
Abstract
Atypical hemolytic uremic syndrome (aHUS) is a rare, genetic, progressive, life-threatening form of thrombotic microangiopathy (TMA) predominantly caused by dysregulation of the alternative pathway of the complement system. Complement-amplifying conditions (CACs), including pregnancy complications [preeclampsia, HELLP (hemolysis, elevated liver enzymes, low platelet count) syndrome], malignant hypertension, autoimmune diseases, transplantation, and others, are associated with the onset of TMA in up to 69 % of cases of aHUS. CACs activate the alternative pathway of complement and may be comorbid with aHUS or may unmask a previously undiagnosed case. In this review, three case reports are presented illustrating the onset and diagnosis of aHUS in the setting of different CACs (pregnancy complications, malignant hypertension, renal transplantation). The report also reviews the evidence for a variety of CACs, including those mentioned above as well as infections and drug-induced TMA, and the overlap with aHUS. Finally, we introduce an algorithm for diagnosis and treatment of aHUS in the setting of CACs. If TMA persists despite initial management for the specific CAC, aHUS should be considered. The terminal complement inhibitor eculizumab should be initiated for all patients with confirmed diagnosis of aHUS, with or without a comorbid CAC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thrombotic microangiopathy (TMA) is a life-threatening syndrome of systemic microvascular occlusions and is characterized by sudden or gradual onset of thrombocytopenia, microangiopathic hemolytic anemia, and renal or other end-organ damage [1, 2]. TMA has been associated with diverse diseases and syndromes, such as systemic infections, cancer, pregnancy complications [e.g. preeclampsia, eclampsia, HELLP (hemolysis, elevated liver enzymes, low platelet count) syndrome], autoimmune disorders [e.g. systemic lupus erythematosus (SLE), systemic sclerosis, antiphospholipid syndrome], hematopoietic stem-cell or organ transplantation, and severe hypertension [1].
The etiologies of TMA also include atypical hemolytic uremic syndrome (aHUS) [1], a rare, progressive, life-threatening form predominantly caused by dysregulation of the complement alternative pathway [3]. aHUS can manifest at any age. While approximately 80 % of patients present with thrombocytopenia, microangiopathic hemolytic anemia, and renal impairment [4], onset may be more gradual in other patients [5]. Because aHUS can affect multiple vascular beds [6], extrarenal manifestations occur in up to 48 % of patients, with frequent neurologic and cardiovascular involvement [7–10].
Patients with aHUS who are untreated remain at lifelong risk of renal impairment, end-stage renal disease, extrarenal complications, and premature death [4, 9]. Management with plasma exchange/plasma infusion (PE/PI) may improve hematologic parameters temporarily [11, 12] but not long-term outcomes [4]. The efficacy and safety of eculizumab (Soliris®, Alexion Pharmaceuticals, Inc., Cheshire, CT, USA), a terminal complement inhibitor and the only approved treatment for aHUS [13, 14], were first established in two prospective, multicenter clinical studies [15, 16], followed by prospective, multicenter studies in pediatric [17] and adult [18] populations. Eculizumab therapy was demonstrated to inhibit complement-mediated TMA and improve hematologic parameters, renal function, and quality of life [15, 17, 18].
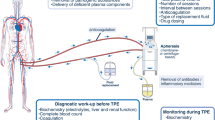
According to the “multiple-hit” hypothesis [19], aHUS is a consequence of both genetic predisposition to alternative complement dysregulation as well as the occurrence of events or conditions that may precipitate TMA by activating complement and/or damaging the endothelium [19, 20]. Complement-amplifying conditions (CACs), such as pregnancy complications (preeclampsia, HELLP), autoimmune diseases and others, may be comorbid with aHUS, unmask a previously undiagnosed case, or lead to a misdiagnosis [3, 21–23]. Malignant hypertension (MHT) is another CAC that may precipitate aHUS or occur secondary to aHUS [21], potentially confounding the diagnosis. In this review, we describe case reports that demonstrate the onset of aHUS in the setting of CACs. We also review the evidence for a number of CACs, including pregnancy complications, MHT, autoimmune diseases, transplantation, infections, and drugs, and the overlap of these disorders with aHUS. Finally, we present an algorithm for diagnosis and treatment of aHUS in the setting of CACs (Fig. 1) [5].

Management algorithm for patients with CACs and TMA. ADAMTS13 a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13, aHUS atypical hemolytic uremic syndrome, CAC complement-amplifying condition, STEC Shiga-like toxin-producing Escherichia coli, TMA thrombotic microangiopathy, TTP thrombotic thrombocytopenic purpura. aThe differential diagnosis section of the algorithm has been adapted from [5]
Case reports
Case 1
A 33-year-old Hispanic woman developed abruptio placentae leading to fetal death at 33 weeks of gestation. She underwent cesarean section and hysterectomy, and a subsequent exploratory laparotomy. The patient had extensive blood loss and received numerous transfusions. She developed thrombocytopenia [39 × 109/L (normal range 150–350 × 109/L)], microangiopathic hemolytic anemia [hemoglobin level 6.7 g/dL (normal range 14.0–17.5 g/dL)]; lactate dehydrogenase (LDH) level, 2670 U/L (normal range at institution, 100–200 U/L); haptoglobin level, 5.8 mg/dL (normal range at institution, 26–185 mg/dL); numerous schistocytes on a blood smear, and renal failure [serum creatinine level, 6.0 mg/dL (normal range 0.6–1.2 mg/dL)] necessitating initiation of hemodialysis. The fibrinogen level as well as prothrombin and partial thromboplastin times were normal. ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) activity testing was ordered and PE was initiated. The patient showed minimal improvement in hematologic parameters (hemoglobin level, 7.0 g/dL; platelet count, 42 × 109/L) and no improvement in renal function (dialysis dependent) after five daily PEs, and the ADAMTS13 activity level was 56 %. Following diagnosis of aHUS, PE was discontinued. After the discontinuation of PE, the patient was vaccinated against meningococcus, antibiotic prophylaxis was started, and eculizumab therapy was initiated. Two weeks later, dialysis was discontinued. Laboratory tests showed a platelet count of 147 × 109/L, hemoglobin level of 8.8 mg/dL, and serum creatinine level of 3.4 mg/dL. At last follow-up after 27 weeks of eculizumab therapy, platelet count (198 × 109/L), hemoglobin level (13.0 g/dL), and serum creatinine level (0.9 mg/dL) were normal. The patient remains on ongoing eculizumab therapy.
