
Abstract
Purpose
Multiple endocrine neoplasia type 2 (MEN2) affects patients with RET proto-oncogene mutations. This cohort study refers to patients who were diagnosed with familial medullary thyroid carcinoma (MTC) and underwent RET genetic testing in Cyprus between years 2002 and 2017.
Methods and patients
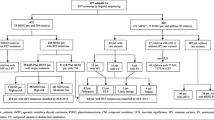
Forty patients underwent RET testing by Sanger sequencing of exons 10–11 and 13–16. Genotyping with STR genetic markers flanking the RET gene along with Y-chromosome genotyping and haplogroup assignment was also performed.
Results
RET mutations were identified in 40 patients from 11 apparently unrelated Cypriot families and two non-familial sporadic cases. Nine probands (69.2%) were heterozygous for p.Cys618Arg, one (7.7%) for p.Cys634Phe, one (7.7%) for the somatic delE632-L633 and two (15.4%) for p.Met918Thr mutations. The mean age at MTC diagnosis of patients carrying p.Cys618Arg was 36.8 ± 14.2 years. The age of pheo diagnosis ranged from 26 to 43 years and appeared simultaneously with MTC in 5/36 (13.9%) cases. The high frequency of the p.Cys618Arg mutation suggested a possible ancestral mutational event. Haplotype analysis was performed in families with and without p.Cys618Arg. Six microsatellite markers covering the RET gene and neighboring regions identified one core haplotype associated with all patients carrying p.Cys618Arg mutation.
Conclusions
The mutation p.Cys618Arg is by far the most prevalent mutation in Cyprus followed by other reported mutations of variable clinical significance. The provided molecular evidence speculates p.Cys618Arg mutation as an ancestral mutation that has spread in Cyprus due to a possible founder effect.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple endocrine neoplasia 2 (MEN2) is an autosomal dominant inherited cancer syndrome categorized into three clinically distinct forms known as MEN2A, MEN2B and Familial medullary thyroid carcinoma (FMTC) [1]. MEN2A is characterized by the association with medullary thyroid carcinoma (MTC) in 95%, pheochromocytoma (pheo) in 50% and parathyroid hyperplasia or adenoma in 15–30% of the cases [2]. MEN2B is the most aggressive type of the MEN2 variants and is characterized by abnormal body proportion seen as decreased upper/lower body ratio, marfanoid habitus and mucosal and intestinal ganglioneuromatosis [3].
MEN2A and MEN2B were first reported to be caused by point mutations in the RET proto-oncogene [4, 5]. The RET proto-oncogene encodes a transmembrane protein tyrosine kinase receptor involved in the transduction of signals for cell growth and differentiation in human neural crest-derived and neuronal tissues, such as Schwann cells, sympathetic ganglia, adrenal medulla, astrocytes and cerebral cortical neurons [6].
Up-to-date, more than 100 gain-of-function RET proto-oncogene mutations have been reported in patients with MTC, including germline mutations in patients with hereditary disease and somatic mutations in patients with sporadic disease [7]. The majority of the RET proto-oncogene mutations are located in exons 10, 11, 13, 14, 15, and 16 [2, 8,9,10,11]. Several germline mutations in exon 10 of the RET proto-oncogene in codons 609, 611, 618, 620 have also been associated in Hirschsprung disease [1, 2]. The clinical expression of MEN2 exhibits variable expression with different amino acid RET substitutions at the same codon. Such mutations are explicitly located in cysteines within this extracellular cysteine-rich domain and give rise to the subtype of MEN2A [12]. Codon 618 with p.Cys618Arg, p.Cys618Gly and p.Cys618Ser and to a lesser extent with codons p.Cys618Phe and p.Cys618Tyr, have been associated with the greatest rates of pheo [13,14,15]. Some studies revealed that the severity of pheo due to mutations in codons 609, 618 and 620 can be as aggressive as the one that is usually shown by the five amino acid substitutions at codon 634. However, the majority of the cases, demonstrated greatest expression for 634, followed in decreasing order by codons 618, 620 and 609 [13, 16, 17]. In recent reports, the spectrum of mutations identified in the RET proto-oncogene in patients with MTC has shifted from the ‘classical’ and most prevalent worldwide mutation at codon 634 in exon 11 to clinically less aggressive forms with mutations in exons 13–15 [18,19,20]. In the present study, we characterized clinically and molecularly Cypriot families with FMTC, MEN2A and MEN2B. The phenomenon of founder effect is not unusual in the population of Cyprus, and several recent reports have documented several founder mutations on the island. The recent evidence of a founder effect unveiled from the genetic population profile of certain endocrinopathies describes the past migration trends in Cyprus [21]. Therefore, we further investigated the possibility of our patients carrying a common allelic haplotype and identified a unique and identical haplotype in all patients carrying the missense p.Cys618Arg. Consequently, this common haplotype is the result of an ancestral mutation that has spread in the island of Cyprus due to a possible founder effect.
