
Abstract
Background
Genetic testing of patients with inherited kidney diseases has emerged as a tool of clinical utility by improving the patients’ diagnosis, prognosis, surveillance and therapy.
Methods
The present study applied a Next Generation Sequencing (NGS)-based panel, named NephroPlex, testing 115 genes causing renal diseases, to 119 individuals, including 107 probands and 12 relatives. Thirty-five (poly)cystic and 72 non (poly)cystic individuals were enrolled. The latter subgroup of patients included Bardet-Biedl syndrome (BBS) patients, as major components.
Results
Disease-causing mutations were identified in 51.5 and 40% of polycystic and non-polycystic individuals, respectively. Autosomal dominant polycystic kidney disease (ADPKD) patients with truncating PKD1 variants showed a trend towards a greater slope of the age-estimated glomerular filtration rate (eGFR) regression line than patients with (i) missense variants, (ii) any PKD2 mutations and (iii) no detected mutations, according to previous findings. The analysis of BBS individuals showed a similar frequency of BBS4,9,10 and 12 mutations. Of note, all BBS4-mutated patients harbored the novel c.332+1G>GTT variant, which was absent in public databases, however, in our internal database, an additional heterozygote carrier was found. All BBS4-mutated individuals originated from the same geographical area encompassing the coastal provinces of Naples.
Discussion
In conclusion, these findings indicate the potential for a genetic panel to provide useful information at both clinical and epidemiological levels.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The development of “Next-Generation Sequencing” (NGS) has determined a revolution in clinical genetics, thus improving the possibility of increasing sequencing content while dramatically reducing costs, due to the simultaneous analysis of multiple genes through one single reaction [1,2,3]. Genetic kidney diseases (GKD) are a heterogeneous group of disorders, accounting for approximately 10% of adult chronic kidney disease (CKD) and up to 30% of pediatric CKD patients [4, 5]. It has recently been shown that genetic analysis of GKD patients has a significant clinical impact in terms of either diagnosis and management, reinforcing the rationale for analyzing these patients at the molecular level [4]. While whole exome sequencing (WES) and whole genome sequencing (WGS) are widely used as the first choice genetic analysis, focused genetic panels still retain unique advantages: they produce higher coverage and better separate genes from pseudogenes, an important clue in genetic kidney diseases, especially in the most common one, i.e., autosomal dominant polycystic kidney disease (ADPKD) [6]. In addition, handling large amounts of data produced by WGS and WES requires significant computing power and storage capacity. We developed a gene panel that includes 115 genes causing kidney disorders, including major genetic loci which account for ~ 85–90% of ADPKD and over 96% of Bardet-Biedl syndrome (BBS), namely PKD1-2 and BBS1-15, respectively [7, 8]. Our study illustrates the potential of using Nephroplex to test GKD patients, demonstrating its utility in the molecular diagnosis of classic, challenging and genetically heterogeneous conditions, such as ADPKD and BBS and showing major challenges in PKD1 analysis.
Methods
Patient recruitment and clinical characterization
One-hundred-nineteen subjects referred to the Units of Nephrology of the University of Campania L. Vanvitelli were studied by Nephroplex. This group of individuals included 107 probands and 12 unaffected relatives. Among the 107 probands, 7 were used as positive controls. All probands fulfilled specific diagnostic criteria. Patients were defined as having (poly)cystic kidney diseases (n = 35) and non-(poly)cystic kidney disease (n = 72) (Tables 1 and 2). The former group of individuals included patients with a clinical diagnosis of ADPKD based on the number of kidney cysts and family history [7]; sporadic cases were included when a clear clinical suspicion based on kidney ultrasound or abdominal CT scan was present. Patients with non-cystic disorders were classified as follows: patients with hypokalemic tubulopathies (N = 12), when documented metabolic alkalosis and hypokalemia were ascertained after excluding gastrointestinal and endocrine causes; patients with a clinical suspicion of Alport Syndrome (AS) (n = 11) were defined according to current guidelines [8]. Other tubulopathies were included, such as cystinuria(n = 1) and distal renal tubular acidosis (dRTA) (n = 3). The diagnosis of dRTA was based on a urine acidification test [9], while the Fanconi syndrome patient was defined by the presence of aminoaciduria, low-molecular weight proteinuria and metabolic acidosis due to urine bicarbonate loss that was detected after the loading test. Twenty-seven patients fulfilled the clinical criteria for the diagnosis of Bardet-Biedl syndrome, according to Beales criteria[10]. Further 8 hypercalciuric patients, 1 individual with diabetes insipidus and 5 patients with congenital anomalies of the kidney and urinary tract were included. One patient with familial drug-resistant hypertension. Patients with a likely immune pathogenesis, and other acquired kidney diseases (such as diabetic nephropathy) were excluded. Clinical and laboratory findings, information on familial segregation, and previous genetic testings were requested for each patient (Zacchia et al, DOI: sfaa182 in Clinical Kidney Journal, in press). All patients provided written informed consent, in accordance with standard procedures.
