MRI identifies early changes in both sacroiliac joints and spine, especially in ERA and AS patients, as the most sensitive indicator of inflammation on these sites. This imaging cannot be used routinely in children, but should be always considered when back pain is present, because demonstration of an earlier involvement of the sacroiliac joints could influence the therapeutic approach.

T1-weighted magnetic resonance imaging of left sacroiliitis in a patient with enthesitis-related arthritis
Ultrasonography has several advantages over other imaging modalities, including noninvasiveness, rapidity of performance, relatively low cost, ability to scan multiple joints at one time, repeatability, safety, and high acceptability among patients. Ultrasonography is well suited for the diagnosis and assessment of synovitis and related abnormalities, with color and power Doppler ultrasonographic techniques being considered superior to grayscale ultrasonography in identifying active disease [59, 60]. However, it is an operator-dependent technique and requires training and a careful interpretation of the abnormalities [61]. Recently, age- and sex-related normal standards in cartilage thickness in small and large joints on ultrasonographic images have been established [62]. The capacity to assess joints dynamically, and in real time, and to capture bone erosions [63, 64], as well as its usefulness to guide local injections into joints, tendons, or other periarticular structures [65], are additional advantages of this technique.
Biomarkers
A number of biomarkers have been tested or are under development for defining JIA subtypes, measuring disease activity, and predicting disease course, response to therapy, or risk for complications [66]. Hunter et al. [67] found remarkable differences in cell frequencies, inflammatory protein levels, and gene expression in the affected joints between children with extended oligoarthritis sampled before extension and children who had a persistent oligoarthritis. Similar results were found by Gibson et al. [68] in the proteome profiles in the synovial fluid of these two subgroups of patients. The serum levels of matrix metalloproteinase-3 (MMP-3), an endopeptidase that may directly damage cartilage and bone, were shown to be higher in children with ERA than in healthy subjects [69] and to correlate with various clinical measures of disease activity, suggesting that this protein may be a marker of disease severity [70] and progression of structural joint damage [71]. Two pro-inflammatory S100 proteins, the S100A8/9 [or myeloid-related protein (MRP) 8/14] and the neutrophil-derived S100A12, were shown to be sensitive measures of disease activity in JIA [72–75] and may help to identify patients who are more likely to respond to antirheumatic therapies, such as methotrexate [76] or IL-1 or tumor necrosis factor (TNF) inhibitors [73]. The presence of higher MRP8/14 concentrations was associated with risk of relapse after treatment discontinuation, which led to the hypothesis that the measurement of these proteins may support the decision to discontinue the medication [77] and predict an earlier disease relapse [78]. IL-18 is a candidate biomarker for response to therapy in systemic JIA [79], as demonstrated by Vastert et al. [80]. Biomarkers may facilitate the diagnosis and prediction of MAS in patients with systemic JIA. The serum levels of soluble interleukin-2 receptor α (sIL-2Rα, also known as CD25) and soluble CD163 (sCD163), which reflect the degree of activation and expansion of T cells and phagocytic macrophages, respectively, were found to represent valuable diagnostic parameters for MAS and to identify patients with subclinical forms [81–83]. Gorelik et al. [84] showed that serum levels of follistatin-like protein 1, a glycoprotein overexpressed in certain inflammatory diseases, were markedly elevated during acute MAS and returned to normal after treatment.
Treatment
The optimal approach to the management of a child with JIA is based on a multidisciplinary team comprising a pediatric rheumatologist, ophthalmologist, orthopedic surgeon, specialist nurse, physical therapist, occupational therapist, and psychologist [2]. Non-pharmacological and pharmacological interventions may aid in the management of JIA patients.
Non-Pharmacological Interventions
An important aim of the management of JIA is to foster the normal psychosocial and social development of the child and to tackle possible difficulties caused by the disease or its consequences on family life [85, 86]. Participation in peer-group activities and regular attendance at school should be strongly encouraged, as well as sporting activities, like swimming and cycling. Appropriate attention to psychosocial issues, with the help of a pediatric psychologist, whenever needed, can have a positive impact on the well-being of the child.
Physiotherapy and occupational therapy, with the aim to keep or restore joint function and alignment as much as possible and to achieve a normal pattern of mobility, are important components of the therapeutic approach to any patients with JIA [85]. Orthotic devices can be useful in selected patients (i.e., those with flexion contractures).
Surgical approaches to irreversible joint contractures, dislocations, or joint replacement may be indicated, although the role of orthopedic surgery in JIA is much more limited than in the past. The long-term outcome of children with joint disease is not altered by prophylactic synovectomy. However, arthroscopic synoviectomy may prolong the duration of remission in a frequently relapsing joint [87].
Pharmacological Interventions
Nonsteroidal anti-inflammatory drugs (NSAIDs) have traditionally been the mainstay treatment for all forms of JIA. However, their use as monotherapy for more than 2 months is discouraged if arthritis is still active [88]. NSAIDs are not disease modifying, but merely symptomatic medications. Only a few NSAIDs are approved for use in children: the most common are naproxen, ibuprofen, and indomethacin. They are usually better tolerated by children than adults, and the role of antiacids and proton pump inhibitors to reduce gastrointestinal complications in pediatric subjects is unclear. Experience with cyclooxygenase (COX)-2 inhibitors in children is scarce [89, 90]. Meloxicam, an inhibitor of both COX-1 and COX-2, has proven to be effective and safe in a controlled trial [89].
Intra-articular corticosteroid (IAC) injections are widely used in the management of children with JIA, particularly in those with oligoarthritis, to induce rapid relief of inflammatory symptoms and for functional improvement as well as to obviate the need for regular systemic therapy [65, 91]. The strategy of performing multiple IAC injections is used by some pediatric rheumatologists in children with polyarticular JIA to induce prompt remission of synovitis, while simultaneously initiating therapy with disease-modifying antirheumatic drugs (DMARDs) and/or a biologic agent [92, 93]. Triamcinolone hexacetonide (TH) is the medication of choice in JIA [65]. Although there are no established guidelines for this practice, most rheumatologists will limit the frequency of reinjections to three times per year. Subcutaneous atrophic skin changes at the site of injection, periarticular calcifications, crystal-induced synovitis, and septic arthritis are potential complications of IACs. The potential role of IAC injections in the hip in causing avascular necrosis of the femoral head is uncertain [94, 95].
