
Abstract
Background
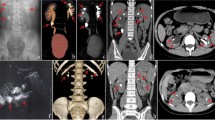
Primary hyperoxaluria (PH) is a rare autosomal recessive disease commonly arising in childhood and presenting with nephrolithiasis, nephrocalcinosis and/or chronic renal failure. Three genes are currently known as responsible: alanine-glyoxylate aminotransferase (AGXT, PH type 1), glyoxylate reductase/hydroxypyruvate reductase (GRHPR, PH type 2), and 4-hydroxy-2-oxoglutarate aldolase (HOGA1, PH type 3). In our Centre, at the end of 2014 molecular diagnosis of PH1 had been performed in 80 patients, while one patient received a PH2 diagnosis.
Materials and methods
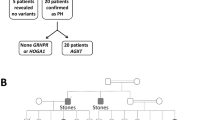
Fifteen patients referred to our Centre and suspected to have PH on clinical grounds were negative for pathogenic variants in the entire coding sequence and exon–intron boundaries of the AGXT gene. Therefore, we extended the analysis to the AGXT promoter region and the GRHPR and HOGA1 genes.
Results
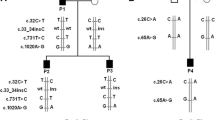
Two patients were heterozygous for two novel AGXT-promoter variants (c.-647C > T, c.-424C > T) that were probably non pathogenic. One patient was homozygous for a novel HOGA1 variant of intron 2 (c.341-81delT), whose pathogenicity predicted by in silico splicing tools was not confirmed by a minigene splicing assay in COS-7 and HEK293T cells.
Conclusion
New genetic subtypes of PH can be hypothesized in our patients, that may be caused by mutations in other gene encoding proteins of glyoxylate metabolism. Alternatively, some kind of mutations (e.g., deletions/duplications, deep intronic splicing regulatory variants) could be missed in a few cases, similarly to other genetic diseases.



Similar content being viewed by others
Abbreviations
- AGT:
-
Alanine-glyoxylate aminotransferase
- ESRD:
-
End stage renal disease
- GFR:
-
Glomerular filtration rate
- GRHPR:
-
Glyoxylate reductase/hydroxypyruvate reductase
- GO:
-
Glycolate oxidase
- HOGA:
-
4-Hydroxy-2-oxoglutarate aldolase
- LDH:
-
Lactate dehydrogenase
- Nv:
-
Normal value
- PH:
-
Primary hyperoxaluria
- PH1:
-
Primary hyperoxaluria type 1
- PH2:
-
Primary hyperoxaluria type 2
- PH3:
-
Primary hyperoxaluria type 3
- Pox:
-
Plasma oxalate
- Uox:
-
Urinary oxalate
- WT:
-
Wild type
References
Beck BB, Hoyer-Kuhn H, Goebel H et al (2013) Hyperoxaluria and systemic oxalosis: an update on current therapy and future directions. Expert Opin Investig Drugs 22(1):117–129
Leumann E, Hoppe B (2001) The primary hyperoxalurias. J Am Soc Nephrol 12(9):1986–1993
Cochat P, Hulton SA, Acquaviva C et al (2012) Primary hyperoxaluria Type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 27(5):1729–1736
Hoppe B, Beck BB, Milliner DS (2009) The primary hyperoxalurias. Kidney Int 75(12):1264–1271
Cregeen DP, Williams EL, Hulton S, Rumsby G (2003) Molecular analysis of the glyoxylate reductase (GRHPR) gene and description of mutations underlying primary hyperoxaluria type 2. Hum Mutat 22(6):497
Milliner DS, Wilson DM, Smith LH (2001) Phenotypic expression of primary hyperoxaluria: comparative features of types I and II. Kidney Int 59(1):31–36
Marangella M, Petrarulo M, Cosseddu D (1994) End-stage renal failure in primary hyperoxaluria type 2. N Engl J Med 330(23):1690
Williams H, Smith L (1968) l-glyceric aciduria: a new genetic variant of primary hyperoxaluria. N Enl J Med 278:233–239
Belostotsky R, Seboun E, Idelson GH et al (2010) Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Gen 87(3):392–399
Beck B, Baasner A, Buerscher A et al (2012) Novel findings in patients with primary hyperoxaluria type III and implications for advanced molecular testing strategies. Eur J Hum Genet 21(2):162–172