The glomerular filtration rate(GFR) was estimated (eGFR) using the CKD-EPI formula : eGFR = 141 × min(serum Creatinine/κ, 1)α × max(SCr /κ, 1)−1.209 × 0.993Age x1.018 [if female] × 1.159 [if Black], according to the literature and using standardized serum creatinine (SCr) [31].
All studies were conducted according to the international guidelines and to the tenets of the 2008 and 2013 Helsinki Declaration. In addition, the study was approved by the Ethics Committee of the University of Campania, L. Vanvitelli.
Gene panel construction and validation
A custom enrichment tool, named Nephroplex, covering all exons and at least ten flanking nucleotides of the 115 genes causing different inherited kidney diseases was built (Supplemental Table 1). Gene selection was conducted based on literature analysis showing the association between chosen genetic loci and human disease. As a strategy for targeting regions of interest, corresponding to 338.809 Kbp, the HaloPlex TM Target Enrichment System (Agilent) was used.
DNA extraction and NGS workflow
DNA samples were extracted from whole blood, using standard procedures. DNA quality and quantity were assessed using both spectrophotometric (Nanodrop ND 1000, Thermo Scientific Inc., Rockford, IL, USA) and fluorometry-based (Qubit 2.0 Fluorometer, Life Technologies, Carlsbad, CA, USA) methods, according to the manufacturer’s instructions (HaloPlex Target Enrichment System for Illumina Sequencing, Agilent Technologies, Santa Clara, CA, USA). For library preparation, 200 ng of genomic DNA was digested in restriction reactions for each individual. The fragments were hybridized to specific probes, as described elsewhere [32]. After the capture of target DNA, fragments were closed by a ligase, captured and amplified by PCR. The enriched target DNA in each library sample was validated and quantified by microfluidics analysis using the Bioanalyzer High Sensitivity DNA Assay kit (Agilent Technologies) and the 2100 Bioanalyzer with the 2100 Expert Software. All samples were analyzed in 4 different experimental sections, with a mean of 30 samples per run. Each group was loaded on a single lane of HIseq1000 Illumina system.
Targeted sequencing analysis
The libraries were sequenced using the HiSeq1000 system (Illumina, San Diego, CA, USA). The generated sequences were analyzed using eXSP, an in-house pipeline designed to automate the analysis workflow, composed of modules performing every step using the appropriate tools available to the scientific community or developed in-house [33]. Paired sequencing reads were aligned to the reference genome (UCSC, hg19 build) using BWA and sorted with SAM tools and Picard (http://picard.sourceforge.net). Post alignment processing (local realignment around insertions-deletions and base recalibration) and SNV and small insertions-deletions (ins-del) calling were performed using the Genome Analysis Toolkit (GATK) [34] with parameters adapted to the haloplex-generated sequences. The called SNV and ins-del variants produced with both platforms were annotated using ANNOVAR [35] with; the relative position in genes using RefSeq [36] gene model, amino acid change, presence in dbSNP v137 [37], frequency in NHLBI Exome Variant Server (http://evs.gs.washington.edu/EVS) and the 1000 genomes large scale projects, multiple cross-species conservation and prediction scores of damaging on protein activity [38]. The annotated variants were then imported into the internal variation database.
Validation of nephroplex
To design the Nephroplex-panel, a straightforward procedure was followed. Briefly, disease genes causing major inherited kidney disorders were selected. The target sequences were enriched by the HaloPlex system (see “Methods” Section). To validate NephroPlex, the analysis included DNA samples belonging to patients with known genetic mutations (n = 7, see Table 1), with 100% specificity. The average read depth of the target region was more than 98% at 20 × and more than 90% at 100 ×. Damaging variants were validated by Sanger sequencing. Primers for PCR were designed using PRIMER3PLUS free software (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) and synthesized by Eurofins Genomics. Sanger sequencing was performed using the BigDye Terminator v1.1 cycle sequencing kit and ABI3130xl, as suggested by the manufacturer (thermoFisher). For validation of PKD1 variants, Long PCR was performed to discriminate the PKD1 gene from the pseudogene overlapping region (exon1- exon34), as reported by Tan YC et al [39].