The administration of systemic corticosteroids is mainly restricted to the management of the extra-articular manifestations of systemic arthritis (high fever unresponsive to NSAIDs, severe anemia, myocarditis or pericarditis, and MAS) [86, 96, 97]. High-dose “pulse” intravenous methylprednisolone (10–30 mg/kg/day to a maximum of 1 g/day on 1–3 consecutive days) is effective in controlling these features, but the effect is often short-lived. Therefore, continued corticosteroid therapy with oral prednisone (1–2 mg/kg/day to a maximum of 60 mg/day in a single or divided daily doses) is frequently necessary. A short course of low-dose prednisone (e.g., 0.5 mg/kg/day) may be considered for severe polyarthritis refractory to other therapies or while awaiting the full therapeutic effect of a recently initiated second-line or biologic agent.
Conventional DMARDs
Methotrexate (MTX) remains the most widely used conventional DMARD in the management of JIA because of its effectiveness at achieving disease control and acceptable toxic effects [98, 99]. Its efficacy was established in a controlled trial in 1992 at a dosage of 10 mg/m2 per week given orally [100]. A subsequent randomized study has shown that MTX exerts its maximum therapeutic effect with parenteral administration of 15 mg/m2 per week. There was no additional advantage in giving higher doses up to 30 mg/m2 per week [101]. MTX can be given both orally and subcutaneously, with some studies reporting no differences in effectiveness [102]. However, there is an increased bioavailability of the subcutaneous route at higher doses [103], and other investigators have found increased efficacy after switching from oral to subcutaneous administration [104]. The greatest efficacy of MTX has been seen in patients with extended oligoarthritis. A decrease in the rate of radiographic progression has been reported in two small uncontrolled studies [105, 106]. Recently, no advantage in prolonging MTX administration for 12 instead of 6 months after the achievement of disease remission was seen [77]. Tests to monitor complete blood counts, liver enzymes, and renal function are recommended during MTX treatment, although the optimal frequency of testing is not established [107]. The supplementation of folic or folinic acid may help to prevent the occurrence of liver enzyme abnormalities, oral ulcerations, and nausea [108].
Leflunomide may have similar effectiveness and safety as MTX and is an alternative option to it in case of intolerance [109]. However, experience with this medication in childhood arthritis is still limited.
Biologic DMARDs
Etanercept, a fully human TNF inhibitor, is the first biologic agent registered for use in JIA. Its efficacy at a dosage of 0.8 mg/kg weekly was demonstrated in a controlled trial on 69 patients refractory or intolerant to MTX [110]. Long-term extension studies of the original trial cohort and several national registries have subsequently confirmed the sustained clinical benefit and acceptable safety profile of the drug [111–113]. Etanercept in JIA has been demonstrated to improve ability and quality of life [114], growth velocity and bone status [115, 116] and reduce the progression of radiographic joint damage [117]. Complete disease quiescence can be achieved in half of the patients [118, 119].
Infliximab, a chimeric TNF-α inhibitor, failed to show a statistically significant difference in its primary outcome at 3 months in a placebo-controlled trial [120]. However, after 1 year the response to infliximab was comparable to that observed with etanercept. Paradoxically, despite similar efficacy, patients treated with 3 mg/kg of infliximab experienced a greater frequency of serious adverse events and autoantibodies than those given 6 mg/kg. Infliximab is not approved for use in JIA.
The efficacy of adalimumab, a recombinant human anti-TNF agent, was established in a controlled trial including patients who were either MTX naive, resistant, or intolerant [121], with 94% of patients treated with MTX responding at week 16, versus 74% who did not receive concomitant MTX. Recently, adalimumab was found to be highly effective in children and adolescents with JIA who had been previously treated with other biologic agents [122]. Adalimumab is registered for use in JIA both in the USA, at a fixed dosage of 20 or 40 mg every 2 weeks for children less than 30 kg or at least 30 kg, respectively, and in Europe, at a dosage of 24 mg/m2 (maximum 40 mg) every 2 weeks.
A clinical trial on a second recombinant human TNF inhibitor, golimumab [123], in 173 children with active arthritis despite MTX therapy for at least 3 months showed a rapid response to the medication after 16 weeks of open-label treatment, resulting in achievement of an American College of Rheumatology (ACR) Pediatric 30 response and the state of inactive disease in 87.3% and 36.1% of the patients, respectively. However, no differences in flare rates between golimumab and placebo arms were seen from week 16 to 48 among responders to golimumab in the open-label phase, and the primary endpoint of the trial was not met. The safety profile was acceptable and injections were well tolerated. This drug has not yet been approved for use in JIA.
TNF inhibitors are more effective if administered early in the disease [124], in combination with MTX [125] and/or prednisone [126]. Recent data indicate that TNF inhibitors are efficacious and safe in juvenile spondyloarthropathies and PsA [127, 128].
Abatacept is a soluble, fully human fusion protein that comprises the extracellular portion of human CTLA4 and a fragment of the Fc region of a human IgG1. The binding between abatacept and the CD80/86 molecules prevents their interaction with the CD28 receptor and, therefore, blocks the second signal necessary for T cell activation [129]. The efficacy of abatacept in JIA has been documented in a double-blind randomized controlled withdrawal trial in 190 patients with polyarticular course JIA and an inadequate response or intolerance to at least one DMARD [130]. During the double-blind treatment, flares of arthritis were observed in 53% patients on placebo versus 20% of patients on abatacept (p = 0.0003), who showed a lower risk of flares (hazard ratio 0.31, 95% CI 0.16–0.95). The median time to flare was 6 months for patients given placebo, while insufficient events occurred in the abatacept group (p = 0.0002). The frequency of adverse events did not differ in the two treatment groups. Drug effectiveness was found to be durable in the long-term open-label extension phase of the trial and was noticed also in patients who were initially nonresponders [131]. The improvement was also recorded in HRQoL [132]. Abatacept is registered for JIA patients older than 6 years at the dosage of 10 mg/kg intravenously every 28 days.
A randomized controlled trial on the IL-6 receptor inhibitor tocilizumab in polyarticular-course JIA [133] has enrolled 188 patients placed on tocilizumab at 10 mg/kg if less than 30 kg or 8 mg/kg if at least 30 kg. In the second part of the study, 163 patients were continued with tocilizumab or switched to placebo. Disease flare occurred in 48.1% of patients on placebo versus 25.6% continuing tocilizumab (p = 0.0024). At the end of the second part, 64.6% and 45.1% of patients receiving tocilizumab had ACR Pediatric 70 and 90 responses, respectively. Infection was the most common serious adverse event (4.9/100 patient/years). Tocilizumab has been approved by the US Food and Drug Administration (FDA) for the treatment of polyarticular JIA in children aged 2 years and older.