Patients and methods
Patients
Forty patients with MEN2 diagnoses (MTC with and without pheo) were screened for exons 10, 11, 13, 14, 15 and 16 of the RET proto-oncogene between January 2002 and September 2017. Except for one female patient of Russian descent the remaining 39 patients of the cohort were exclusively of Cypriot origin. Although defining and separating familial MTC from MEN2A and MEN2B has been challenging, the subjects were classified based on the symptoms presented so far. The specific clinical parameters used prior to sending for genetic testing included MTC, pheo, hyperparathyroidism and cutaneous lichen amyloidosis [1]. Informed consent for this study was obtained from all adult patients. Moreover, molecular testing for pre-symptomatic diagnosis of all minors was performed following informed consent obtained by the parents after appropriate genetic counselling and advice by a Pediatric Endocrinologist and a Nuclear Medicine Specialist.
Oligonucleotides, PCR conditions and direct sequencing of the RET proto-oncogene
Genomic DNA was isolated from peripheral blood leukocytes using a kit from QIAGEN (QIAGEN, GmbH D-40724, Hilden, Germany). The primers and conditions for PCR amplification and direct sequencing of exons 10, 11, 13, 14, 15 and 16 were as described previously [15, 22].
Genotyping with STR genetic markers flanking the RET proto-oncogene
In the present study, six STR markers flanking the RET proto-oncogene and according to GRCh38.p7 primary assembly reference sequence: NC_000010.11 were used. These genetic markers included STR1 (43207304–43207530 bp), STR2 (43664013–43664241 bp) and STR3 (43143426–43143640 bp) all located upstream of the RET proto-oncogene and the STR4 (43015181–43015361 bp), D10S469 (43445299–43445435 bp) and D10S681 (42897992–42897871 bp) all located downstream of the RET proto-oncogene. The forward primer of each genetic marker was labeled with 6-FAM fluorescent dye at 5′. The primer sequence pairs included STR1_F: 5′- CTTCAGTGCCATGACAGGAC-3′ & STR1_R: 5′-AGGAAACCTTTAGGTGTTTGGTG-3′; STR2_F: 5′- CTTTTCCAGACTCTCTCAAAGAGCAG-3′ & STR2_R: 5′-TATGGCAGACGTGGGTGCT-3′; STR3_F: 5′- GGAAAGTGGAATTGTGAATTGCTG- 3′ & STR3_R: 5′-GGCATAACACTTCACCTCTCAG-3′; STR4_F: 5′-GTTATTGTAGCCTCAGAGGC-3′ & STR4_R 5′-ACACTCTTCAAATCCATTCTGCG-3′; D10S469_F: 5′-GCAACAAGTGTGAGAGTCCAT-3′ & D10S469_R: 5′-GGATGTTCTGTCTCTCCACAGT-3′; D10S681_F: 5′- GACCAGAGGAAAGGCTAATGC-3′ & D10S681_R: 5′-GAAGCGGCCAGAACTTAGC- 3′. PCR was performed using 50 ng of genomic DNA, 1 × Amplitaq Gold Buffer (Applied Biosystems), 200 mΜ each dNTP (Sigma), 1,25 units of Amplitaq Gold DNA Polymerase (Applied Biosystems), 10 µM of each primer and double distilled water to a final volume of 25 μl. The amplification reaction was initialized at 95 °C for 10 min, followed by 35 cycles for 30 s at 95 °C, 60 s at 58 °C; 60 s at 72 °C and a final extension for 7 min at 72 °C. Each of the PCR products was diluted tenfold using ddH2O. Analysis of PCR products was performed by capillary electrophoresis on an ABI 3130xl Genetic Analyzer (Applied Biosystems) preceded by a denaturation step. A mix of 8.5 μl of HiDi™ formamide (Applied Biosystems) and 0.5 μl of GeneScan-600 LIZ size standard (Applied Biosystems) was added to 3 μl of diluted PCR product and denatured at 95 °C for 5 min. The mixture was immediately transferred on ice for 5 min and loaded for electrophoresis onto the ABI 3130xl genetic analyzer (Applied Biosystems). PCR products were visualized and analyzed with GeneMapper™ v4.1 Software (Applied Biosystems).