Variant interpretation
To provide clinically relevant data, a multidisciplinary board consisting of geneticists and nephrologists reviewed the analysis in the context of clinical data. To identify causal variants, the latter were first prioritized based on frequency in public databases (http://www.broadinstitute.org/) and in the internal database, using a minimum allele frequency (MAF)<1% as the cut-off . Then, among the rare variants, we selected exonic and splicing mutations. These variants were searched for in public databases, such as CLINVAR (https://www.ncbi.nlm.nih.gov/clinvar/), HGMD (http://www.hgmd.cf.ac.uk/ac/index.php), and, with regard to ADPKD patients, also in the MAYO CLINIC Database. All variants were classified into the five categories defined by ACMG standards: pathogenic (P), likely pathogenic (LP), uncertain significance (US), likely benign (LB) and benign (B). P variants were defined as such when reported in the literature as deleterious or when they resulted in protein truncation. LP variants were defined in the same way as those previously established as LP in the literature; LB and B variants were those defined as such by other articles or that were predicted not to be damaging by in silico programs, such as SIFT and Polyphen.
Statistics
To compare the effect of genetic mutations (truncating PKD1 vs non-truncating) on the eGFR decrease as a function of age we used covariance analysis with eGFR as the dependent variable, age as covariate and type of mutation (truncating vs. non truncating) as factor. Statistical significance was accepted for p<0.05.
Results
Patient cohort
One-hundred-nineteen subjects were enrolled in the study, including 107 probands and 12 relatives. Probands underwent genetic analysis to address the molecular basis of the following clinical pictures: inherited polycystic diseases (N = 35) (Table 1); among non-cystic patients, the following categories were included in the study; hypokalemic metabolic alkalosis tubulopathies (n = 12), Fanconi syndrome (n = 1), cystinuria (n = 1), renal glycosuria (n = 3), distal renal tubular acidosis /dRTA (n = 2), hypercalciuria (n = 8), diabetes insipidus (n = 1), Alport Syndrome/ AS(n = 11), congenital anomalies of kidney and urinary tract (CAKUT)(N = 5), Bardet-Biedl Syndrome/ BBS (n = 27), and resistant hypertension (n = 1) (Table 2). Most patients were analyzed as single/sporadic cases (97 out of 107 patients), and ten as familial.
Molecular analysis of polycystic patients
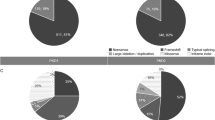
Thirty-five individuals underwent genetic analysis due to the clinical suspicion of inherited kidney cystic diseases. A causative mutation was found in over 51% of patients studied; the remaining patients showed either variants of uncertain significance (VUS) or no putative genetic mutations (Fig. 1a). Thirty-six variants in PKD1 and PKD2 were found in 25 individuals; twenty-two variants were novel, the remaining were already described in the literature. PKD1 variants occurred with higher frequency than PKD2 (83.4% vs 16.6%, respectively). We detected damaging PKD1-2 variants in 15 individuals; 12 in PKD1 and 3 in PKD2, respectively (Figs. 1b and C). Twelve pathogenic variants were truncating variants, while the remaining were missense variants. Seven out of 12 damaging PKD1 mutations were located in duplicated regions. Finally, seven patients did not show rare variants in the genes of interest. Interestingly, analysis of covariance of eGFR using age as covariate and type of mutation (truncating PKD1 variants vs. all others, including no detected variants) as factor revealed significant effects for age (F = 5.87, p = 0.027) and borderline age x mutation type interaction effect (F = 3.1, p = 0.09). This was due to the greater slope of the age-eGFR regression line in the group with PKD1- truncating patients, as indicated by previous studies [40, 41]. Indeed, regression analysis in this group showed that each year of age led to a mean loss of eGFR of 2.54 ml/min/1.73m2, whereas in the non-truncating group the loss of eGFR was of 0.73ml/min/1.73m2 (Supplemental Fig. 1). Moreover, we found a frameshift hemizygote OFD1 mutation in a young female patient, one patient with compound heterozygote PKHD1 variants, and one patient with a frameshift MUC1 variant (Fig. 1b and c).