A growing body of evidence suggests that in active systemic JIA the proinflammatory cytokines that play a major pathogenic role are IL-6 [134, 135] and IL-1 [136], rather than TNF-α. Excellent responses of patients with the systemic subtype of JIA to the IL-1 receptor antagonist anakinra have been observed in uncontrolled studies [136, 137]. Despite the efficacy of the drug in the adult equivalent of systemic JIA (Still’s disease), anakinra has not been registered for the treatment of systemic JIA yet. Two double-blind placebo-controlled trials of canakinumab, a novel monoclonal antibody against IL-1, in children with systemic JIA and active systemic features, have been completed, which showed good efficacy and safety [138, 139]. Canakinumab has been approved for the treatment of active systemic JIA in children aged 2 years and older both in Europe and the USA. A 24-week randomized trial of another IL-1 antagonist, rilonacept, in 71 children with active arthritis in at least two joints demonstrated a shorter time to drug response and good tolerance [140]. A potential advantage of canakinumab and rilonacept over anakinra, which has a short half-life and requires a daily injection, is a longer half-life, which enables the administration at longer intervals (every 4 weeks and weekly, respectively). A retrospective analysis of 46 patients who received anakinra as first-line therapy led to the conclusion that introduction of anti-IL-1 therapy early in the course of systemic JIA may help to prevent refractory arthritis [141]. In a prospective cohort study of 20 patients with new-onset systemic JIA, excellent responses were seen in nearly all patients within 3 months of anakinra as first-line therapy. In the majority of responding patients, treatment could be stopped within 1 year, with remission being maintained during follow-up [80]. Based on these observations as well as on data from animal studies, a biphasic model of systemic JIA has been theorized. It has been speculated that early treatment with biologics may take advantage of this “window of opportunity”, in which disease pathophysiology may be altered to prevent chronic arthritis [142].
Uncontrolled studies [143, 144] and a controlled withdrawal trial performed in Japan [145] have shown impressive clinical responses to the administration of the IL-6 blocker tocilizumab in patients with refractory systemic JIA. These findings were confirmed in a double-blind controlled trial of tocilizumab against placebo in patients with or without systemic manifestations, which showed at the end of the 12-week double-blind phase as an ACR Pediatric 30 response plus absence of fever in 85% of patients on tocilizumab and in 24% of patients on placebo (p < 0.001) [146].
Anecdotal studies have reported the effectiveness of rituximab, a humanized chimeric monoclonal antibody to the B lymphocyte CD20 antigen, in severe resistant systemic JIA [147]. However, so far the information on the use of this agent is very limited.
Safety of Biologics
Most data on the safety of etanercept come from a drug-specific registry [112] and several national registries [113, 148, 149]. In the 594 patients included in the drug-specific registry, the rates of adverse events (AEs), serious adverse events (SAEs), medically important infections, and autoimmune events were similar in those treated with MTX alone, etanercept alone, and MTX and etanercept in combination [112]. No case of TB, demyelinating disease, malignancy, or death was observed. However, the ongoing national registries have reported less favorable data. Among 322 patients who received etanercept in the German registry (592 patient-years of exposure) there were 12 SAEs and treatment was permanently stopped due to AEs in 11 patients, of whom one developed thyroid carcinoma and one demyelination. No opportunistic infection or lupus-like syndrome was detected [113]. Five malignancies out of 1260 patients treated with etanercept were reported in a subsequent publication from the same registry [149]. In the Dutch registry, which included 146 patients (313 patient-years of exposure), nine SAEs, and six permanent discontinuations due to AEs were recorded. Three new-onset autoimmune diseases (sarcoidosis, Crohn’s disease, and ulcerative colitis) and one case of TB were noticed, but there was no demyelinating disease, opportunistic infection, malignancy, or death [150]. Twenty-one of the 483 patients enrolled in the British registry (941 patient-years of exposure) discontinued etanercept because of toxic events: five had infections, 10 central nervous system adverse manifestations, and six other events. One patient developed inflammatory bowel disease but no opportunistic infections or deaths were observed [148].
Fewer data are available on the safety of adalimumab. In the registrative trial of 171 patients who received this medication for up to 104 weeks, 14 patients had SAEs, including seven major infections, and 12 patients were discontinued from therapy because of toxicity. No malignancies, TB, opportunistic infections or demyelinating diseases, new autoimmune diseases, or deaths were reported [121]. Twenty-six SAEs, including six serious infections, were observed in a randomized controlled trial of infliximab [120]. The greater frequency of SAEs, infusion reactions, antibodies to infliximab, and newly produced ANA and anti-DNA antibodies in patients treated with 3 mg/kg rather than in those who were given 6 mg/kg has been discussed above.
In clinical practice, it is important to consider that the administration of anti-TNF agents has been associated with an increasing risk of TB infection onset or reactivation. For this reason, an accurate screening for TB during baseline assessment and a careful monitoring for the entire duration of treatment are mandatory [151].
The potential of anti-TNF agents to induce malignancy is still uncertain. In 2010, the US FDA reported 48 malignancies in pediatric patients who had been treated with TNF inhibitors [152]. However, only 19 of the 48 cases had chronic arthritis and the study was affected by a number of confounding biases, which hampered the interpretation of its findings [153]. The subsequent studies suggested that JIA itself is associated with an increased risk of malignancy and that treatment with TNF blockers does not augment this risk [154–157]. A more definite answer to these safety concerns will be provided by a large-scale effort aimed at collecting safety data related to biologic agents in a multinational population of children with JIA, which is underway.
SAEs reported for other biologics mostly include serious non-opportunistic infections for abatacept, and reaction in the injection site and cases of hepatitis [158] for anakinra. The tolerability profile of tocilizumab has been studied in different trials over the last few years, but a pivotal role is to attribute to the TENDER and CHERISH studies for systemic JIA and polyarticular JIA, respectively [133, 146]. The TENDER trial showed that most of the AEs during tocilizumab treatment are mild or moderate in intensity, not depending on the different dosage, and mostly represented by infections with a rate of 3.4 per patient-year with tocilizumab versus 2.9 with placebo. Streptococcal sepsis, pulmonary hypertension, neutropenia, thrombocytopenia, and high transaminases were also reported. A few years later Yokota et al. described similar AEs, also reporting two cases of MAS possibly due to tocilizumab [159], although further studies are necessary to clarify the real correlation with the biologic treatment. Similar results were reported in polyarticular JIA by Brunner et al. [133] and Imagawa et al. [160]. The safety data collected in the two trials by Ruperto et al. in 2012 [139] confirmed the good safety profile of canakinumab in systemic JIA patients, with the rate of infection during treatment similar to the placebo group. Transient neutropenia and thrombocytopenia were reported, with no higher risk of infections in patients. Seven cases of MAS were reported, with two associated deaths. The mortality rate was not increased compared to other systemic JIA patients.