Y-chromosome genotyping and haplogroup assignment
Since in Cyprus the gene pool is characterized by deep inbreeding and high degree of conservation, we decided to perform Y-STR haplotype analysis for all male patients carrying p.Cys618Arg. The methodology used was similar to the one of a recent paper [23]. This methodology allows the simultaneous determination of short tandem repeats (STRs) on 23 loci located on the Y-chromosome by the PowerPlex® Y23 System (Promega). Subsequently, the 23 loci Y-STR haplotypes were input to the online Whit Athey’s Haplogroup Predictor tool [24] which generates probabilities for assignment to one of the major Y-DNA haplogroups.
Results
Clinical and genetic findings
RET mutations were identified in a total of 40 patients from 11 unrelated Cypriot families, one sporadic case of Cypriot descent and lastly one sporadic case of Russian descent. The cohort included 23 females (57.5%) and 17 males (42.5%). Among the 11 families, 9 probands (69.2%) were heterozygous for the missense p.Cys618Arg, one (7.7%) for the p.Cys634Phe, one (7.7%) for the somatic p.Glu632_Leu633del (delE632-L633) and two (15.4%) for the p.Met918Thr mutation. The mean age at MTC diagnosis in the group of patients carrying p.Cys618Arg was 36.8 ± 14.2 years (range 18–62 years). The age of pheo diagnosis in the same group ranged between 26 and 62 years and appeared simultaneously with MTC in 6/36 (16.7%) cases. The clinical and genetic findings of all patients and relatives diagnosed between 2002 and 2017 are summarized in Table 1.
Screening of family members from all nine MEN2A families carrying the p.Cys618Arg mutation showed the presence of the same genetic defect in thirty-six individuals of various ages, 24 of whom underwent prophylactic thyroidectomy (Table 1). Family histories of five out of nine MEN2A families with the p.Cys618Arg mutation reveal that they originated from the same village at the north-western end of the Limassol province in Cyprus (Fig. 1).

Geographical representation of the common origin of the five out of nine families carrying the p.Cys618Arg mutation. The island of Cyprus located in the eastern Mediterranean Sea. The village is located at the north-western end of the Limassol province and is indicated with red colour. The common haplotype which is segregated with the p.Cys618Arg mutation is demonstrated at the right bottom of the figure
Only eight out of the total forty patients with MEN2 were clinically reported as having both MTC and pheo. Six of these patients belonged to the group identified with the missense p.Cys618Arg mutation in exon 10 of the RET proto-oncogene. Unfortunately, two of the patients with MEN2A carrying p.Cys618Arg passed away and no clinical follow-up as far as the development of pheo and their clinical status was available. Thus far, from the remaining MEN2A patients carrying the same p.Cys618Arg, sixteen manifested only MTC and fourteen were asymptomatic (Table 1). Therefore, a strong manifestation of MTC with a wide range of ages was observed in these families (Table 1).
The patients identified with the sporadic p.Cys634Phe and the somatic p.Glu632_Leu633del (delE632-L633) mutation also causing MEN2A only had MTC at the time of diagnosis. Lastly, two discrete patients were both identified with the severe p.Met918Thr MEN2B causing mutation. The first patient is a male who unfortunately passed away at the age of 39 and who underwent total thyroidectomy when he was clinically diagnosed with MTC and pheo at the age of 25. The second patient is currently a 16-year-old female who up to now exhibited only MTC. Recently, a year after her initial diagnosis she underwent total thyroidectomy with central node dissection (Table 1).
Haplotype analysis of families with the RET proto-oncogene p.Cys618Arg missense mutation
In the present study, a common haplotype was demonstrated to be present in a cohort of nine and apparently non-related Cypriot MEN2A families that all carried the same missense p.Cys618Arg (Fig. 2). This specific haplotype was not found in the rest of the examined Cypriot MEN2 probands and their families identified with other than the specific p.Cys618Arg defects (Table 1).