Case 2
A 43-year-old Caucasian woman with a history of migraine headaches since childhood presented with severe headaches and visual impairment lasting for several days. The examination showed a blood pressure of 300/185 mmHg resulting in immediate hospitalization. Fundoscopic examination revealed papilledema, and a subsequent cerebral magnetic resonance tomography showed alterations consistent with posterior reversible encephalopathy syndrome. Laboratory tests including hemoglobin level of 10.8 g/dL, LDH level of 447 U/L (normal range at institution, <250 U/L) and schistocytes on a blood smear revealed microangiopathic hemolytic anemia; the platelet count was normal. Acute kidney injury [serum creatinine level, 3.4 mg/dL (normal range at institution, 0.5–1.0 mg/dL); proteinuria] also was evident. PE was initiated because thrombotic thrombocytopenic purpura (TTP) could not be ruled out initially, but was discontinued after the ADAMTS13 activity was determined to be 64 %. The patient’s hypertension was managed with intravenous and oral antihypertensive medications resulting in the resolution of neurological symptoms. Stool examination showed no Shiga toxin-producing Escherichia coli (STEC). A kidney biopsy revealed severe obliterative arteriolosclerosis, ischemic glomerular collapses, and extensive acute tubular injury. Together with typical signs of hypertensive retinopathy and echocardiographic evidence of hypertensive heart disease, the patient was considered to have MHT. However, despite adequate blood pressure control and resolution of hemolysis (LDH, 163 U/L), there was no improvement in anemia (hemoglobin, 10.7 g/dL) and renal function (serum creatinine level, 3.3 mg/dL) over approximately 5.5 weeks from presentation. Therefore, aHUS was diagnosed with MHT as a presenting sign. No complement gene mutations were identified. After meningococcal vaccination and antibiotic prophylaxis, initiation of eculizumab therapy resulted in gradual improvement of renal function. After 9 months of therapy, the patient’s hemoglobin level was 12.2 g/dL and serum creatinine level was stable at 2.1 mg/dL. After 11 months, the hemoglobin and serum creatinine levels were 12.9 g/dL and 2.0 mg/dL, respectively. The patient discontinued from eculizumab therapy after 1 year.
Case 3
A 37-year-old Caucasian female hemodialysis patient with a 14-month history of end-stage renal disease due to recurrent pyelonephritis underwent living-related donor kidney transplantation. Excellent graft function was noted immediately following the surgery, and the serum creatinine level decreased to 0.9 mg/dL. Over the subsequent days, however, urine output gradually decreased and serum creatinine levels increased (1.85 mg/dL on day 5 post-surgery). Humoral rejection was suspected (increasing titer of donor-specific antibodies), and the patient was treated with high-dose corticosteroids and PI. However, the patient developed anuria. Doppler ultrasound showed near-absent graft perfusion. In addition, TMA was suggested by laboratory values including the presence of schistocytes, platelet count of 33 × 109/L, hemoglobin level of 11.7 g/dL, LDH of 675 U/L (normal range at institution, <250 U/L), serum creatinine of 3.5 mg/dL, and heavy proteinuria (6701 mg/g creatinine). The patient was started on hemodialysis because of volume overload and progressive renal dysfunction. On post-transplant day 8, a diagnosis of aHUS was made. Eculizumab therapy, along with antibiotic prophylaxis for meningococcal infection, was initiated, leading to gradual resolution of hemolysis and improved renal function. A renal allograft biopsy revealed TMA consistent with the clinical diagnosis of aHUS. Immunostaining demonstrated C4d staining of peritubular capillaries consistent with humoral rejection. Immunoabsorption was performed for 3 days followed by two doses of intravenous immunoglobulins. Eculizumab treatment was continued with improvement in renal function without the need for further renal replacement therapy. The patient received meningococcal vaccination following discharge. At a follow-up of 6 months, platelet count continues to be stable at 213 × 109/L, hemoglobin level at 11.9 g/dL, LDH level at 273 U/L, and serum creatinine level at 1.7 mg/dL. The patient continues to receive eculizumab therapy. Genetic testing did not reveal any complement gene mutations.
Discussion
These case reports illustrate aHUS in the setting of three CACs: pregnancy complications, MHT, and renal transplantation. In all three cases, a CAC preceded the onset of TMA. Importantly, the standard management of the individual CAC (i.e. cesarean section and subsequent hysterectomy after pregnancy complications, antihypertensive medications for MHT, and corticosteroid therapy for humoral allograft rejection) did not resolve TMA. Each patient had a thorough evaluation for potential underlying causes of TMA. After prompt diagnosis of TMA and recognition of aHUS in each case, treatment with eculizumab was associated with improvement in both hematologic parameters and renal function.
Accumulating evidence shows that patients with underlying complement dysregulations are particularly prone to develop TMA when experiencing a CAC. Chronic complement dysregulation, both in aHUS and other disorders, leaves patients predisposed to TMA [24]. When patients are unable to regulate complement, onset or exacerbation of CACs may precipitate aHUS or cause additional manifestations, resulting in persistent TMA despite treatment of CAC symptoms [25]. Findings from a large observational study of patients with aHUS showed that 69 % of the patients had their first TMA manifestations while experiencing a CAC [9].