Genetic analysis of cystic patients. 1a Genetic diagnosis was obtained for 51.5% of patients, the remaining showed either variants of uncertain significance (VUS) or no causative variants. 1b. Genetic analysis confirmed the diagnosis of autosomal dominant polycystic kidney dissease (ADPKD) in 15 individuals, Oro-facio-digital type 1 Syndrome (OFD-1, n = 1), autosomal dominant tubulointerstitial kidney disease (ADTKD, n = 1) and autosomal recessive polycystic kidney disease (ARPKD, n = 1). 1c. Among the pathogenic variants, our results showed that the main mutations occurred in PKD1, followed by PKD2. OFD1, MUC1 and PKHD1 mutations were less frequent
Molecular analysis of non (poly)cystic patients
BBS individuals
Two of the 27 BBS individuals were studied as trios (K73 and K128). Nine patients showed homozygote variants and five patients had compound heterozygote variants in known BBS genes. Six patients showed only heterozygote BBS variants, while 7 patients did not show any alteration in the genes of interest (Table 2). Major variants were predicted as likely pathogenic or pathogenic (see Table 2). The most common mutations were detected in BBS10, BBS12, BBS4, and BBS9 genes.
Alport Syndrome patients
Eleven patients (3 males and 8 females) with a clinical suspicion of AS were analyzed. Six indexes showed pathogenic variants: all mutations were in the COL4A5 gene. Among these patients, four related individuals showed the known COL4A5c.520G>C pathogenic variant, while two sisters showed the novel COL4A5 c.3032deIC variant, resulting in a frameshift mutation (Table 2).
Tubulopathies and CAKUT patients
Patients with hypokalemic metabolic alkalosis of renal origin made up the most substantial subgroup of patients with a clinical suspicion of tubulopathies, accounting for 12 individuals. Six individuals showed either pathogenic or likely pathogenic homozygote or composite heterozygote variants in SLC12A3 (n = 5) or CLCKNB (n = 1). One patient showed only a heterozygote SLC12A3 variant and the remaining 5 patients showed no mutations in genes of interest. Two out of three patients with renal glycosuria showed variants of uncertain significance in SLC5A2; hypercalciuric patients, as well as patients with CAKUT were all unsolved. Major genetic findings of non-cystic individuals are summarized in Fig. 2 and table 2.

Genetic analysis of non-cystic patients. a Forty percent of patients were solved. b Classes of disorders and relative number of solved and unsolved individuals. c Major pathogenic variants detected in this category of individuals. BBS Bardet-Biedl syndrome; AS Alport syndrome; Hypok hypokalemic; RTA renal tubular acidosis; Fanconi S Fanconi syndrome; DI diabetes insipidus
Frequency of BBS4c.332 + 1G > GTT in the patients’ cohort
We found the c.332+1G>GTT variant in the BBS4 gene in five unrelated BBS individuals. The variant was homozygote in three BBS patients, while two patients were heterozygote. The predicted effect of genetic mutation of protein function is depicted in Fig. 3. One of the two heterozygote patients showed a second BBS4 variant, described in the literature as pathogenic. The other patient did not show additional variants in BBS4, thus was unsolved. Given the high frequency of this variant, we searched for the variant in our internal database, accounting for 4,000 individuals: besides the cases reported above, it was detected in an additional subject. The latter underwent genetic analysis for the suspicion of AS (K121). The BBS4 c.332+1G>GTT variant was heterozygote and, was consistent with the autosomal recessive inheritance of BBS. This patient did not show clinical signs of the disorder and was considered an unaffected carrier. Interestingly, individuals harboring the variant showed restricted geographic origin.

Schematic representation of the BBS4 c. 332 + 1G > GTT variant. The figure shows the possible effects of the genetic variants, according to in silico program: (1) retention of the enthrone; (2) activation of a cryptic site of splicing, with the resulting protein encountering a premature stop codon
PKD1 variants in non-cystic individuals
During the analysis of PKD1 variants in non-cystic individuals, a high prevalence of PKD1 variants was observed. We detected a total of 28 rare PKD1-2 variants in 21 out of 75 adult individuals with non-cystic phenotype (28%). Five variants were detected in PKD2, while the remaining variants were found in PKD1 (18% vs 82%, respectively). Supplemental Table 2 shows the position of the variants and whether they have been previously reported in major public databases, such as Clinvar and/or the Mayo Clinic database. All detected variants were predicted by in silico program as benign or likely benign variants, with two exceptions. Patients K7 and K17 underwent genetic analysis due to the clinical suspicion of hypercalciuria and Gitelman syndrome, respectively. K7 was unsolved, while K17 showed a homozygote SLC12A3 variant, explaining the phenotype. Moreover, our analysis revealed that both individuals carried a PKD1 variant: a frameshift PKD1 mutation (K7), predicted as pathogenic, and a missense PKD1 variant predicted as likely pathogenic (K17), respectively. Both mutations were located in duplicated PKD1 regions, as well as in 77.3% of detected PKD1 variants in this subgroup of individuals. To further analyze whether our findings might have been the result of contamination by pseudogenes, we performed ClustalW alignment of PKD1 with all pseudogene sequences to localize the position of the ‘incidental variants’. Our analysis revealed that all the variants detected in exon 10-33 were located in overlapped regions with almost one pseudogene (see Supplemental Table 2 and additional supplemental material). These findings suggest possible contamination.