Pedigree and disease-haplotype segregation of families with the RET proto-oncogene p.Cys618Arg missense mutation. Blackened symbols represent affected individuals with MEN2A phenotype. White symbols represent individuals with a normal phenotype. Circles and squares indicate females and males, respectively. Red characters represent the mutant haplotype which segregates with the p.Cys618Arg missense mutation
Y-haplogroup assignment
A total of seven different major Y-haplogroups (G2a, J1, R1b, R1a, I2a, E1b1a, J2b) were predicted amongst the 10 typed male Cypriot patients all carrying p.Cys618Arg mutation. The most frequent Y-haplogroup was G2a (40%) and was followed by J1, R1b, R1a, I2a, E1b1a and J2b at 10%, each.
Discussion
The distribution of genetic defects in our cohort differed from that of other populations in the context that the RET p.Cys618Arg mutation was found to be the most prevalent (69.2%). According to the latest ATA Management Guidelines, mutations at RET codon C618 are categorized as ATA moderate (MOD) level and carry a lower risk for aggressive MTC (ATA levels are categorized as moderate (MOD), high (H) and highest risk (HST) [1].
The risk of MTC has been stratified in three types according to the mutations of the RET proto-oncogene. Children with MEN2B due to defects in codon 883, 918 and 922 have the greatest risk of aggressive MTC (HST) and ought to undergo a total thyroidectomy with central node dissection, within the first 6 months [1]. Children bearing any of the RET codons 611, 618, 620 are prone to MOD risk of MTC, while those bearing 634 the majority of times exhibit a high (H) risk. Thus, a total thyroidectomy should be carried out before the age of 5 years. Children with defects in the RET codons 609, 768, 790,791, 804 and 891 have in general a less aggressive MTC and thyroidectomy may be performed at a later stage [1, 25].
In 85% of MEN2A patients, RET codon 634 (p.Cys634Arg) has been reported as the most prevalent in European and non-European families [18, 20, 26]. Several other smaller and larger multicenter studies mainly from groups around Europe demonstrated codon 533 as the prevailing in Greece [27], 611 in Portugal and Denmark [28, 29], 618 in Cyprus [15], 790 in Germany [18] and 804 and 891 in Italy [19].
It is estimated that more than 50% of familial MTC cases occur in patients with defects in codon 618. According to the most recent consensus guidelines, aberrations in codon 618 are classified as moderate (MOD) for MTC for which prophylactic thyroidectomy is recommended prior to the age of 5 years [1, 30]. All nine probands of the current study carrying the missense p.Cys618Arg and several of their close relatives also identified with the specific defect underwent prophylactic thyroidectomy (Table 1). Pheo is a distinct constituent of MEN2 syndrome and as reported it occurs in 15–20% of MEN2 patients, depending on age, follow-up time and type of RET mutation [10, 17, 31]. The coexistence of MTC and pheo usually establishes the diagnosis of either MEN2A or MEN2B [1] and in the present study, it was observed in 8/40 patients identified with RET mutations. Generally, pheo penetrance and age of diagnosis highly correlate with MTC aggressiveness based on the RET mutation status. The coexistence of pheo and MTC was only observed in one of the two patients with MEN2B due to p.Met918Thr, a 25-year-old male who underwent thyroidectomy. The other MEN2B female patient is currently 16 years old and up to now only manifested MTC. Since penetrance of pheo progressively increases with age an at risk-patient such as the young female of the present study identified with the MEN2B as a result of p.Met918Thr, recently underwent total thyroidectomy with central node dissection. MTC is a calcitonin making tumor of the parafollicular cells of the thyroid gland, which is preceded by multifocal C cell hyperplasia. The timing of the progression of MTC in relation to the incidence of pheo is variable and may take many years and not all mutations have the same effect on how aggressive the MTC is [32]. In general, RET mutations occurring in the extracellular cysteine domains (e.g., C611, C618, C620) of the gene have weaker transforming ability as a result of being more distant from the cell membrane when compared to those located close to the cell membrane such as p.Met918Thr [30, 33]. This phenomenon explains the varied ages from 15 to 62 years at which the probands and their relatives were diagnosed. The aggressiveness of MTC correlates with pheo penetrance and age at diagnosis and is usually associated with RET mutation codon status [34]. Prevention or therapy of MTC is successfully achieved with thyroidectomy once the diagnosis is made or before the age of likely malignant progression [1, 34]. On the basis of our genetic screening and clinical evaluation performed during a follow-up of 1–15 years, we report a higher prevalence of Familial MTC in the group of patients identified with the same missense p.Cys618Arg mutation.