Proper diagnosis may be particularly challenging in the setting of aHUS and CACs due to overlapping comorbidities [1]. Patients may not necessarily present with the classic triad of microangiopathic hemolytic anemia, thrombocytopenia, and renal impairment [3]; in particular, thrombocytopenia may be absent or mild in MHT [26]. In a large observational study of patients with aHUS, 16 % of patients did not have thrombocytopenia at disease onset [4]. In the described case with MHT, the patient had a normal platelet count at presentation. It is possible that some patients may develop thrombocytopenia relative to earlier laboratory tests, although all values may remain in the normal range. Elevated LDH levels and the presence of schistocytes may also be considered important diagnostic features of microangiopathic hemolytic anemia [5].
Review of CACs
Pregnancy complications
TMA occurs in approximately 1 per 25,000 pregnancies [27]. Pregnancy-related aHUS (P-aHUS) may account for approximately 7 % of total aHUS cases [9] and up to 20 % of cases in adult females [4, 28]. Complement activation may be augmented during pregnancy, when the placenta may be subject to attack by the complement and immune system [28]. In addition, the complement pathway may be activated postpartum due to maternal circulation of fetal cells, infections, and hemorrhage [28]. Recently, increased complement activation was identified in a subset of women with preeclampsia and HELLP syndrome [29].
In addition to microangiopathic hemolytic anemia, thrombocytopenia, and renal insufficiency, general signs and symptoms of P-aHUS may include fatigue, headache, nausea, and vomiting. Diagnosis may be difficult because of similarities between P-aHUS and more common pregnancy complications such as preeclampsia and HELLP [27, 30]. A recent study of 21 women with P-aHUS showed that most cases occurred postpartum and during second pregnancies [28]. Clinical conditions could rapidly deteriorate, resulting in poor maternal outcomes [27]. Hypertension and chronic kidney disease were frequent long-term complications [27]. End-stage renal disease occurred in 76 % of patients. In severe cases, death may occur within hours to days after the onset of P-aHUS [31].
P-aHUS case reports were first published more than 40 years ago [32]. Delmas et al. [33] were the first to show the beneficial effects of eculizumab on hematologic and renal parameters in a patient with postpartum aHUS. More recent case studies also documented the efficacy of eculizumab in the treatment of P-aHUS, including normalization of hematologic parameters and renal function (Table 1) [33–41].
Emerging evidence shows the safety of eculizumab during pregnancy despite potential placental transfer to the fetus. In a study of 75 pregnancies in 61 women with paroxysmal nocturnal hemoglobinuria (PNH) treated with eculizumab during pregnancy and postpartum, fetal mortality rates were not increased [38]. In these patients, eculizumab was present at low levels in 35 % of cord blood samples, but not in breast milk [42]. Similarly, recently reported case series involving pregnant PNH patients treated with eculizumab demonstrated low levels of the drug in cord blood, but not in breast milk [43, 44]. There were no adverse effects on the newborns noted.
Malignant hypertension
MHT can be associated with TMA [45, 46]. Many patients with aHUS first present with hypertension, potentially with high severity and/or MHT [7, 9, 10]. In a retrospective study of 45 children with aHUS, 71 % presented with hypertension [10]. In a large observational study, 8 % of patients with aHUS also had MHT [9].
The role of the endothelium as a pathogenic link between MHT and aHUS was recently reviewed [47]. TMA may occur following fluid shear stress on endothelial cells and subsequent vascular injury (i.e. fibrinoid necrosis, thrombosis, and luminal narrowing), leading to red blood cell fragmentation and platelet consumption [45, 46, 48]. Aldosterone has been implicated as a potential mediator of vascular endothelial damage in hypertension [21, 49, 50]. In one study, serum aldosterone levels were found to be higher in MHT patients with TMA versus those without TMA [21]. In addition, hypertensive crises are known to be prothrombotic, leading to platelet aggregation, thrombin generation, and fibrinolysis [51].
Patients with MHT may present with microangiopathic hemolytic anemia, renal impairment, and thrombocytopenia [46], although the latter may be modest and/or resolve quickly [26, 52]. Differentiation between MHT-associated TMA and TTP may be particularly difficult because both are associated with neurologic symptoms; however, renal dysfunction may be more common in TMA caused by MHT [26]. Prior history of hypertension and/or relatively high arterial pressure, signs of hypertensive heart disease, relatively high platelet count, and retinopathy are suggestive of MHT-associated TMA [26]. To that end, imaging techniques may be useful in confirming congestive heart disease and/or neurologic involvement due to MHT.
Without proper diagnosis and adequate treatment, patients with aHUS and hypertension and/or MHT may have severe symptoms and poor outcomes, including death [53, 54]. Because standard therapies for MHT do not address underlying complement dysregulation, TMA may persist despite such treatment. In a retrospective analysis of 21 patients with TMA and severe/malignant hypertension, 86 % did not recover normal renal function despite antihypertensive therapy [55]. It has been proposed that a diagnosis of aHUS should be suspected in patients with difficult-to-control MHT who demonstrate persistent TMA [52]. In such patients, treatment with eculizumab should be considered. Indeed, case studies have demonstrated the efficacy and safety of eculizumab, with or without antihypertensive agents, in the treatment aHUS in patients with MHT (Table 1) [52, 56–62].
Renal transplantation
Recurrent and de novo aHUS following renal transplantation have been reviewed in detail previously [5, 63]. The availability of eculizumab has substantially changed the landscape of renal transplantation in patients with aHUS [5, 63]. In the pre-eculizumab era, renal transplantation in aHUS was associated with poor graft survival and high rates of disease recurrence [64]. In contrast, a large series of 22 patients demonstrated that eculizumab therapy was effective in preventing and treating aHUS recurrence post-transplant [65]. Patients at high risk for recurrence are now candidates for renal transplantation [5]. Additionally, living-non-related donor transplantation may now be considered for certain patients [63]. Expert groups recommend that patients, especially with moderate or high risk for disease recurrence following renal transplantation, receive prophylactic eculizumab therapy [5, 63]. Eculizumab should also be considered for patients with de novo aHUS following renal transplantation [5].