Conclusions
Over the past 15 years, there have been major advances in the management of JIA, particularly the introduction of the biologic medications, which have dramatically improved the prognosis for children with this disease. Although the studies performed so far have shown that biologic agents are generally safe, only large-scale data collections will define their long-term safety profiles, in particular the risk of malignancy. The genetic and immunologic research that is ongoing will help link the immunopathogenesis to the clinical phenotypes, which should aid in the revision of classification criteria. The identification of new biomarkers, together with the development of more effective outcome measures and the refinement of imaging techniques, may foster the implementation of targeted therapies and personalized therapeutic interventions, with the ultimate goals of improving the remission rates while minimizing disease damage and treatment-related side effects.
References
Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767–78.
Petty RE, Cassidy JT. Textbook of pediatric rheumatology. Philadelphia: Saunders Elsevier; 2011.
Szer I, Kimura Y, Malleson P, Southwood T. Arthritis in children and adolescents. Oxford: Oxford University Press; 2006.
Fujikawa S, Okuni M. Clinical analysis of 570 cases with juvenile rheumatoid arthritis: results of a nationwide retrospective survey in Japan. Acta Paediatr Jpn. 1997;39:245–9.
Consolaro A, Ruperto N, Filocamo G, et al. Seeking insights into the EPidemiology, treatment and Outcome of Childhood Arthritis through a multinational collaborative effort: introduction of the EPOCA study. Pediatr Rheumatol Online J. 2012;10:39.
Consolaro A, Ravelli A. Unraveling the phenotypic variability of juvenile idiopathic arthritis across races or geographic areas - key to understanding etiology and genetic factors? J Rheumatol. 2016;43:683–5.
Petty RE, Southwood TR, Manners P, et al. International League of Associations for rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390–2.
Fink CW. Proposal for the development of classification criteria for idiopathic arthritides of childhood. J Rheumatol. 1995;22:1566–9.
Petty RE, Southwood TR, Baum J, et al. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol. 1998;25:1991–4.
Ramsey SE, Bolaria RK, Cabral DA, Malleson PN, Petty RE. Comparison of criteria for the classification of childhood arthritis. J Rheumatol. 2000;27:1283–6.
Foeldvari I, Bidde M. Validation of the proposed ILAR classification criteria for juvenile idiopathic arthritis. International League of Associations for Rheumatology. J Rheumatol. 2000;27:1069–72.
Fantini F. Classification of chronic arthritides of childhood (juvenile idiopathic arthritis): criticisms and suggestions to improve the efficacy of the Santiago-Durban criteria. J Rheumatol. 2001;28:456–9.
Krumrey-Langkammerer M, Häfner R. Evaluation of the ILAR criteria for juvenile idiopathic arthritis. J Rheumatol. 2001;28:2544–7.
Berntson L, Fasth A, Andersson-Gäre B, et al. Construct validity of ILAR and EULAR criteria in juvenile idiopathic arthritis: a population based incidence study from the Nordic countries. International League of Associations for Rheumatology. European League Against Rheumatism. J Rheumatol. 2001;28:2737–43.
Berntson L, Fasth A, Andersson-Gäre B, et al. The influence of heredity for psoriasis on the ILAR classification of juvenile idiopathic arthritis. J Rheumatol. 2002;29:2454–8.
Burgos-Vargas R, Rudwaleit M, Sieper J. The place of juvenile onset spondyloarthropathies in the Durban 1997 ILAR classification criteria of juvenile idiopathic arthritis. International League of Associations for Rheumatology. J Rheumatol. 2002;29:869–74.
Martini A. Are the number of joints involved or the presence of psoriasis still useful tools to identify homogeneous disease entities in juvenile idiopathic arthritis? J Rheumatol. 2003;30:1900–3.
Ravelli A, Felici E, Magni-Manzoni S, et al. Patients with antinuclear antibody-positive juvenile idiopathic arthritis constitute a homogeneous subgroup irrespective of the course of joint disease. Arthritis Rheum. 2005;52:826–32.
Ravelli A, Varnier GC, Oliveira S, et al. Antinuclear antibody-positive patients should be grouped as a separate category in the classification of juvenile idiopathic arthritis. Arthritis Rheum. 2011;63:267–75.
Martini A. It is time to rethink juvenile idiopathic arthritis classification and nomenclature. Ann Rheum Dis. 2012;71:1437–9.
Martini A. Systemic juvenile idiopathic arthritis. Autoimmun Rev. 2012;12:56–9.
Ravelli A, Magni-Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005;146:598–604.
Davì S, Minoia F, Pistorio A, et al. Performance of current guidelines for diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2014;66:2871–80.
Davì S, Consolaro A, Guseinova D, et al. An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2011;38:764–8.
Minoia F, Davì S, Bovis F, et al. Development of new classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Pediatr Rheumatol. 2014;12(Suppl 1). doi:10.1002/art.39332.
Ravelli A, Minoia F, Davì S, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2016;68:566–76.
Ravelli A, Minoia F, Davì S, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Iniative. Ann Rheum Dis. 2016;75(3):481-9.
Ansell BM. Juvenile chronic arthritis. Scand J Rheumatol Suppl. 1987;66:47–50.
Al-Matar MJ, Petty RE, Tucker LB, Malleson PN, Schroeder ML, Cabral DA. The early pattern of joint involvement predicts disease progression in children with oligoarticular (pauciarticular) juvenile rheumatoid arthritis. Arthritis Rheum. 2002;46:2708–15.
Felici E, Novarini C, Magni-Manzoni S, et al. Course of joint disease in patients with antinuclear antibody-positive juvenile idiopathic arthritis. J Rheumatol. 2005;32:1805–10.
Tsitsami E, Bozzola E, Magni-Manzoni S, et al. Positive family history of psoriasis does not affect the clinical expression and course of juvenile idiopathic arthritis patients with oligoarthritis. Arthritis Rheum. 2003;49:488–93.
Ravelli A, Consolaro A, Schiappapietra B, Martini A. The conundrum of juvenile psoriatic arthritis. Clin Exp Rheumatol. 2015;33:S40–3.
Ravelli A, Viola S, Ruperto N, Corsi B, Ballardini G, Martini A. Correlation between conventional disease activity measures in juvenile chronic arthritis. Ann Rheum Dis. 1997;56:197–200.
Filocamo G, Davì S, Pistorio A, et al. Evaluation of 21-numbered circle and 10-centimeter horizontal line visual analog scales for physician and parent subjective ratings in juvenile idiopathic arthritis. J Rheumatol. 2010;37:1534–41.
Filocamo G, Consolaro A, Ferrari C, Ravelli A. Introducing new tools for assessment of parent- and child-reported outcomes in paediatric rheumatology practice: a work in progress. Clin Exp Rheumatol. 2013;31:964–8.