In the present study, the common haplotype was exclusively found in the cohort of Cypriot MEN2A families carrying the missense p.Cys618Arg mutation. Based on the fact that this specific haplotype was not found in the remaining Cypriot MEN2 probands identified with other than the specific p.Cys618Arg mutation, speculates that it is the result of an ancestral mutation that has spread in the island of Cyprus due to a possible founder effect (Fig. 2).
In addition to the probable founder effect suggested here, similar phenomena have been projected for a number of other mutations or diseases among the population of Cyprus such as the predominance of the IVS1-2A > G mutation in the 5α Steroid Reductase type 2 (SRD5A2) gene in patients with 5 Alpha Reductase deficiency [35], the frequency of 1:7–1:10 for Friedreich ataxia [36] and the reported novel COL4A4 gene mutation p.Gly871Cys in a cohort of Greek-Cypriot families with thin membrane nephropathy and focal segmental glomerulosclerosis [37].
As reported in the early seventeenth century, in a village listed as property of the Venetian Government’ at the north-western end of the Limassol province in Cyprus, an infectious disease plunged this village and nearby settlements (Fig. 1). We speculate that the reported disease of that time was the result of a founder mutation such as p.Cys618Arg. Based on the genetic data produced in the present study, we have now speculation that this founder mutation was introduced to the locals by an invader or a settler during the Venetian era between 1489 and 1570 or prior to that period during the Crusades and the Lusignan Period 1191–1489. To investigate whether a common Y-haplogroup could be demonstrated in male individuals harboring the p.Cys618Arg mutation, we extended our study by performing a Y-chromosome analysis on male patients identified with the abovementioned mutation. In the cohort of male patients heterozygous for the p.Cys618Arg mutation, seven different major Y-haplogroups (G2a, J1, R1b, R1a, I2a, E1b1a, J2b) were found which were previously identified in Y-chromosomal studies of the Cypriot population [23, 38]. The different observations regarding the inheritance of the p.Cys618Arg mutation (suggesting a founder effect) and the genetic variety of Y-haplogroups in these individuals from Cyprus may indicate dissimilarities in the inheritance patterns between autosomal and Y-chromosome markers due to genetic drift, gene flow bias and other population level events.
In conclusion, the distribution of RET proto-oncogene mutations in Cyprus differs from that of other populations. The missense mutation p.Cys618Arg in codon 10 is by far the most prevalent mutation followed by other reported mutations of variable clinical significance. Additionally, the present study is the most extensive study ever accomplished addressing the ancestral RET proto-oncogene p.Cys618Arg mutation origin and including a large number of Cypriot families. The provided molecular evidence speculates that the most prevalent p.Cys618Arg mutation could be the result of an ancestral mutation that has spread in the island of Cyprus due to a possible founder effect.
References
Wells SA Jr, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, Raue F, Frank-Raue K, Robinson B, Rosenthal MS, Santoro M, Schlumberger M, Shah M, Waguespack SG, American Thyroid Association Guidelines Task Force on Medullary Thyroid C (2015) Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25(6):567–610. https://doi.org/10.1089/thy.2014.0335
Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, van Amstel HK, Lips CJ, Nishisho I, Takai SI, Marsh DJ, Robinson BG, Frank-Raue K, Raue F, Xue F, Noll WW, Romei C, Pacini F, Fink M, Niederle B, Zedenius J, Nordenskjold M, Komminoth P, Hendy GN, Mulligan LM et al (1996) The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. Int RET Mutat Consort Anal JAMA 276(19):1575–1579