Discussion
In the present study, we set-up and validated a gene panel, named Nephroplex, that includes 115 genes causing inherited kidney disorders, with the aim to define the genetic landscape of a cohort of individuals with kidney cystic and non-cystic phenotype. Recently, WES and WGS have entered into clinical use in several fields. However, there has been a great deal of speculation concerning the perceived advantages and limitations of these studies as compared to focused panels. Costs, time to results, coverage and scalability are major considerations. Given the reduction of costs of NGS, the latter is not a crucial discriminator in choosing sequencing strategies. Moreover, focused gene panels still retain some advantages when used for diagnostic purposes. WGS produces massive amounts of data, requiring intense computational analysis and adequate instrumentation that few clinical laboratories have embraced. The generation of so much sequence data per patient causes low coverage compared with targeted panels, even though this limitation has been overcome in recent studies [42]. Thus, while gene panels and WGS provide similar diagnostic yield, a more laborious analysis is required to handle WGS data. Clearly, WGS offers the advantage of re-analysis paralleling the advances in knowledge and the possibility to discover novel disease, risk and modifier genes, when probands are studied as trios and when data are validated properly.
In our study, the group of polycystic individuals consisted mainly of ADPKD patients. The genetic panel included the two most common genes causing ADPKD, namely PKD1 and PKD2 [43, 44]. The study is in line with data from the literature suggesting the superiority of NGS compared with Sanger in analyzing the PKD1, which is a large gene consisting of 46 exons[45, 46]. Molecular screening is unusually difficult, as exons 1–33 have six copies of this region presenting as pseudogenes (PKD1P1-P6), located ~ 13–16Mb proximal to PKD1, on the short arm of chromosome 16 [47]. These pseudogenes have early stop codons, so they do not generate large protein products and are 98–99% identical to PKD1 in homologous regions. This complexity makes molecular diagnosis challenging. Comprehensive screening of well-characterized ADPKD patients has revealed definite (truncating) mutations in up to 61% of affected families, and in-frame changes in ~ 26%, all of which were scored as pathogenic [48, 49]. Screening for larger rearrangements using multiplex ligation-dependent probe amplification detected mutations in a further ~ 4% of families [50]. Non-definite mutations were found in 26% of patients, and ~ 9% of individuals showed no mutations in either PKD1 or PKD2 [48]. There are several explanations for this: missed mutations in PKD1 gene due to technical limitations; PKD1 pseudogenes; intronic mutations; gene promoter changes; mosaicism; other genes, as recently suggested [51]. In our study, we found a higher prevalence of PKD1 than PKD2 mutations in ADPKD patients, just as reported in the literature. Interestingly, in the study we encountered the greatest difficulties during ADPKD molecular diagnosis : (1) the high incidence of private mutations; (2) the large prevalence of missense variants. As largely addressed by experts, the classification of missense variants remains cumbersome given the technical difficulties of performing functional studies. In this scenario, the high allelic heterogeneity of PKD1 and PKD2 in non-cystic individuals further complicates molecular diagnosis, as we showed in our cohort. Most variants found in subjects with no clinical ADPKD phenotype were missense variants. Only two patients showed a pathogenic and a likely pathogenic variant, respectively, according to prediction tools. However, the majority of PKD1 variants in the cohort of non-cystic individuals were located in duplicated regions, including the ones defined as pathogenic: our alignment studies suggest that they may be the result of contamination (PKD1 gene vs pseudogenes?).
BBS was the second most represented disease in our cohort. The analysis revealed a diagnostic rate of 44%. Interestingly, the study showed a surprisingly high prevalence of BBS4 variants. BBS1, 2, and 10 are known to constitute nearly 50% of diagnoses [52, 53]. One possible explanation is that patients were selected from a cohort consisting of over 60 well-characterized BBS individuals, with most of them possessing a genetic diagnosis at basal. Thus, several BBS1-mutated patients were excluded from the study. The high prevalence of BBS4 mutations in our study was peculiar and attracted our attention. All patients harbored the same BBS4c.332+1G>GTT variant. The latter was homozygote in three unrelated BBS individuals. A fourth patient showed two BBS4 mutations. An additional BBS subject with no complete molecular diagnosis showed the heterozygote BBS4c.332+1G>GTT variant. The identified mutation is predicted to determine defective splicing. Interestingly, all individuals were from the same region of Southern Italy. Three of 5 BBS4-patients were from an area south of Naples, between Torre del Greco and Castellammare di Stabia. The remaining two BBS individuals were from Naples city. A review of both the public and of our own internal database showed no evidence of the variant, except for one additional individual who was identified in the internal database. The patient was a woman born in Naples, undergoing genetic analysis for the clinical suspicion of AS(K121). She showed the heterozygote BBS4c.332+1G>GTT variant, in the absence of any signs of BBS, as expected. Considering the rarity of the disease, with a prevalence of 1:160,000 individuals, the detected BBS4 variant shows a striking prevalence in Naples, indicating a possible founder mutation. These observations provide the rationale for a cost- and time-efficient screening of this limited geographic area to determine allele frequency distribution and to estimate the risk of BBS occurrence.