Brunner HI, Ravelli A. Developing outcome measures for paediatric rheumatic diseases. Best Pract Res Clin Rheumatol. 2009;23:609–24.
Brunner HI, Giannini EH. Health-related quality of life in children with rheumatic diseases. Curr Opin Rheumatol. 2003;15:602–12.
Luca NJC, Feldman BM. Health outcomes of pediatric rheumatic diseases. Best Pract Res Clin Rheumatol. 2014;28:331–50.
Feldman BM, Grundland B, McCullough L, Wright V. Distinction of quality of life, health related quality of life, and health status in children referred for rheumatologic care. J Rheumatol. 2000;27:226–33.
Pincus T, Yazici YSC. Quality control of a medical history: improving accuracy with patient participation, supported by a four-page version of the multidimensional health assessment questionnaire (MDHAQ). Rheum Dis Clin North Am. 2009;35:851–60.
Consolaro A, Giancane G, Schiappapietra B, et al. Clinical outcome measures in juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2016;14:23.
Consolaro A, Negro G, Chiara Gallo M, et al. Defining criteria for disease activity states in nonsystemic juvenile idiopathic arthritis based on a three-variable juvenile arthritis disease activity score. Arthritis Care Res (Hoboken). 2014;66:1703–9.
Ravelli A. The time has come to include assessment of radiographic progression in juvenile idiopathic arthritis clinical trials. J Rheumatol. 2008;35:553–7.
Reed MH, Wilmot DM. The radiology of juvenile rheumatoid arthritis. A review of the English language literature. J Rheumatol Suppl. 1991;31:2–22.
Van Rossum MA, Zwinderman AH, Boers M, et al. Radiologic features in juvenile idiopathic arthritis: a first step in the development of a standardized assessment method. Arthritis Rheum. 2003;48:507–15.
Magni-Manzoni S, Rossi F, Pistorio A, et al. Prognostic factors for radiographic progression, radiographic damage, and disability in juvenile idiopathic arthritis. Arthritis Rheum. 2003;48:3509–17.
Rossi F, Di Dia F, Galipò O, et al. Use of the sharp and larsen scoring methods in the assessment of radiographic progression in juvenile idiopathic arthritis. Arthritis Care Res. 2006;55:717–23.
Ravelli A, Ioseliani M, Norambuena X, et al. Adapted versions of the Sharp/van der Heijde score are reliable and valid for assessment of radiographic progression in juvenile idiopathic arthritis. Arthritis Rheum. 2007;56:3087–95.
Doria AS, de Castro CC, Kiss MHB, et al. Inter- and intrareader variability in the interpretation of two radiographic classification systems for juvenile rheumatoid arthritis. Pediatr Radiol. 2003;33:673–81.
Mason T, Reed AM, Nelson AM, Thomas KB. Radiographic progression in children with polyarticular juvenile rheumatoid arthritis: a pilot study. Ann Rheum Dis. 2005;64:491–3.
Van Rossum MAJ, Boers M, Zwinderman AH, et al. Development of a standardized method of assessment of radiographs and radiographic change in juvenile idiopathic arthritis: introduction of the Dijkstra composite score. Arthritis Rheum. 2005;52:2865–72.
Cannizzaro E, Schroeder S, Müller LM, Kellenberger CJ, Saurenmann R. Temporomandibular joint involvement in children with juvenile idiopathic arthritis. J Rheumatol. 2011;38:510–5.
Pedersen TK, Kuseler A, Gelineck J, Herlin T. A prospective study of magnetic resonance and radiographic imaging in relation to symptoms and clinical findings of the temporomandibular joint in children with juvenile idiopathic arthritis. J Rheumatol. 2008;35:1668–75.
Argyropoulou MI, Fanis SL, Xenakis T, Efremidis SC, Siamopoulou A. The role of MRI in the evaluation of hip joint disease in clinical subtypes of juvenile idiopathic arthritis. Br J Radiol. 2002;75:229–33.
Nistala K, Babar J, Johnson K, et al. Clinical assessment and core outcome variables are poor predictors of hip arthritis diagnosed by MRI in juvenile idiopathic arthritis. Rheumatology. 2007;46:699–702.
Malattia C, Damasio MB, Basso C, et al. Dynamic contrast-enhanced magnetic resonance imaging in the assessment of disease activity in patients with juvenile idiopathic arthritis. Rheumatology (Oxford). 2010;49:178–85.
McQueen FM. Bone marrow edema and osteitis in rheumatoid arthritis: the imaging perspective. Arthritis Res Ther. 2012;14:224.
Müller L, Avenarius D, Damasio B, et al. The paediatric wrist revisited: redefining MR findings in healthy children. Ann Rheum Dis. 2011;70:605–10.
Walther M, Harms H, Krenn V, Radke S, Kirschner S, Gohlke F. Synovial tissue of the hip at power Doppler US: correlation between vascularity and power Doppler US signal. Radiology. 2002;225:225–31.
Albrecht K, Müller-Ladner U, Strunk J. Quantification of the synovial perfusion in rheumatoid arthritis using Doppler ultrasonography. Clin Exp Rheumatol. 2006;25:630–8.
Brown AK, Quinn MA, Karim Z, et al. Presence of significant synovitis in rheumatoid arthritis patients with disease-modifying antirheumatic drug-induced clinical remission: evidence from an imaging study may explain structural progression. Arthritis Rheum. 2006;54:3761–73.
Spannow AH, Pfeiffer-Jensen M, Andersen NT, Herlin T, Stenbøg E. Ultrasonographic measurements of joint cartilage thickness in healthy children: age- and sex-related standard reference values. J Rheumatol. 2010;37:2595–601.
Grassi W, Filippucci E, Farina A, Salaffi F, Cervini C. Ultrasonography in the evaluation of bone erosions. Ann Rheum Dis. 2001;60:98–103.
Lanni S, Wood M, Ravelli A, et al. Towards a role of ultrasound in children with juvenile idiopathic arthritis. Rheumatology (Oxford). 2013;52:413–20.
Scott C, Meiorin S, Filocamo G, et al. A reappraisal of intra-articular corticosteroid therapy in juvenile idiopathic arthritis. Clin Exp Rheumatol. 2010;28:774–81.
Consolaro A, Varnier GC, Martini A, Ravelli A. Advances in biomarkers for paediatric rheumatic diseases. Nat Rev Rheumatol. 2014;11:1–11.
Hunter PJ, Nistala K, Jina N, et al. Biologic predictors of extension of oligoarticular juvenile idiopathic arthritis as determined from synovial fluid cellular composition and gene expression. Arthritis Rheum. 2010;62:896–907.