Castinetti F, Moley J, Mulligan LM, Waguespack SG (2017) A comprehensive review on MEN 2B. Endocr Relat Cancer. https://doi.org/10.1530/ERC-17-0209
Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L et al (1993) Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 363(6428):458–460. https://doi.org/10.1038/363458a0
Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, Howe JR, Moley JF, Goodfellow P, Wells SA Jr (1993) Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 2(7):851–856
Durbec P, Marcos-Gutierrez CV, Kilkenny C, Grigoriou M, Wartiowaara K, Suvanto P, Smith D, Ponder B, Costantini F, Saarma M et al (1996) GDNF signalling through the Ret receptor tyrosine kinase. Nature 381(6585):789–793. https://doi.org/10.1038/381789a0
Margraf RL, Crockett DK, Krautscheid PM, Seamons R, Calderon FR, Wittwer CT, Mao R (2009) Multiple endocrine neoplasia type 2 RET protooncogene database: repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Hum Mutat 30(4):548–556. https://doi.org/10.1002/humu.20928
Mulligan LM, Eng C, Healey CS, Clayton D, Kwok JB, Gardner E, Ponder MA, Frilling A, Jackson CE, Lehnert H et al (1994) Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat Genet 6(1):70–74. https://doi.org/10.1038/ng0194-70
Voss RK, Feng L, Lee JE, Perrier ND, Graham PH, Hyde SM, Nieves-Munoz F, Cabanillas ME, Waguespack SG, Cote GJ, Gagel RF, Grubbs EG (2017) Medullary thyroid carcinoma in MEN2A: ATA moderate- or high-risk RET mutations do not predict disease aggressiveness. J Clin Endocrinol Metab 102(8):2807–2813. https://doi.org/10.1210/jc.2017-00317
Castinetti F, Qi XP, Walz MK, Maia AL, Sanso G, Peczkowska M, Hasse-Lazar K, Links TP, Dvorakova S, Toledo RA, Mian C, Bugalho MJ, Wohllk N, Kollyukh O, Canu L, Loli P, Bergmann SR, Biarnes Costa J, Makay O, Patocs A, Pfeifer M, Shah NS, Cuny T, Brauckhoff M, Bausch B, von Dobschuetz E, Letizia C, Barczynski M, Alevizaki MK, Czetwertynska M, Ugurlu MU, Valk G, Plukker JT, Sartorato P, Siqueira DR, Barontini M, Szperl M, Jarzab B, Verbeek HH, Zelinka T, Vlcek P, Toledo SP, Coutinho FL, Mannelli M, Recasens M, Demarquet L, Petramala L, Yaremchuk S, Zabolotnyi D, Schiavi F, Opocher G, Racz K, Januszewicz A, Weryha G, Henry JF, Brue T, Conte-Devolx B, Eng C, Neumann HP (2014) Outcomes of adrenal-sparing surgery or total adrenalectomy in phaeochromocytoma associated with multiple endocrine neoplasia type 2: an international retrospective population-based study. Lancet Oncol 15(6):648–655. https://doi.org/10.1016/S1470-2045(14)70154-8
Eng C, Smith DP, Mulligan LM, Nagai MA, Healey CS, Ponder MA, Gardner E, Scheumann GF, Jackson CE, Tunnacliffe A et al (1994) Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum Mol Genet 3(2):237–241
Accardo G, Conzo G, Esposito D, Gambardella C, Mazzella M, Castaldo F, Di Donna C, Polistena A, Avenia N, Colantuoni V, Giugliano D, Pasquali D (2017) Genetics of medullary thyroid cancer: an overview. Int J Surg 41(Suppl 1):S2–S6. https://doi.org/10.1016/j.ijsu.2017.02.064
Lindskog S, Nilsson O, Jansson S, Nilsson B, Illerskog AC, Ysander L, Ahlman H, Tisell LE (2004) Phenotypic expression of a family with multiple endocrine neoplasia type 2A due to a RET mutation at codon 618. Br J of Surg 91(6):713–718. https://doi.org/10.1002/bjs.4457
Raue F, Frank-Raue K (2007) Multiple endocrine neoplasia type 2: 2007 update. Horm Res 68(Suppl 5):101–104. https://doi.org/10.1159/000110589
Neocleous V, Skordis N, Portides G, Efstathiou E, Costi C, Ioannou N, Pantzaris M, Anastasiadou V, Deltas C, Phylactou LA (2011) RET proto-oncogene mutations are restricted to codon 618 in Cypriot families with multiple endocrine neoplasia 2. J Endocrinol Invest 34(10):764–769. https://doi.org/10.3275/7605