Additional non cystic patients in the study included patients suffering from tubulopathies and CAKUT. Fifty percent of patients with hypokalemic metabolic alkalosis were solved as Gitelman Syndrome or Bartter syndrome type 3. Conversely, patients with hypercalciuria and CAKUT were all unsolved. The scarce knowledge of the genetic landscape of these disorders and the contribution of acquired factors to their pathogenesis account at least in part for these results [54, 55].
The present study demonstrates the potential of a kidney focused gene-panel in the diagnosis of renal inherited disorders. In the era of WGS and WES, the potential of focused genetic panels is still of clinical utility and scientific interest, providing advantages when studying inherited kidney disorders in terms of both diagnostic purpose and identification of allele frequency in a restricted geographic area, a pre-requisite to address the risk of occurrence of genetic disorders.
References
Bamshad MJ, Ng SB, Bigham AW et al (2011) Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12:745–755. https://doi.org/10.1038/nrg3031
Ku C-S, Naidoo N, Pawitan Y (2011) Revisiting mendelian disorders through exome sequencing. Hum Genet 129:351–370. https://doi.org/10.1007/s00439-011-0964-2
Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE (2013) Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet 14:681–691. https://doi.org/10.1038/nrg3555
Jayasinghe K, Stark Z, Kerr PG et al (2020) Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genet Med. https://doi.org/10.1038/s41436-020-00963-4
Hildebrandt F (2010) Genetic kidney diseases. Lancet 375:1287–1295. https://doi.org/10.1016/S0140-6736(10)60236-X
Alvarez WJ, Lee I-H, Hernandez-Ferrer C et al (2018) WEScover : selection of whole exome sequencing vs. gene panel testing. Bioinformatics 71:650
Pei Y, Obaji J, Dupuis A et al (2009) Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol 20:205–212. https://doi.org/10.1681/ASN.2008050507
Savige J, Ariani F, Mari F et al (2018) Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr Nephrol 34:1175–1189. https://doi.org/10.1007/s00467-018-3985-4
Walsh SB, Shirley DG, Wrong OM et al (2007) Urinary acidificazion assessed by simultaneous furosemide and fludrocortisone treatment: an alternatve to ammonium chloride. Kidney Int 71:1310–1316. https://doi.org/10.1038/sj.ki.5002220
Beales PL, Elcioglu N, Woolf AS et al (1999) New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet 36:437–446
Audrezet MP, Cornec-Le Gall E, Chen JM et al (2012) Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat 33:1239–1250. https://doi.org/10.1002/humu.22103
National Center for Biotechnology Information. ClinVar; [VCV000374187.3], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000374187.3. Accessed 11 Sept 2014
Bataille S, Berland Y, Fontes M et al (2011) High Resolution Melt analysis for mutation screening in PKD1 and PKD2. BMC Nephrol 12:57. https://doi.org/10.1186/1471-2369-12-57
National Center for Biotechnology Information. ClinVar; [VCV000618796.1], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000618796.1. Accessed 10 Apr 2011
National Center for Biotechnology Information. ClinVar; [VCV000440071.4], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000440071.4. Accessed Sept 2010
Rossetti S, Chauveau D, Walker D et al (2002) A complete mutation screen of the ADPKD genes by DHPLC. Kidney Int 61:1588–1599. https://doi.org/10.1046/j.1523-1755.2002.00326.x
Burtey S, Lossi AM, Bayle J et al (2002) Mutation screening of the PKD1 transcript by RT-PCR. J Med Gen 39:422–429. https://doi.org/10.1136/jmg.39.6.422
Rossetti S, Strmecki L, Gamble V et al (2001) Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am J Hum Genet 68:46–63. https://doi.org/10.1086/316939
National Center for Biotechnology Information. ClinVar; [VCV000041117.4], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000041117.4. Accessed July 2012
Dulfer E, Hoefsloot LH, Timmer A et al (2010) Two sibs with Bardet-Biedl syndrome due to mutations in BBS12: No clues for modulation by a third mutation in BBS10. Am J Med Genet. https://doi.org/10.1002/ajmg.a.33650