Gibson DS, Finnegan S, Jordan G, et al. Stratification and monitoring of juvenile idiopathic arthritis patients by synovial proteome analysis. J Proteom Res. 2009;8:5601–9.
Myles A, Aggarwal A. Expression of Toll-like receptors 2 and 4 is increased in peripheral blood and synovial fluid monocytes of patients with enthesitis-related arthritis subtype of juvenile idiopathic arthritis. Rheumatology (Oxford). 2011;50:481–8.
Viswanath V, Myles A, Dayal R, Aggarwal A. Levels of serum matrix metalloproteinase-3 correlate with disease activity in the enthesitis-related arthritis category of juvenile idiopathic arthritis. J Rheumatol. 2011;38:2482–7.
Aoki C, Inaba Y, Choe H, et al. Discrepancy between clinical and radiological responses to tocilizumab treatment in patients with systemic-onset juvenile idiopathic arthritis. J Rheumatol. 2014;41:1171–7.
Foell D, Roth J. Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheum. 2004;50:3762–71.
Holzinger D, Frosch M, Kastrup A, et al. The Toll-like receptor 4 agonist MRP8/14 protein complex is a sensitive indicator for disease activity and predicts relapses in systemic-onset juvenile idiopathic arthritis. Ann Rheum Dis. 2012;71:974–80.
De Jager W, Hoppenreijs EP, Wulffraat NM, Wedderburn LR, Kuis W, Prakken BJ. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross-sectional study. Ann Rheum Dis. 2007;66:589–98.
Lotito APN, Campa A, Silva CAA, Kiss MHB, Mello SBV. Interleukin 18 as a marker of disease activity and severity in patients with juvenile idiopathic arthritis. J Rheumatol. 2007;34:823–30.
Moncrieffe H, Ursu S, Holzinger D, et al. A subgroup of juvenile idiopathic arthritis patients who respond well to methotrexate are identified by the serum biomarker MRP8/14 protein. Rheumatology (Oxford). 2013;52:1467–76.
Foell D, Wulffraat N, Wedderburn LR, et al. Methotrexate withdrawal at 6 vs 12 months in juvenile idiopathic arthritis in remission: a randomized clinical trial. JAMA. 2010;303:1266–73.
Gerss J, Roth J, Holzinger D, et al. Phagocyte-specific S100 proteins and high-sensitivity C reactive protein as biomarkers for a risk-adapted treatment to maintain remission in juvenile idiopathic arthritis: a comparative study. Ann Rheum Dis. 2012;71:1991–7.
Vastert S, Prakken B. Update on research and clinical translation on specific clinical areas: from bench to bedside: how insight in immune pathogenesis can lead to precision medicine of severe juvenile idiopathic arthritis. Best Pract Res Clin Rheumatol. 2014;28:229–46.
Vastert SJ, de Jager W, Noordman BJ, et al. Effectiveness of first-line treatment with recombinant interleukin-1 receptor antagonist in steroid-naive patients with new-onset systemic juvenile idiopathic arthritis: results of a prospective cohort study. Arthritis Rheumatol (Hoboken). 2014;66:1034–43.
Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34:1133–8.
Bleesing J, Prada A, Siegel DM, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56:965–71.
Reddy VV, Myles A, Cheekatla SS, Singh S, Aggarwal A. Soluble CD25 in serum: a potential marker for subclinical macrophage activation syndrome in patients with active systemic onset juvenile idiopathic arthritis. Int J Rheum Dis. 2014;17:261–7.
Gorelik M, Fall N, Altaye M, et al. Follistatin-like protein 1 and the ferritin/erythrocyte sedimentation rate ratio are potential biomarkers for dysregulated gene expression and macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2013;40:1191–9.
Häfner R, Truckenbrodt H, Spamer M. Rehabilitation in children with juvenile chronic arthritis. Baillieres Clin Rheumatol. 1998;12:329–61.
Wallace CA. Current management of juvenile idiopathic arthritis. Best Pract Res Clin Rheumatol. 2006;20:279–300.
Toledo MMM, Martini G, Gigante C, Da Dalt L, Tregnaghi A, Zulian F. Is there a role for arthroscopic synovectomy in oligoarticular juvenile idiopathic arthritis? J Rheumatol. 2006;33:1868–72.
Beukelman T, Patkar NM, Saag KG, et al. 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: initiation and safety monitoring of therapeutic agents for the treatment of arthritis and systemic features. Arthritis Care Res (Hoboken). 2011;63:465–82.
Ruperto N, Nikishina I, Pachanov ED, et al. A randomized, double-blind clinical trial of two doses of meloxicam compared with naproxen in children with juvenile idiopathic arthritis: short- and long-term efficacy and safety results. Arthritis Rheum. 2005;52:563–72.
Reiff A, Lovell DJ, Van Adelsberg J, et al. Evaluation of the comparative efficacy and tolerability of rofecoxib and naproxen in children and adolescents with juvenile rheumatoid arthritis: a 12-week randomized controlled clinical trial with a 52-week open-label extension. J Rheumatol. 2006;33:985–95.
Cleary AG, Murphy HD, Davidson JE. Intra-articular corticosteroid injections in juvenile idiopathic arthritis. Arch Dis Child. 2003;88:192–6.
Lanni S, Bertamino M, Consolaro A, et al. Outcome and predicting factors of single and multiple intra-articular corticosteroid injections in children with juvenile idiopathic arthritis. Rheumatology. 2011;50:1627–34.
Papadopoulou C, Gonzalez MI, Nieto JC, et al. Delineating the role of multiple corticosteroid joint injections in the management of juvenile idiopathic arthritis in the biologic ERA. Arthritis Rheum. 2012;64:S503–S503.
Neidel J, Boehnke M, Küster RM. The efficacy and safety of intraarticular corticosteroid therapy for coxitis in juvenile rheumatoid arthritis. Arthritis Rheum. 2002;46:1620–8.
Tynjälä P, Honkanen V, Lahdenne P. Intra-articular steroids in radiologically confirmed tarsal and hip synovitis in juvenile idiopathic arthritis. Clin Exp Rheumatol. 2004;22:643–8.
Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767–78.
Hashkes PJ, Laxer RM. Medical treatment of juvenile idiopathic arthritis. JAMA. 2005;294:1671–84.
Ravelli A, Martini A. Methotrexate in juvenile idiopathic arthritis: answers and questions. J Rheumatol. 2000;27:1830–3.
Gutiérrez-Suárez R, Burgos-Vargas R. The use of methotrexate in children with rheumatic diseases. Clin Exp Rheumatol. 2010;28:S122–7.