Autoimmune diseases
Systemic lupus erythematosus
SLE is characterized by the formation of immune complexes that activate complement, leading to cellular injury [66]. Dysregulation of the terminal complement activation has been implicated in the pathogenesis and prognosis of SLE and lupus nephritis [66–68]. Complement gene mutations have been identified in patients with SLE and are associated with disease susceptibility [69] and earlier disease onset [69, 70]. In patients with SLE, TMA is associated with increased SLE activity, intercurrent infections [71], reduced long-term renal function, and poor overall survival [72–76]. Although TMA is typically Coombs-negative [5], patients with SLE and aHUS may have positive Coombs tests [77].
Recent case studies of SLE comorbid with aHUS have reported varying outcomes with standard SLE therapies (e.g. cyclophosphamide, high-dose steroids, mycophenolate mofetil). Patients may have slow recovery [78], no response [79–81], or remain dialysis-dependent [82]. Findings from reports of patients with SLE and aHUS treated with eculizumab have demonstrated the terminal complement inhibitor to be well tolerated and associated with improvement in symptoms, hematologic laboratory values, and renal function (Table 1) [79, 80, 83].
Scleroderma
Progressive scleroderma, or systemic sclerosis, can be complicated by chronic kidney disease associated with hypertension and mild proteinuria as well as by scleroderma renal crisis (SRC). SRC is the most severe form of renal disease in progressive scleroderma and carries a high mortality. SRC manifests as MHT, TMA, and rapid renal failure [84]. Diagnosis can be complicated by the lack of skin changes in some cases making renal histology and serology results the primary basis for appropriate diagnosis [85]. The pathogenesis of progressive scleroderma and its relation to aHUS is not well understood; it is believed that systemic vasoconstriction leads to ischemic injury and organ dysfunction [86].
Several cases of scleroderma-related aHUS have been reported in the literature [86–90]. Overall, outcomes were poor, including death within months of onset in one case [89]. The effects of eculizumab have not been documented in scleroderma-related aHUS. However, in a recent case report of SRC, in which diagnosis of aHUS was not ruled out or substantiated, treatment with eculizumab was associated with improvement in renal function, hypertension, and other symptoms [91].
Ulcerative colitis
Diagnosis of aHUS in patients with gastrointestinal symptoms may require differentiation from inflammatory bowel disorders such as ulcerative colitis (UC) [92]. Interestingly, alternative complement activation may also contribute to the pathogenesis of inflammatory bowel disorders. For example, it has been postulated that the upregulation of complement components may contribute to local inflammation and tissue damage in Crohn’s disease [93]. Inflammatory bowel disorders have been associated with upregulation of C3, which was strongly correlated with mucosal inflammation [94]. Deposition of C3b and the terminal complement complex have also been demonstrated in mucosal tissue from patients with UC [95].
Only 2 cases of UC-associated aHUS have been reported in the literature (Table 1) [96, 97]. In the case reported by Green et al. [96], the patient was found to have complement factor H autoantibodies. Both patients were treated with eculizumab and had favorable outcomes with improvement in renal function and hematologic parameters [96, 97].
Drug-induced TMA
aHUS and other TMAs may develop subsequent to the use of certain medications and have been reviewed elsewhere [5]. Data from a recent systematic review showed that nine medications account for 76 % of TMA cases: quinine, tacrolimus, cyclosporine, interferons, gemcitabine, mitomycin, clopidogrel, estrogen/progesterone, and ticlopidine [98]. The pathogenesis of drug-induced TMA involves two distinct mechanisms: immune mediated and direct toxicity. Evidence shows that quinine-, ticlopidine-, and clopidogrel-induced TMAs occur via an immune-mediated reaction, which is typically characterized by severe systemic manifestations including anuric acute kidney injury. TMA caused by cyclosporine, gemcitabine, and mitomycin occurs through a toxicity-mediated mechanism that is dose dependent and may also lead to renal impairment [98, 99]. For TMA caused by cancer medications, onset may or may not be dose related and timing can vary from immediately following therapy initiation to a 12-month delay [100]. Gemcitabine- and mitomycin-related TMA occur in <1 and 2‒15 % of patients, respectively, and outcomes may be quite poor: renal failure and/or mortality have been reported in up to 70–75 % of cases [101].
It is recommended that patients discontinue the medication if it is suspected to be the cause of TMA [1]. This approach should result in TMA resolution. Patients with TMA caused by immunosuppressants (e.g. cyclosporine, tacrolimus) should receive reduced doses or switch to another agent [102]. However, TMA may continue to progress despite the removal of an offending agent, as has been documented with both mitomycin- [103] and tacrolimus-induced TMA [104]. The optimal management strategy and timing for drug-induced TMA has not been established, but it has been suggested that drug avoidance and supportive care may be the only beneficial options [1]. However, the role for PE/PI is unclear [100], and outcomes have included lack of improvement in or worsening renal function in mitomycin- [104], tacrolimus- [104, 105], and gemcitabine-induced TMA [106]. In these example cases, eculizumab therapy, sometimes in addition to anti-inflammatory agents, led to improvements in both hematologic parameters and renal function [103–106]. More studies are necessary to determine a potential role for eculizumab in drug-induced TMA.
Infection-induced TMA
Infections, particularly of the respiratory and gastrointestinal tract, precede aHUS in approximately half of cases [4, 9]. Common bacterial and viral infections associated with TMA have been reviewed elsewhere [5, 63] and include Streptococcus pneumoniae, cytomegalovirus, H1N1 influenza, human immunodeficiency virus, and parvovirus. Such infections are thought to activate the alternative pathway of complement and may be associated with increased production of C5 and deposition of C5b-9 [63]. Clinical symptoms of some infections, including diarrhea, may complicate the diagnostic process [3, 5].