National Center for Biotechnology Information. ClinVar; [VCV000100595.4]. https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000100595.4. Accessed Sept 2016
Katsanis N, Eichers ER, Ansley SJ et al (2002) BBS4 is a minor contributor to Bardet-Biedl syndrome and may also participate in Triallelic inheritance. Am J Hum Genet 71:22–29. https://doi.org/10.1086/341031
National Center for Biotechnology Information. ClinVar; [VCV000265988.1], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000265988.1. Accessed Feb 2018
Billingsley G, Bin J, Fieggen KJ, Duncan JL et al (2010) Mutations in chaperonin-like BBS genes are a major contributor to disease development in a multiethnic Bardet-Biedl syndrome patient population. J Med Gen 47:453–463. https://doi.org/10.1136/jmg.2009.073205
Barker DF, Denison JC, Atkin C et al (2001) Efficient detection of Alport syndrome COL4A5 mutations with multiplex genomic PCR-SSCP. Am J Med Genet 98:148–160
Gorski MM, Lecchi A, Fermia EA et al (2019) Complications of whole-exome sequencing for causal gene discovery in primary platelet secretion defects. Plat Biol Dis. https://doi.org/10.3324/haematol.2018.204990
Syren ML, Tedeschi S, Cesareo L et al (2002) Identification of fifteen novel mutations in the SLC12A3 gene encoding the Na-Cl Co-transporter in Italian patients with Gitelman syndrom. Human Mut 20:78. https://doi.org/10.1002/humu.9045
Riveira-Munoz E, Chang Q, Godefroid N et al (2007) Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome. J Am Soc Nephrol 18:1271–1283. https://doi.org/10.1681/ASN.2006101095
Seys E, Andrini O, Keck M et al (2017) Clinical and genetic spectrum of Bartter syndrome type 3. J Am Soc Nephrol 28:2540–2552. https://doi.org/10.1681/ASN.2016101057
Rhodes H, Yarram-Smith L, Rice SJ et al (2015) Clinical and genetic analysis of patients with cystinuria in the United Kingdom. Clin J Am Soc Nephrol 10:1235–1245. https://doi.org/10.2215/CJN.10981114
Levey AS, Stevens LA, Schmid CH et al (2009) A new equation to estimate glomerular filtration rate. Ann Intern Med 150:604–612
Savarese M, Di Fruscio G, Mutarelli M et al (2014) MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathol Commun 2:100. https://doi.org/10.1186/s40478-014-0100-3
Mutarelli M, Marwah V, Rispoli R et al (2014) A community-based resource for automatic exome variant-calling and annotation in Mendelian disorders. BMC Genom 15:S5. https://doi.org/10.1186/1471-2164-15-S3-S5
DePristo MA, Banks E, Poplin R et al (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. https://doi.org/10.1038/ng.806
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164–e164. https://doi.org/10.1093/nar/gkq603
Pruitt KD, Tatusova T, Klimke W, Maglott DR (2009) NCBI reference sequences: current status, policy and new initiatives. Nucleic Acids Res 37:D32–D36. https://doi.org/10.1093/nar/gkn721
Sherry ST (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29:308–311. https://doi.org/10.1093/nar/29.1.308
The 1000 Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073. https://doi.org/10.1038/nature09534
Tan YC, Michaeel A, Blumenfeld J et al (2012) D A novel long-range PCR sequencing method for genetic analysis of the entire PKD1 gene. J Mol Diagn 14:305–313. https://doi.org/10.1016/j.jmoldx.2012.02.007
Cornec-Le Gall E, Audrezet MP, Chen JM et al (2013) Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol 24:1006–1013. https://doi.org/10.1681/ASN2012070650
Heyer CM, Sundsbak JM, Abebe KZ et al (2016) Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27:2872–2884. https://doi.org/10.1681/ASN.2015050583
Cirino AL, Lakdawala NK, McDonough B et al (2017) A comparison of whole genome sequencing to multi-gene panel testing in hypertrophic cardiomyopathy patients. Circ Cardiovasc Genet 10:e001768. https://doi.org/10.1161/CIRCGENETICS.117.001768
Viggiano D, Zacchia M, Simonelli F et al (2018) The renal lesions in Bardet-Biedl syndrome: history before and after the discovery of BBS genes. G Ital Nefrol Organo Uff Della Soc Ital Nefrol 35:95–100
Schönauer R, Baatz S, Nemitz-Kliemchen M et al (2020) Matching clinical and genetic diagnoses in autosomal dominant polycystic kidney disease reveals novel phenocopies and potential candidate genes. Genet Med. https://doi.org/10.1038/s41436-020-0816-3