Giannini EH, Brewer EJ, Kuzmina N, et al. Methotrexate in resistant juvenile rheumatoid arthritis. Results of the USA-USSR double-blind, placebo-controlled trial. The Pediatric Rheumatology Collaborative Study Group and The Cooperative Children’s Study Group. N Engl J Med. 1992;326:1043–9.
Ruperto N, Murray KJ, Gerloni V, et al. A randomized trial of parenteral methotrexate comparing an intermediate dose with a higher dose in children with juvenile idiopathic arthritis who failed to respond to standard doses of methotrexate. Arthritis Rheum. 2004;50:2191–201.
Klein A, Kaul I, Foeldvari I, Ganser G, Urban A, Horneff G. Efficacy and safety of oral and parenteral methotrexate therapy in children with juvenile idiopathic arthritis: an observational study with patients from the German Methotrexate Registry. Arthritis Care Res (Hoboken). 2012;64:1349–56.
Tuková J, Chládek J, Němcová D, Chládková J, Doležalová P. Methotrexate bioavailability after oral and subcutaneous administration in children with juvenile idiopathic arthritis. Clin Exp Rheumatol. 2009;27:1047–53.
Alsufyani K, Ortiz-Alvarez O, Cabral DA, Tucker LB, Petty RE, Malleson PN. The role of subcutaneous administration of methotrexate in children with juvenile idiopathic arthritis who have failed oral methotrexate. J Rheumatol. 2004;31:179–82.
Ravelli A, Viola S, Ramenghi B, Beluffi G, Zonta LA, Martini A. Radiologic progression in patients with juvenile chronic arthritis treated with methotrexate. J Pediatr. 1998;133:262–5.
Harel L, Wagner-Weiner L, Poznanski AK, Spencer CH, Ekwo E, Magilavy DB. Effects of methotrexate on radiologic progression in juvenile rheumatoid arthritis. Arthritis Rheum. 1993;36:1370–4.
Ortiz-Alvarez O, Morishita K, Avery G, et al. Guidelines for blood test monitoring of methotrexate toxicity in juvenile idiopathic arthritis. J Rheumatol. 2004;31:2501–6.
Ravelli A, Migliavacca D, Viola S, Ruperto N, Pistorio A, Martini A. Efficacy of folinic acid in reducing methotrexate toxicity in juvenile idiopathic arthritis. Clin Exp Rheumatol. 1999;17:625–7.
Silverman E, Mouy R, Spiegel L, et al. Leflunomide or methotrexate for juvenile rheumatoid arthritis. N Engl J Med. 2005;352:1655–66.
Lovell DJ, Giannini EH, Reiff A, et al. Etanercept in children with polyarticular juvenile rheumatoid arthritis. N Engl J Med. 2000;342:763–9.
Quartier P, Taupin P, Bourdeaut F, et al. Efficacy of etanercept for the treatment of juvenile idiopathic arthritis according to the onset type. Arthritis Rheum. 2003;48:1093–101.
Giannini EH, Ilowite NT, Lovell DJ, et al. Long-term safety and effectiveness of etanercept in children with selected categories of juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:2794–804.
Horneff G, Schmeling H, Biedermann T, et al. The German etanercept registry for treatment of juvenile idiopathic arthritis. Ann Rheum Dis. 2004;63:1638–44.
Prince FHM, Geerdink LM, Borsboom GJJM, et al. Major improvements in health-related quality of life during the use of etanercept in patients with previously refractory juvenile idiopathic arthritis. Ann Rheum Dis. 2010;69:138–42.
Giannini EH, Ilowite NT, Lovell DJ, et al. Effects of long-term etanercept treatment on growth in children with selected categories of juvenile idiopathic arthritis. Arthritis Rheum. 2010;62:3259–64.
Billiau AD, Loop M, Le PQ, et al. Etanercept improves linear growth and bone mass acquisition in MTX-resistant polyarticular-course juvenile idiopathic arthritis. Rheumatology. 2010;49:1550–8.
Nielsen S, Ruperto N, Gerloni V, et al. Preliminary evidence that etanercept may reduce radiographic progression in juvenile idiopathic arthritis. Clin Exp Rheumatol. 2008;26:688–92.
Otten MH, Prince FHM, Armbrust W, et al. Factors associated with treatment response to etanercept in juvenile idiopathic arthritis. JAMA. 2011;306:2340–7.
Solari N, Palmisani E, Consolaro A, et al. Factors associated with achievement of inactive disease in children with juvenile idiopathic arthritis treated with etanercept. J Rheumatol. 2013;40:192–200.
Ruperto N, Lovell DJ, Cuttica R, et al. A randomized, placebo-controlled trial of infliximab plus methotrexate for the treatment of polyarticular-course juvenile rheumatoid arthritis. Arthritis Rheum. 2007;56:3096–106.
Lovell DJ, Ruperto N, Goodman S, et al. Adalimumab with or without methotrexate in juvenile rheumatoid arthritis. N Engl J Med. 2008;359:810–20.
Schmeling H, Minden K, Foeldvari I, Ganser G, Hospach T, Horneff G. Efficacy and safety of adalimumab as the first and second biologic agent in juvenile idiopathic arthritis: the German Biologics JIA Registry. Arthritis Rheumatol. 2014;66:2580–9.
Brunner H, Ruperto N, Tzaribachev N, et al. A148: a multi-center, double-blind, randomized-withdrawal trial of subcutaneous golimumab in pediatric patients with active polyarticular course juvenile idiopathic arthritis despite methotrexate therapy: week 48 results. Arthritis Rheumatol. 2014;66:S191–2.
Tynjala P, Vahasalo P, Tarkiainen M, et al. Aggressive combination drug therapy in very early polyarticular juvenile idiopathic arthritis (ACUTE-JIA): a multicentre randomised open-label clinical trial. Ann Rheum Dis. 2011;70:1605–12.
Horneff G, De Bock F, Foeldvari I, et al. Safety and efficacy of combination of etanercept and methotrexate compared to treatment with etanercept only in patients with juvenile idiopathic arthritis (JIA): preliminary data from the German JIA Registry. Ann Rheum Dis. 2009;68:519–25.
Wallace CA, Giannini EH, Spalding SJ, et al. Trial of early aggressive therapy in polyarticular juvenile idiopathic arthritis. Arthritis Rheum. 2012;64:2012–21.
Hugle B, Burgos-Vargas R, Inman RD, et al. Long-term outcome of anti-tumor necrosis factor alpha blockade in the treatment of juvenile spondyloarthritis. Clin Exp Rheumatol. 2014;32:424–31.