Quayle FJ, Fialkowski EA, Benveniste R, Moley JF (2007) Pheochromocytoma penetrance varies by RET mutation in MEN 2A. Surgery 142(6):800–805. https://doi.org/10.1016/j.surg.2007.09.013 (discussion 805 e801)
Mucha L, Leidig-Bruckner G, Frank-Raue K, Bruckner T, Kroiss M, Raue F, German Study Group for Rare Thyroid C (2017) Phaeochromocytoma in multiple endocrine neoplasia type 2: RET codon-specific penetrance and changes in management during the last four decades. Clin Endocrinol 87(4):320–326. https://doi.org/10.1111/cen.13386
Machens A, Lorenz K, Sekulla C, Hoppner W, Frank-Raue K, Raue F, Dralle H (2013) Molecular epidemiology of multiple endocrine neoplasia 2: implications for RET screening in the new millenium. Eur J Endocrinol 168(3):307–314. https://doi.org/10.1530/EJE-12-0919
Romei C, Mariotti S, Fugazzola L, Taccaliti A, Pacini F, Opocher G, Mian C, Castellano M, degli Uberti E, Ceccherini I, Cremonini N, Seregni E, Orlandi F, Ferolla P, Puxeddu E, Giorgino F, Colao A, Loli P, Bondi F, Cosci B, Bottici V, Cappai A, Pinna G, Persani L, Verga U, Boscaro M, Castagna MG, Cappelli C, Zatelli MC, Faggiano A, Francia G, Brandi ML, Falchetti A, Pinchera A, Elisei R, Ita MENn (2010) Multiple endocrine neoplasia type 2 syndromes (MEN 2): results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur J Endocrinol 163(2):301–308. https://doi.org/10.1530/eje-10-0333
Romei C, Tacito A, Molinaro E, Agate L, Bottici V, Viola D, Matrone A, Biagini A, Casella F, Ciampi R, Materazzi G, Miccoli P, Torregrossa L, Ugolini C, Basolo F, Vitti P, Elisei R (2015) Twenty years of lesson learning: how does the RET genetic screening test impact the clinical management of medullary thyroid cancer? Clin Endocrinol (Oxf) 82(6):892–899. https://doi.org/10.1111/cen.12686
Shammas C, Neocleous V, Toumba M, Costi C, Phedonos AA, Efstathiou E, Kyriakou A, Phylactou LA, Skordis N (2012) Overview of genetic defects in endocrinopathies in the island of Cyprus; evidence of a founder effect. Genet Test Mol Biomark 16(9):1073–1079. https://doi.org/10.1089/gtmb.2011.0381
Neocleous V, Passalaris T, Spanou E, Kitsios P, Skordis N, Deltas CC (2004) Description of the first two seemingly unrelated Greek Cypriot families with a common C618R RET proto-oncogene mutation. Genet Test 8(2):163–168. https://doi.org/10.1089/gte.2004.8.163
Heraclides A, Bashiardes E, Fernandez-Dominguez E, Bertoncini S, Chimonas M, Christofi V, King J, Budowle B, Manoli P, Cariolou MA (2017) Y-chromosomal analysis of Greek Cypriots reveals a primarily common pre-Ottoman paternal ancestry with Turkish Cypriots. PLoS ONE 12(6):e0179474. https://doi.org/10.1371/journal.pone.0179474
Athey TW (2006) Haplogroup prediction from Y-STR values Using a Bayesian-Allele-frequency approach. J Genet Geneal 2:34–39
American Thyroid Association Guidelines Task F, Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, Moley JF, Pacini F, Ringel MD, Schlumberger M, Wells SA Jr (2009) Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 19(6):565–612. https://doi.org/10.1089/thy.2008.0403
Wang J, Zhang B, Liu W, Zhang Y, Di X, Yang Y, Yan D (2016) Screening of RET gene mutations in Chinese patients with medullary thyroid carcinoma and their relatives. Fam Cancer 15(1):99–104. https://doi.org/10.1007/s10689-015-9828-6
Sarika HL, Papathoma A, Garofalaki M, Saltiki K, Pappa T, Pazaitou-Panayiotou K, Anastasiou E, Alevizaki M (2015) Genetic screening of patients with medullary thyroid cancer in a referral center in Greece during the past two decades. Eur J Endocrinol 172(4):501–509. https://doi.org/10.1530/EJE-14-0817
Prazeres HJ, Rodrigues F, Figueiredo P, Naidenov P, Soares P, Bugalho MJ, Lacerda M, Campos B, Martins TC (2006) Occurrence of the Cys611Tyr mutation and a novel Arg886Trp substitution in the RET proto-oncogene in multiple endocrine neoplasia type 2 families and sporadic medullary thyroid carcinoma cases originating from the central region of Portugal. Clin Endocrinol (Oxf) 64(6):659–666. https://doi.org/10.1111/j.1365-2265.2006.02524.x