Diagnosis and management of TMA in patients with CACs
aHUS can be clinically identical to other TMAs, including STEC-HUS and TTP. No testing is available for definitive diagnosis of aHUS [5]. In the setting of CACs and TMA, it may be particularly challenging to rule out other potential causes of TMA, including the existing CAC, to arrive at a diagnosis of aHUS [63]. It is possible that at the onset of aHUS, some patients with existing CACs may not present with all of the classic signs (i.e. thrombocytopenia, microangiopathic hemolytic anemia, acute renal failure) [53].
An algorithm for the management of patients with CACs and TMA is presented in Fig. 1. In a patient presenting with TMA and a specific CAC [9, 20, 24, 28, 53], clinicians should first initiate specific management for the identified CAC, in order to treat underlying causes of TMA. If renal or extrarenal TMA arose from the CAC in the absence of complement dysregulation, TMA should resolve rapidly. As the French Study Group for aHUS/C3G (C3 glomerulopathies) has recommended, resolution of renal TMA can be defined as normalization of platelet count and LDH level, and a decrease in serum creatinine by ≥25 % [63].
The persistence of TMA despite specific management for the CAC strongly suggests that the CAC is lowering the threshold for manifestations and unmasking aHUS [5, 24, 35, 53]. In these cases, differential diagnosis of TMA is required [5]. All patients with TMA should have a thorough evaluation for underlying causes [5]. STEC-HUS can be ruled out with a negative stool test for STEC. ADAMTS13 activity <5 % (depending on the assay used) indicates TTP [5]. Genetic investigations may help determine long-term prognosis of aHUS but are not required for diagnosis [5]. Complement gene mutations or factor H autoantibodies are identified in approximately 50–70 % of patients with aHUS [4, 9]. The number of mutations characterized has been increasing steadily over recent years [107], and others may be identified in the future.
Overall, clinicians should have a strong suspicion for aHUS in patients with ADAMTS13 activity ≥5 %, negative STEC test, and persistent TMA despite treatment of the CAC. It should be noted that testing results for ADAMTS13 activity level and STEC may not be rapidly available. Thus, some patients may initiate PE during the differential diagnosis period [5] to temporarily maintain hematologic parameters, although it does not inhibit the underlying complement-mediated pathogenesis of aHUS [12] or prevent end-stage renal disease or mortality [9].
For patients with diagnosed aHUS, with or without a comorbid CAC, clinicians should initiate eculizumab therapy immediately according to established guidelines [5, 63, 108]. Clinical studies have shown that earlier intervention with eculizumab is associated with better renal outcomes for patients with aHUS [15]. The specific effects of eculizumab in aHUS comorbid with individual CACs (e.g. autoimmune diseases) will be demonstrated as evidence accumulates in the literature.
The optimal treatment duration for patients with aHUS and specific CACs has not been established. Expert and regulatory guidance notes that ongoing treatment may prevent risks of potentially life-threatening TMA that may occur following therapy discontinuation [5, 13, 14].
Conclusions
CACs are increasingly identified in the medical literature as being comorbid with aHUS or unmasking previously undiagnosed cases. The presented case studies illustrate potential complexities in disease onset and differential diagnosis when both a CAC and aHUS are present, as well as the benefits of eculizumab treatment for these patients. Overall, clinicians should consider a diagnosis of aHUS if TMA persists despite specific management for the CAC. Once aHUS is diagnosed, eculizumab should be initiated promptly to halt target organ injury and improve outcomes related to TMA.
References
George JN, Nester CM (2014) Syndromes of thrombotic microangiopathy. N Engl J Med 371:654–666
Moake JL (2002) Thrombotic microangiopathies. N Engl J Med 347:589–600
Noris M, Remuzzi G (2009) Atypical hemolytic-uremic syndrome. N Engl J Med 361:1676–1687
Fremeaux-Bacchi V, Fakhouri F, Garnier A et al (2013) Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8:554–562
Campistol JM, Arias M, Ariceta G et al (2015) An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia 35:421–447
Nayer A, Asif A (2016) Atypical hemolytic-uremic syndrome: a clinical review. Am J Ther 23:e151–e158
Neuhaus TJ, Calonder S, Leumann EP (1997) Heterogeneity of atypical haemolytic uraemic syndromes. Arch Dis Child 76:518–521
Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA et al (2007) Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol 18:2392–2400
Noris M, Caprioli J, Bresin E et al (2010) Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 5:1844–1859
Geerdink LM, Westra D, van Wijk JA et al (2012) Atypical hemolytic uremic syndrome in children: complement mutations and clinical characteristics. Pediatr Nephrol 27:1283–1291
Ariceta G, Besbas N, Johnson S et al (2009) Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol 24:687–696
Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T (2010) Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 36:673–681
US Food and Drug Administration (2015) Soliris (eculizumab) [prescribing information]. Alexion Pharmaceuticals, Inc., Cheshire, CT