Horneff G, Burgos-Vargas R, Constantin T, et al. Efficacy and safety of open-label etanercept on extended oligoarticular juvenile idiopathic arthritis, enthesitis-related arthritis and psoriatic arthritis: part 1 (week 12) of the CLIPPER study. Ann Rheum Dis. 2014;73:1114–22.
Kremer JM. Cytotoxic T-lymphocyte antigen 4-immunoglobulin in rheumatoid arthritis. Rheum Dis Clin North Am. 2004;30:381–91, viii.
Ruperto N, Lovell DJ, Quartier P, et al. Abatacept in children with juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled withdrawal trial. Lancet. 2008;372:383–91.
Ruperto N, Lovell DJ, Quartier P, et al. Long-term safety and efficacy of abatacept in children with juvenile idiopathic arthritis. Arthritis Rheum. 2010;62:1792–802.
Ruperto N, Lovell DJ, Li T, et al. Abatacept improves health-related quality of life, pain, sleep quality, and daily participation in subjects with juvenile idiopathic arthritis. Arthritis Care Res (Hoboken). 2010;62:1542–51.
Brunner HI, Ruperto N, Zuber Z, et al. Efficacy and safety of tocilizumab in patients with polyarticular-course juvenile idiopathic arthritis: results from a phase 3, randomised, double-blind withdrawal trial. Ann Rheum Dis. 2015;74:1110–7.
De Benedetti F, Massa M, Pignatti P, Albani S, Novick D, Martini A. Serum soluble interleukin 6 (IL-6) receptor and IL-6/soluble IL-6 receptor complex in systemic juvenile rheumatoid arthritis. J Clin Invest. 1994;93:2114–9.
De Benedetti F, Martini A. Is systemic juvenile rheumatoid arthritis an interleukin 6 mediated disease? J Rheumatol. 1998;25:203–7.
Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–86.
Verbsky JW, White AJ. Effective use of the recombinant interleukin 1 receptor antagonist anakinra in therapy resistant systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2004;31:2071–5.
Ruperto N, Quartier P, Wulffraat N, et al. A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 2012;64:557–67.
Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2396–406.
Ilowite NT, Prather K, Lokhnygina Y, et al. Randomized, double-blind, placebo-controlled trial of the efficacy and safety of rilonacept in the treatment of systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2014;66:2570–9.
Nigrovic PA, Mannion M, Prince FHM, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63:545–55.
Nigrovic PA. Review: is there a window of opportunity for treatment of systemic juvenile idiopathic arthritis? Arthritis Rheumatol (Hoboken). 2014;66:1405–13.
Yokota S, Miyamae T, Imagawa T, et al. Therapeutic efficacy of humanized recombinant anti-interleukin-6 receptor antibody in children with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2005;52:818–25.
Woo P, Wilkinson N, Prieur A-M, et al. Open label phase II trial of single, ascending doses of MRA in Caucasian children with severe systemic juvenile idiopathic arthritis: proof of principle of the efficacy of IL-6 receptor blockade in this type of arthritis and demonstration of prolonged cli. Arthritis Res Ther. 2005;7:R1281–8.
Yokota S, Imagawa T, Mori M, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;371:998–1006.
De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385–95.
Alexeeva EI, Valieva SI, Bzarova TM, et al. Efficacy and safety of repeat courses of rituximab treatment in patients with severe refractory juvenile idiopathic arthritis. Clin Rheumatol. 2011;30:1163–72.
Southwood TR, Foster HE, Davidson JE, et al. Duration of etanercept treatment and reasons for discontinuation in a cohort of juvenile idiopathic arthritis patients. Rheumatology. 2011;50:189–95.
Horneff G, Foeldvari I, Minden K, Moebius D, Hospach T. Report on malignancies in the German juvenile idiopathic arthritis registry. Rheumatology. 2011;50:230–6.
Prince FHM, Twilt M, ten Cate R, et al. Long-term follow-up on effectiveness and safety of etanercept in juvenile idiopathic arthritis: the Dutch national register. Ann Rheum Dis. 2009;68:635–41.
Dekker L, Armbrust W, Rademaker CM, Prakken B, Kuis W, Wulffraat NM. Safety of anti-TNFalpha therapy in children with juvenile idiopathic arthritis. Clin Exp Rheumatol. 2004;22:252–8.
Diak P, Siegel J, La Grenade L, Choi L, Lemery S, McMahon A. Tumor necrosis factor alpha blockers and malignancy in children: forty-eight cases reported to the Food and Drug Administration. Arthritis Rheum. 2010;62:2517–24.
Lehman TJ. Should the Food and Drug Administration warning of malignancy in children receiving tumor necrosis factor alpha blockers change the way we treat children with juvenile idiopathic arthritis? Arthritis Rheum. 2010;62:2183–4.
Beukelman T, Haynes K, Curtis JR, et al. Rates of malignancy associated with juvenile idiopathic arthritis and its treatment. Arthritis Rheum. 2012;64:1263–71.
Nordstrom BL, Mines D, Gu Y, Mercaldi C, Aquino P, Harrison MJ. Risk of malignancy in children with juvenile idiopathic arthritis not treated with biologic agents. Arthritis Care Res (Hoboken). 2012;64:1357–64.
Cron RQ, Beukelman T. Guilt by association—what is the true risk of malignancy in children treated with etanercept for JIA? Pediatr Rheumatol Online J. 2010;8:23.
Ruperto N, Martini A. Juvenile idiopathic arthritis and malignancy. Rheumatology (Oxford). 2014;53(6):968–74.
Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70:747–54.
Yokota S, Tanaka T, Kishimoto T. Efficacy, safety and tolerability of tocilizumab in patients with systemic juvenile idiopathic arthritis. Ther Adv Musculoskelet Dis. 2012;4:387–97.
Imagawa T, Yokota S, Mori M, et al. Safety and efficacy of tocilizumab, an anti-IL-6-receptor monoclonal antibody, in patients with polyarticular-course juvenile idiopathic arthritis. Mod Rheumatol. 2012;22:109–15.
Acknowledgments
Parts of this review are based on sections of the Handbook of Juvenile Idiopathic Arthritis, also authored by Prof. Angelo Ravelli. No funding or sponsorship was received for this study or publication of this article. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Gabriella Giancane, Alessandro Consolaro, Stefano Lanni, Sergio Davì, Benedetta Schiappapietra, and Angelo Ravelli have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on a review of previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/89E4F060559BF6A3.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Giancane, G., Consolaro, A., Lanni, S. et al. Juvenile Idiopathic Arthritis: Diagnosis and Treatment. Rheumatol Ther 3, 187–207 (2016). https://doi.org/10.1007/s40744-016-0040-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-016-0040-4