Allard L, Cochat P, Leclere AL et al (2015) Renal function can be impared in children with primary hyperoxaluria type 3. Pediatr Nephrol 30(10):1807–1813
Milliner DS (2005) The primary hyperoxalurias: an algorithm for diagnosis. Am J Nephrol 25(2):154–160
Cavalieri S, Pozzi E, Gatti AR, Brusco A (2012) Deep intronic ATM mutation detected by genomic resequencing and corrected in vitro by antisense morpholino oligonucleotide (AMO). Eur J Hum Genet 21(7):774–778
Mancini C, Vaula G, Scalzitti L et al (2012) Megalencephalic leukoencephalopathy with subcortical cysts type 1 (MLC1) due to a homozygous deep intronic splicing mutation (c.895-226T > G) abrogated in vitro using an antisense morpholino oligonucleotide. Neurogenetics 13(3):205–214
SpliceSiteFinder (SSF) http://www.genet.sickkids.on.ca/~ali/splicesitefinder.html. Accessed 29 July 2015
GeneSplicer http://www.cbcb.umd.edu/software/GeneSplicer/gene_spl.shtml. Accessed 29 July 2015
NNSplice http://www.fruitfly.org/seq_tools/splice.html. Accessed 29 July 2015
HumanSplicingFinder (HSF) http://www.umd.be/HSF/. Accessed 25 July 2015
MaxEntScan http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html. Accessed 29 July 2015
Hopp K, Cogal AG, Bergstrahl EJ et al (2015) Phenotype–genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol 26(10):2559–2570
Williams EL, Bagg EA, Mueller M et al (2015) Performance evaluation of Sanger sequencing for the diagnosis of primary hyperoxaluria and comparison with targeted next generation sequencing. Mol Genet Genomic Med 3(1):69–78
Sato M, Toné S, Ishikawa T et al (2002) Functional analysis of the 5′-Flanking region of the human alanine:glyoxylate aminotransferase gene AGXT. Biochim Biophys Acta 1574(2):205–209
Bunker RD, Loomes KM, Baker EN (2012) Purification, crystallization and preliminary crystallographic analysis of human dihydrodipicolinate synthase-like protein (DHDPSL). Acta Cryst 68(Pt 1):59–62
Clifford-Mobley O, Hewitt L, Rumsby G (2015) Simultaneous analysis of urinary metabolites for preliminary identification of primary hyperoxaluria. Ann Clin Biochem. doi:10.1177/0004563215606158
Primary hyperoxaluria mutation database. www.uclh.nhs.uk/OURSERVICES/SERVICEA-Z/PATHBIOMED/CBIO/Pages/Phmdatabase.aspx. Accessed 10 Dec 2015
Fu Y, Rope R, Fargue S et al (2014) A mutation creating an out-of-frame alternative translation initiation site in the GRHPR 5′UTR causing primary hyperoxaluria type II. Clin Genet 88(5):494–498
Mdluli K, Booth MP, Brady RL, Rumsby G (2005) A preliminary account of the properties of recombinant human glyoxylate reductase (GRHPR), LDHA and LDHB with glyoxylate, and their potential roles in its metabolism. Biochim Biophys Acta 1753(2):209–216
Frishberg Y, Zeharia A, Lyakhovetsky R et al (2014) Mutations in HAO1 encoding glycolate oxidase cause isolated glycolic aciduria. J Med Genet 51(8):526–529
Monico CG, Rossetti S, Belostotsky R et al (2011) Primary hyperoxaluria type III gene HOGA1 (formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. Clin J Am Soc Nephrol 6(9):2289–2295
Mandrile G, van Woerden CS, Berchialla P et al (2014) Data from a large European study indicate that the outcome of primary hyperoxaluria type 1 correlates with the AGXT mutation type. Kidney Int 86(6):1197–1204
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pelle, A., Cuccurullo, A., Mancini, C. et al. Updated genetic testing of Italian patients referred with a clinical diagnosis of primary hyperoxaluria. J Nephrol 30, 219–225 (2017). https://doi.org/10.1007/s40620-016-0287-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-016-0287-4