Mathiesen JS, Kroustrup JP, Vestergaard P, Stochholm K, Poulsen PL, Rasmussen AK, Feldt-Rasmussen U, Gaustadnes M, Orntoft TF, Rossing M, Nielsen FC, Albrechtsen A, Brixen K, Godballe C, Frederiksen AL (2017) Founder effect of the RET(C611Y) mutation in multiple endocrine neoplasia 2A in Denmark: a nationwide study. Thyroid. https://doi.org/10.1089/thy.2017.0404
Frank-Raue K, Rybicki LA, Erlic Z, Schweizer H, Winter A, Milos I, Toledo SP, Toledo RA, Tavares MR, Alevizaki M, Mian C, Siggelkow H, Hufner M, Wohllk N, Opocher G, Dvorakova S, Bendlova B, Czetwertynska M, Skasko E, Barontini M, Sanso G, Vorlander C, Maia AL, Patocs A, Links TP, De Groot JW, Kerstens MN, Valk GD, Miehle K, Musholt TJ, Biarnes J, Damjanovic S, Muresan M, Wuster C, Fassnacht M, Peczkowska M, Fauth C, Golcher H, Walter MA, Pichl J, Raue F, Eng C, Neumann HP, International RETEC (2011) Risk profiles and penetrance estimations in multiple endocrine neoplasia type 2A caused by germline RET mutations located in exon 10. Hum Mutat 32(1):51–58. https://doi.org/10.1002/humu.21385
Crona J, Taieb D, Pacak K (2017) New perspectives on pheochromocytoma and paraganglioma: toward a molecular classification. Endocr Rev 38(6):489–515. https://doi.org/10.1210/er.2017-00062
Cosci B, Vivaldi A, Romei C, Gemignani F, Landi S, Ciampi R, Tacito A, Molinaro E, Agate L, Bottici V, Cappagli V, Viola D, Piaggi P, Vitti P, Pinchera A, Elisei R (2011) In silico and in vitro analysis of rare germline allelic variants of RET oncogene associated with medullary thyroid cancer. Endocr Relat Cancer 18(5):603–612. https://doi.org/10.1530/ERC-11-0117
Machens A, Hauptmann S, Dralle H (2009) Modification of multiple endocrine neoplasia 2A phenotype by cell membrane proximity of RET mutations in exon 10. Endocr Relat Cancer 16(1):171–177. https://doi.org/10.1677/ERC-08-0096
Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW, Tomassetti P, Tonelli F, Wells SA Jr, Marx SJ (2001) Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86(12):5658–5671. https://doi.org/10.1210/jcem.86.12.8070
Skordis N, Neocleous V, Kyriakou A, Efstathiou E, Sertedaki A, Philibert P, Phylactou LA, Lumbroso S, Sultan C (2010) The IVS1-2A > G mutation in the SRD5A2 gene predominates in Cypriot patients with 5alpha reductase deficiency. J Endocrinol Invest 33(11):810–814. https://doi.org/10.1007/bf03350347
Dean G, Chamberlain S, Middleton L (1988) Friedreich’s ataxia in Kathikas-Arodhes, Cyprus. Lancet 1(8585):587
Voskarides K, Patsias C, Pierides A, Deltas C (2008) COL4A3 founder mutations in Greek-Cypriot families with thin basement membrane nephropathy and focal segmental glomerulosclerosis dating from around 18th century. Genet Test 12(2):273–278. https://doi.org/10.1089/gte.2007.0110
Voskarides K, Mazieres S, Hadjipanagi D, Di Cristofaro J, Ignatiou A, Stefanou C, King RJ, Underhill PA, Chiaroni J, Deltas C (2016) Y-chromosome phylogeographic analysis of the Greek-Cypriot population reveals elements consistent with Neolithic and Bronze Age settlements. Investig Genet 7:1. https://doi.org/10.1186/s13323-016-0032-8
Acknowledgements
This work was supported by the A.G. Leventis Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Bioethics approval was received from the Cyprus National Ethics Committee.
Informed consent
Informed consent was obtained from all individual participants in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fanis, P., Skordis, N., Frangos, S. et al. Multiple endocrine neoplasia 2 in Cyprus: evidence for a founder effect. J Endocrinol Invest 41, 1149–1157 (2018). https://doi.org/10.1007/s40618-018-0841-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-018-0841-0