European Medicines Agency (2015) Soliris (eculizumab) [summary of product characteristics]. Alexion Europe SAS, Paris
Legendre CM, Licht C, Muus P et al (2013) Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368:2169–2181
Licht C, Greenbaum LA, Muus P et al (2015) Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int 87:1061–1073
Greenbaum LA, Fila M, Ardissino G et al (2016) Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int 89:701–711
Fakhouri F, Hourmant M, Campistol JM et al (2016) Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single-arm, open-label trial. Am J Kidney Dis 68:84–93
Riedl M, Fakhouri F, Le Quintrec M et al (2014) Spectrum of complement-mediated thrombotic microangiopathies: pathogenetic insights identifying novel treatment approaches. Semin Thromb Hemost 40:444–464
Kavanagh D, Goodship THJ, Richards A (2006) Atypical haemolytic uraemic syndrome. Br Med Bull 77 and 78:5–22
Akimoto T, Muto S, Ito C et al (2011) Clinical features of malignant hypertension with thrombotic microangiopathy. Clin Exp Hypertens 33:77–83
Barbour T, Johnson S, Cohney S, Hughes P (2012) Thrombotic microangiopathy and associated renal disorders. Nephrol Dial Transplant 27:2673–2685
Nester CM, Thomas CP (2012) Atypical hemolytic uremic syndrome: what is it, how is it diagnosed, and how is it treated? Hematology Am Soc Hematol Educ Program 2012:617–625
Noris M, Mescia F, Remuzzi G (2012) STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol 8:622–633
Kavanagh D, Goodship TH, Richards A (2013) Atypical hemolytic uremic syndrome. Semin Nephrol 33:508–530
Khanal N, Dahal S, Upadhyay S, Bhatt VR, Bierman PJ (2015) Differentiating malignant hypertension-induced thrombotic microangiopathy from thrombotic thrombocytopenic purpura. Ther Adv Hematol 6:97–102
Dashe JS, Ramin SM, Cunningham FG (1998) The long-term consequences of thrombotic microangiopathy (thrombotic thrombocytopenic purpura and hemolytic uremic syndrome) in pregnancy. Obstet Gynecol 91:662–668
Fakhouri F, Roumenina L, Provot F et al (2010) Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol 21e:859–867
Vaught AJ, Gavriilaki E, Hueppchen N et al (2016) Direct evidence of complement activation in HELLP syndrome: a link to atypical hemolytic uremic syndrome. Exp Hematol 44:390–398
Shrivastava M, Modi G, Singh RK, Navaid S (2011) Early diagnosis and management of postpartum hemolytic uremic syndrome with plasma exchange. Transfus Apher Sci 44:257–262
Mu J, Zhang J, Sunnassee A, Dong H (2015) A case report of undiagnosed postpartum hemolytic uremic syndrome. Diagn Pathol 10:89
Calvert GD (1972) Postpartum haemolytic uraemic syndrome: case report and brief review. J Obstet Gynaecol Br Commonw 79:244–249
Delmas Y, Bordes C, Loirat C, Fremeaux-Bacchi V, Combe C (2013) Post-partum atypical haemolytic-uraemic syndrome treated with eculizumab: terminal complement activity assessment in clinical practice. Clin Kidney J 6:243–244
Ardissino G, Wally Ossola M, Baffero GM, Rigotti A, Cugno M (2013) Eculizumab for atypical hemolytic uremic syndrome in pregnancy. Obstet Gynecol 122:487–489
Carr R, Cataland SR (2013) Relapse of aHUS after discontinuation of therapy with eculizumab in a patient with aHUS and factor H mutation. Ann Hematol 92:845–846
Zschiedrich S, Prager EP, Kuehn EW (2013) Successful treatment of the postpartum atypical hemolytic uremic syndrome with eculizumab. Ann Intern Med 159:76
Canigral C, Moscardo F, Castro C et al (2014) Eculizumab for the treatment of pregnancy-related atypical hemolytic uremic syndrome. Ann Hematol 93:1421–1422
Mussoni MP, Veneziano FA, Boetti L, et al. (2014) Innovative therapeutic approach: sequential treatment with plasma exchange and eculizumab in a pregnant woman affected by atypical hemolytic-uremic syndrome. Transfus Apher Sci 51:134–136
De Sousa Amorim E, Blasco M, Quintana L, Sole M, de Cordoba SR, Campistol JM (2015) Eculizumab in pregnancy-associated atypical hemolytic uremic syndrome: insights for optimizing management. J Nephrol 28:641–645
Saad AF, Roman J, Wyble A, Pacheco LD (2016) Pregnancy-associated atypical hemolytic-uremic syndrome. AJP Rep 6:e125–128
Tsai HM, Kuo E (2016) From gestational hypertension and preeclampsia to atypical hemolytic uremic syndrome. Obstet Gynecol 127:907–910
Kelly RJ, Hochsmann B, Szer J et al (2015) Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med 373:1032–1039
Hallstensen RF, Bergseth G, Foss S et al (2015) Eculizumab treatment during pregnancy does not affect the complement system activity of the newborn. Immunobiology 220:452–459
Miyasaka N, Miura O, Kawaguchi T et al (2016) Pregnancy outcomes of patients with paroxysmal nocturnal hemoglobinuria treated with eculizumab: a Japanese experience and updated review. Int J Hematol 103:703–712
Shibagaki Y, Fujita T (2005) Thrombotic microangiopathy in malignant hypertension and hemolytic uremic syndrome (HUS)/thrombotic thrombocytopenic purpura (TTP): can we differentiate one from the other? Hypertens Res 28:89–95
van den Born BJ, Honnebier UP, Koopmans RP, van Montfrans GA (2005) Microangiopathic hemolysis and renal failure in malignant hypertension. Hypertension 45:246–251
Mathew RO, Nayer A, Asif A (2016) The endothelium as the common denominator in malignant hypertension and thrombotic microangiopathy. J Am Soc Hypertens 10:352–359
Kincaid-Smith P (1982) Renal pathology in hypertension and the effects of treatment. Br J Clin Pharmacol 13:107–115