Hughes J, Ward CJ, Peral B et al (1995) The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 10:151–160. https://doi.org/10.1038/ng0695-151
Zacchia M, Marchese E, Trani EM et al (2019) Proteomics and metabolomics studies exploring the pathophysiology of renal dysfunction in autosomal dominant polycystic kidney disease and other ciliopathies. Nephrol Dial Transpl. https://doi.org/10.1093/ndt/gfz121
Martin J, Han C, Gordon LA et al (2004) The sequence and analysis of duplication-rich human chromosome 16. Nature 432:988–994. https://doi.org/10.1038/nature03187
Tan Y-C, Blumenfeld JD, Anghel R et al (2009) Novel method for genomic analysis of PKD1 and PKD2 mutations in autosomal dominant polycystic kidney disease. Hum Mutat 30:264–273. https://doi.org/10.1002/humu.20842
Garcia-Gonzalez MA, Jones JG, Allen SK et al (2007) Evaluating the clinical utility of a molecular genetic test for polycystic kidney disease. Mol Genet Metab 92:160–167. https://doi.org/10.1016/j.ymgme.2007.05.004
Consugar MB, Wong WC, Lundquist PA et al (2008) Characterization of large rearrangements in autosomal dominant polycystic kidney disease and the PKD1/TSC2 contiguous gene syndrome. Kidney Int 74:1468–1479. https://doi.org/10.1038/ki.2008.485
Harris PC, Rossetti S (2010) Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol 6:197–206. https://doi.org/10.1038/nrneph.2010.18
Esposito G, Testa F, Zacchia M et al (2017) Genetic characterization of Italian patients with Bardet-Biedl syndrome and correlation to ocular, renal and audio-vestibular phenotype: identification of eleven novel pathogenic sequence variants. BMC Med Genet 18:10. https://doi.org/10.1186/s12881-017-0372-0
Zacchia M, Zacchia E, Zona E et al (2016) Renal phenotype in Bardet-Biedl syndrome: a combined defect of urinary concentration and dilution is associated with defective urinary AQP2 and UMOD excretion. Am J Physiol-Ren Physiol 311:F686–F694. https://doi.org/10.1152/ajprenal.00224.2016
Capone V, Morello W, Taroni F, Montini G (2017) Genetics of congenital anomalies of the kidney and urinary tract: the current state of play. Int J Mol Sci 18:796. https://doi.org/10.3390/ijms18040796
Zacchia M, Capasso G (2007) Parvalbumin: a key protein in early distal tubule NaCl reabsorption. Nephrol Dial Transplant 23:1109–1111. https://doi.org/10.1093/ndt/gfm886
Acknowledgements
This work is generated within the European Reference Network for Rare Kidney Diseases (ERKNet).
Funding
Open access funding provided by Università degli Studi della Campania Luigi Vanvitelli within the CRUI-CARE Agreement. The manuscript was partially funded by a grant from Università degli Studi della Campania, L. Vanvitelli, Progetto VALERE to MZ. VN received funding from Horizon 2020 under grant agreement No. 779257 (Solve‐RD).
Author information
Authors and Affiliations
Contributions
Study concepts and design: MZ, FDVB, FT, GC and VN; data acquisition: FDVB, AT, GB, VN; data analysis/ interpretation: MZ, FDVB, FT, GC, AM, RP, VDI, DV and FS; manuscript preparation: MZ, FDVB, GDB, AT and AM; manuscript editing: G.P, VN, AP, GC. Manuscript revision and final version approval: all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Ethical approval
All studies were conducted according to the international guidelines and to the tenets of the 2008 and 2013 Helsinki Declaration. In addition, the study was approved by the Ethics Committee of the University of Campania, L. Vanvitelli.
Informed consent
Informed consent was obtained from all participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
40620_2021_1048_MOESM1_ESM.pdf
Supplementary file1 PKD1 alignment with PKD1P1- P6. The position of detected variants in non-cystic individuals is marked in yellow. (PDF 216 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zacchia, M., Blanco, F.D.V., Trepiccione, F. et al. Nephroplex: a kidney-focused NGS panel highlights the challenges of PKD1 sequencing and identifies a founder BBS4 mutation. J Nephrol 34, 1855–1874 (2021). https://doi.org/10.1007/s40620-021-01048-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-021-01048-4