Farquharson CA, Struthers AD (2002) Aldosterone induces acute endothelial dysfunction in vivo in humans: evidence for an aldosterone-induced vasculopathy. Clin Sci (Lond) 103:425–431
Cachofeiro V, Miana M, de Las Heras N et al (2008) Aldosterone and the vascular system. J Steroid Biochem Mol Biol 109:331–335
van den Born BJH, Lowenberg EC, van der Hoeven NV et al (2011) Endothelial dysfunction, platelet activation, thrombogenesis and fibrinolysis in patients with hypertensive crisis. J Hypertens 29:922–927
Tsai HM (2016) Does anticomplement therapy have a role in the management of malignant hypertension? J Clin Hypertens (Greenwich) 18:359–360
Totina A, Iorember F, El-Dahr SS, Yosypiv IV (2013) Atypical hemolytic-uremic syndrome in a child presenting with malignant hypertension. Clin Pediatr (Phila) 52:183–186
Rafiq A, Tariq H, Abbas N, Shenoy R (2015) Atypical hemolytic-uremic syndrome: a case report and literature review. Am J Case Rep 16:109–114
Zhang B, Xing C, Yu X, Sun B, Zhao X, Qian J (2008) Renal thrombotic microangiopathies induced by severe hypertension. Hypertens Res 31:479–483
Al-Akash SI, Almond PS, Savell VH Jr, Gharaybeh SI, Hogue C (2011) Eculizumab induces long-term remission in recurrent post-transplant HUS associated with C3 gene mutation. Pediatr Nephrol 26:613–619
Garjau M, Azancot M, Ramos R, Sanchez-Corral P, Montero MA, Seron D (2012) Early treatment with eculizumab in atypical haemolytic uraemic syndrome. Clin Kidney J 5:31–33
Besbas N, Gulhan B, Karpman D et al (2013) Neonatal onset atypical hemolytic uremic syndrome successfully treated with eculizumab. Pediatr Nephrol 28:155–158
Sajan T, Vinay S, Sonu N, Alan P (2014) How atypical can atypical hemolytic uremic syndrome be? Clin Case Rep 2:57–59
Ohta T, Urayama K, Tada Y et al (2015) Eculizumab in the treatment of atypical hemolytic uremic syndrome in an infant leads to cessation of peritoneal dialysis and improvement of severe hypertension. Pediatr Nephrol 30:603–608
Sevinc M, Basturk T, Sahutoglu T et al (2015) Plasma resistant atypical hemolytic uremic syndrome associated with a CFH mutation treated with eculizumab: a case report. J Med Case Rep 9:92
Sharma S, Pradhan M, Meyers KE, Le Palma K, Laskin BL (2015) Neonatal atypical hemolytic uremic syndrome from a factor H mutation treated with eculizumab. Clin Nephrol 84:181–185
Zuber J, Fakhouri F, Roumenina LT, Loirat C, Fremeaux-Bacchi V (2012) Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol 8:643–657
Le Quintrec M, Zuber J, Moulin B et al (2013) Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant 13:663–675
Zuber J, Le Quintrec M, Krid S et al (2012) Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant 12:3337–3354
Ornstein BW, Atkinson JP, Densen P (2012) The complement system in pediatric systemic lupus erythematosus, atypical hemolytic uremic syndrome, and complocentric membranoglomerulopathies. Curr Opin Rheumatol 24:522–529
Leffler J, Bengtsson AA, Blom AM (2014) The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis 73:1601–1606
Birmingham DJ, Hebert LA (2015) The complement system in lupus nephritis. Semin Nephrol 35:444–454
Zhao J, Wu H, Khosravi M et al (2011) Association of genetic variants in complement factor H and factor H-related genes with systemic lupus erythematosus susceptibility. PLoS Genet 7:e1002079
Jonsen A, Nilsson SC, Ahlqvist E et al (2011) Mutations in genes encoding complement inhibitors CD46 and CFH affect the age at nephritis onset in patients with systemic lupus erythematosus. Arthritis Res Ther 13:R206
Chen MH, Chen MH, Chen WS et al (2011) Thrombotic microangiopathy in systemic lupus erythematosus: a cohort study in North Taiwan. Rheumatology (Oxford) 50:768–775
Bridoux F, Vrtovsnik F, Noel C et al (1998) Renal thrombotic microangiopathy in systemic lupus erythematosus: clinical correlations and long-term renal survival. Nephrol Dial Transplant 13:298–304
Song D, Wu LH, Wang FM et al (2013) The spectrum of renal thrombotic microangiopathy in lupus nephritis. Arthritis Res Ther 15:R12
Musio F, Bohen EM, Yuan CM, Welch PG (1998) Review of thrombotic thrombocytopenic purpura in the setting of systemic lupus erythematosus. Semin Arthritis Rheum 28:1–19
Banfi G, Bertani T, Boeri V et al (1991) Renal vascular lesions as a marker of poor prognosis in patients with lupus nephritis. Gruppo Italiano per lo Studio della Nefrite Lupica (GISNEL). Am J Kidney Dis 18:240–248
Jain R, Chartash E, Susin M, Furie R (1994) Systemic lupus erythematosus complicated by thrombotic microangiopathy. Semin Arthritis Rheum 24:173–182
Giannouli S, Voulgarelis M, Ziakas PD, Tzioufas AG (2006) Anaemia in systemic lupus erythematosus: from pathophysiology to clinical assessment. Ann Rheum Dis 65:144–148
Samson M, Audia S, Leguy V et al (2012) Haemolytic-uraemic syndrome during severe lupus nephritis: efficacy of plasma exchange. Intern Med J 42:95–98
Coppo R, Peruzzi L, Amore A et al (2015) Dramatic effects of eculizumab in a child with diffuse proliferative lupus nephritis resistant to conventional therapy. Pediatr Nephrol 30:167–172
El-Husseini A, Hannan S, Awad A, Jennings S, Cornea V, Sawaya BP (2015) Thrombotic microangiopathy in systemic lupus erythematosus: efficacy of eculizumab. Am J Kidney Dis 65:127–130
Ramachandran R, Sakhuja V, Jha V, Kohli HS, Rathi M (2012) Plasmapheresis in systemic lupus erythematosus with thrombotic microangiopathy. Intern Med J 42:734
Gharbi C, Bourry E, Rouvier P et al (2010) Rapidly progressive lupus nephritis and concomitant thrombotic microangiopathy. Clin Exp Nephrol 14:487–491
Hadaya K, Ferrari-Lacraz S, Fumeaux D et al (2011) Eculizumab in acute recurrence of thrombotic microangiopathy after renal transplantation. Am J Transplant 11:2523–2527
Stratta P, Besso L, Ferrero S et al (1996) Scleroderma renal crisis is still a life-threatening syndrome. Ren Fail 18:567–574
Zwettler U, Andrassy K, Waldherr R, Ritz E (1993) Scleroderma renal crisis as a presenting feature in the absence of skin involvement. Am J Kidney Dis 22:53–56
Yamanaka K, Mizutani H, Hashimoto K, Nishii M, Shimizu M (1997) Scleroderma renal crisis complicated by hemolytic uremic syndrome in a case of elderly onset systemic sclerosis. J Dermatol 24:184–188
Ricker DM, Sharma HM, Nahman NS Jr (1989) Acute renal failure with glomerular thrombosis in a patient with chronic scleroderma. Am J Kidney Dis 14:524–526
Meyrier A, Becquemont L, Weill B, Callard P, Rainfray M (1991) Hemolytic-uremic syndrome with anticardiolipin antibodies revealing paraneoplastic systemic scleroderma. Nephron 59:493–496
Haviv YS, Safadi R (1998) Normotensive scleroderma renal crisis: case report and review of the literature. Ren Fail 20:733–736
Chen WS, Young AH, Wang HP, Huang DF (2009) Hemolytic uremic syndrome with ischemic glomerulonephropathy and obliterative vasculopathy in a systemic sclerosis patient treated with cyclosporine-A. Rheumatol Int 29:821–824
Thomas CP, Nester CM, Phan AC, Sharma M, Steele AL, Lenert PS (2015) Eculizumab for rescue of thrombotic microangiopathy in PM-Scl antibody-positive autoimmune overlap syndrome. Clin Kidney J 8:698–701
Craner GE, Burdick GE (1976) Acute colitis resembling ulcerative colitis in the hemolytic-uremic syndrome. Am J Dig Dis 21:74–76
Ahrenstedt O, Knutson L, Nilsson B, Nilsson-Ekdahl K, Odlind B, Hallgren R (1990) Enhanced local production of complement components in the small intestines of patients with Crohn’s disease. N Engl J Med 322:1345–1349
Sugihara T, Kobori A, Imaeda H et al (2010) The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clin Exp Immunol 160:386–393
Halstensen TS, Mollnes TE, Garred P, Fausa O, Brandtzaeg P (1992) Surface epithelium related activation of complement differs in Crohn’s disease and ulcerative colitis. Gut 33:902–908
Green H, Harari E, Davidovits M et al (2014) Atypical HUS due to factor H antibodies in an adult patient successfully treated with eculizumab. Ren Fail 36:1119–1121
Webb TN, Griffiths H, Miyashita Y et al (2015) Atypical hemolytic uremic syndrome and chronic ulcerative colitis treated with eculizumab. Int J Med Pharm Case Reports 4:105–112
Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN (2015) Drug-induced thrombotic microangiopathy: a systematic review of published reports. Blood 125:616–618
Reese JA, Bougie DW, Curtis BR et al (2015) Drug-induced thrombotic microangiopathy: experience of the Oklahoma registry and the BloodCenter of Wisconsin. Am J Hematol 90:406–410
Izzedine H, Perazella MA (2015) Thrombotic microangiopathy, cancer, and cancer drugs. Am J Kidney Dis 66:857–868
Blake-Haskins JA, Lechleider RJ, Kreitman RJ (2011) Thrombotic microangiopathy with targeted cancer agents. Clin Cancer Res 17:5858–5866
Pisoni R, Ruggenenti P, Remuzzi G (2001) Drug-induced thrombotic microangiopathy: incidence, prevention and management. Drug Saf 24:491–501
Faguer S, Huart A, Fremeaux-Bacchi V, Ribes D, Chaveau D (2013) Eculizumab and drug-induced haemolytic-uraemic syndrome. Clin Kidney J 6:484–485
Safa K, Logan MS, Batal I, Gabardi S, Rennke HG, Abdi R (2015) Eculizumab for drug-induced de novo posttransplantation thrombotic microangiopathy: a case report. Clin Nephrol 83:125–129
Nurnberger J, Philipp T, Witzke O et al (2009) Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med 360:542–544
Starck M, Wendtner CM (2014) Use of eculizumab in refractory gemcitabine-induced thrombotic microangiopathy. Br J Haematol 164:894–896
Noris M, Remuzzi G (2013) Managing and preventing atypical hemolytic uremic syndrome recurrence after kidney transplantation. Curr Opin Nephrol Hypertens 22:704–712
Loirat C, Fakhouri F, Ariceta G et al (2016) An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 31:15–39
Acknowledgments
The authors would like to acknowledge Erin Harvey, MS, CMPP, of Alexion Pharmaceuticals, Inc., and Kristen W. Quinn, PhD, of Peloton Advantage, LLC, who provided medical writing/editorial support with funding from Alexion Pharmaceuticals, Inc.
Funding
Medical writing and editorial support was provided by Peloton Advantage, LLC, and funded by Alexion Pharmaceuticals, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
AA has served as a member of the advisory board and speakers bureau for Alexion Pharmaceuticals, Inc. AN has served as a speaker for Alexion Pharmaceuticals, Inc. CSH has received travel grants and honoraria as a speaker for Alexion Pharmaceuticals, Inc.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors. The case reports presented in this paper have not been published previously in whole or part.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Asif, A., Nayer, A. & Haas, C.S. Atypical hemolytic uremic syndrome in the setting of complement-amplifying conditions: case reports and a review of the evidence for treatment with eculizumab. J Nephrol 30, 347–362 (2017). https://doi.org/10.1007/s40620-016-0357-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-016-0357-7